

1. Introduction: Prenylated Indole Alkaloids
The diverse family of prenylated indole alkaloids containing a bicyclo[2.2.2]diazaoctane core isolated from both terrestrial and marine fungi have been the subject of intense research efforts due to their complex molecular structures and often potent biological activities (Figure 1).1–2 Members of this family of prenylated indole alkaloids have been reported to display insecticidal, antitumor, anthelmintic, calmodulin inhibitory, and antibacterial properties.3,4,5 In addition to the diverse biological activity exhibited by these natural products, their structural diversity is also notable. Two distinct stereochemistries have been observed with respect to the relative configuration at the C19-stereogenic center (sclerotiamide numbering), either an anti-configuration (e.g. 9 and 10) or a syn-configuration (e.g. 1 and 2).
Figure 1.

Various members of the family of prenylated indole alkaloids
Despite this structural diversity, all of these compounds share a bicyclo[2.2.2]diazaoctane core, which is thought to arise biosynthetically through a hetero-Diels-Alder reaction in Nature. Sammes and Birch originally proposed that an intramolecular Diels-Alder reaction of a 5-hydroxypyrazin-2(1H)-one could be responsible for the bioynthesis of this structural framework in the natural product brevianamide A (9) (Scheme 1),6,7 but others doubted that the unactivated dienophile would undergo such a transformation. Through extensive synthetic work carried out in the Williams research group, an extensive body of provocative indirect and corroborating experimental evidence to support a biosynthetic Diels-Alder reaction has been accumulated and intensive research is still devoted to determining whether this transformation is enzyme-catalyzed.
Scheme 1.

Proposed biosynthetic Diels-Alder reaction
This review will cover the major synthetic strategies developed over the last three decades to prepare the bicyclo[2.2.2]diazaoctane core found in the vast number of these prenylated alkaloids. Described will be a number of key disconnections for assembling the bicyclo[2.2.2]diazaoctane core, and these will be presented in essentially chronological order. The first major strategy developed for the synthesis of this common core structure was the intramolecular SN2′ cyclization, and this method enabled the syntheses of a number of complex paraherquamide and stephacidin alkaloids. This was followed by the biomimetic Diels-Alder disconnection, and recent efforts have focused on other bond connections mostly involving radical cyclizations.
2. Intramolecular SN2′ Cyclization
The Williams research group developed an intramolecular SN2′ cyclization reaction that proved to be useful in the preparation of a number of prenylated indole alkaloids. The first generation of this technology was employed in the synthesis of brevianamide B (10) (Scheme 2).8 Starting with the known allylated proline derivative 12, nucleophilic ring opening provided the PMB-protected amide 13. Subsequent acylation of the secondary amine and ring closure installed the diketopiperazine ring in 14. The double bond of the allyl group was then oxidatively cleaved with ozone followed by a Wittig olefination and aldehyde reduction to give the primary alcohol 16. Protection of the alcohol as the corresponding silyl ether and deprotonation of the diketopiperazine followed by quenching with methyl chloroformate led to the key methyl ester 17. When the methyl ester 17 was treated with gramine and PBu3 under the conditions of Somei and Kamatani, the coupled product 18 was obtained, and a straightforward four-step sequence was used to remove the methyl ester and transform the protected primary alcohol to the analogous primary halide 19. This compound served as the key intramolecular SN2′ cyclization substrate, and extensive optimization was required to obtain the bicyclo[2.2.2]diazaoctane core in a highly stereoselective fashion. When the precursor 19 was treated with NaH in DMF at room temperature, a 2:1 ratio of 20 and its epimer were obtained in 62% combined yield. Alternatively, simply changing the solvent to hot benzene and using NaH again as the base led to a complete reversal in selectivity giving the epimer as the major product in a 97:3 ratio and 82% combined yield. Interestingly, the same conditions (benzene, NaH) with the addition of 18-crown-6 gave the desired epimer 20 as the major product in a 4:1 ratio and 56% combined yield. The authors propose that the product ratio is governed by the ability of the solvent or crown ether to adequately solvate the sodium cation of the amide enolate. Without solvent stabilization, the chloride and sodium cation are closely associated in the transition state leading to the syn-product as the major isomer. Alternatively, when the sodium cation is stabilized by DMF or 18-crown-6, this creates an unfavorable steric interaction between the solvent complex and the chloride resulting in a preference for the opposite transition state. With the development of an anti-selective route to the bicyclo[2.2.2]diazaoctane core 20 in hand, the completion of the synthesis of brevianamide B (10) was undertaken. Treatment of the compound 20 with concentrated HCl led to indole deprotection and olefin/cation cyclization to give the hexacyclic indole compound 21, and transformation to the corresponding indoxyl 22 was effected by sequential treatment of 21 with mCPBA and NaOMe. Deprotection of the PMB-group proved to be difficult, and standard oxidation protocols to remove this protecting group were ineffective. Fortunately, deprotonation of the PMB-group in the benzyllic position with tBuLi followed by quenching with oxygen gave brevianamide B (10) in modest yield.
Scheme 2.

Williams’ total syntheis of brevianamide B.
Williams and coworkers later applied the same intramolecular SN2′ cyclization strategy to complete the synthesis of paraherquamide B (28) (Scheme 3).9 The allylic chloride 23 was prepared in a similar fashion to the key cyclization substrate 19 above. Since paraherquamide B (28) contains an syn-bicyclo[2.2.2]diazaoctane core as opposed to the anti-configuration in brevianamide B (10), the syn-selective intramolecular SN2′ cyclization conditions (NaH, benzene) were employed. In this case, exclusive syn-selectivity was observed regardless of the orientation of the starting material, and 24 was obtained as a single enantiomer. Unfortunately, due to the increased functionality present in paraherquamide B (28), the strong protic acid conditions used to cyclize the indole substrate 20 in the above synthesis of brevianamide B (10) were not effective. Many protic and Lewis acids were screened, and none gave any of the cyclized compound 25. However, Pd(II)-mediated cyclization was effective to provide 25,10 and under these conditions the lactim ether was also cleaved. Selective reduction of the tertiary amide in the presence of the secondary amide followed by methylation of the secondary amide and global deprotection gave 26. The spriooxidation was accomplished by treatment of the indole 26 with tBuOCl to give an intermediate chloroindolenine, which underwent hydration in the presence of TsOH to give the spirooxindole 27. Dehydration of 27 with MTPI and DMPU gave (+)-paraherquamide B (28). A similar strategy was deployed by the Williams laboratory in the first asymmetric total synthesis of paraherquamide A.11
Scheme 3.

Application of the intramolecular SN2′ cyclization to paraherquamide B.
The Williams group once again used an intramolecular SN2′ cyclization to prepare stephacidin A (3), avrainvillamide (39), and stephacidin B (4) (Scheme 4), and these syntheses allowed the development of a new strategy to install the allylic chloride utilizing a key cross-metathesis.12 From the advanced protected tryptophan derivative 29, standard amino acid coupling with allyl proline 30 followed by microwave heating led to Boc-deprotection and amide formation to provide 32. After amide protection as a lactim ether and indole protection as the Boc-carbamate 33, they were poised to explore the cross-metathesis. Thus, when 33 was treated with methacrolein and 5 mol% of the Hoveyda-Grubbs 2nd generation catalyst 34,13 the aldehyde 35 was obtained in excellent yield. Interestingly, microwave heating greatly accelerated the rate of this reaction; conventional heating required 48 hours whereas the same yield was obtained in 20 minutes under microwave heating. By employing a cross metathesis to install this aldehyde, the synthesis was significantly streamlined when compared to the previously described syntheses of brevianamide B (10) and paraherquamide B (28). Reduction of the aldehyde and conversion to the allylic chloride gave 36, which underwent a syn-selective intramolecular SN2′ cyclization to give 37. In all of these highly syn-diastereoselective intramolecular SN2′ cyclization reaction s, the authors have proposed a tight, intramolecular contact ion pair-driven closed transition state model (A) to explain the observed syn-selectivity. In the brevianamide B synthesis, which gave a modest anti-diastereoselectivity in the SN2′ cyclization, 18-Crown-6 ether was added that effectively broke open this closed transition state to effect cyclizations from the other face of the allylic π-system. In this context, the authors proposed that the crown ether weakly coordinates to the enolate counterion (Na+ in that case) providing a sterically demanding environment in the sphere of the enolate oxygen atom and thus favoring cyclization from the alternate rotamer of the allylic halide.
Scheme 4.

Intramolecular SN2′ cyclization approach toward the stephacidins.
Palladium-mediated cyclization from 37 gave the heptacycle 38 and thermal removal of the indole Boc-group gave (−)-stephacidin A (3). This remarkably concise and efficient synthesis required a mere seventeen steps compared to Baran’s synthesis of 3 in twenty-nine steps (vide infra). The key to the brevity of this synthesis was the cross-metathesis strategy to install the allylic chloride, and the efficient and well-established intramolecular SN2′ cyclization technology. Following chemistry originally discovered by Baran and Myers, synthetic stephacidin A was used to prepare totally synthetic avrainvillamide (39) and totally synthetic stephacidin B (4) in two and three steps respectively.
3. Biomimetic Diels-Alder Reactions
The first application of a biomimetic Diels-Alder reaction in the context of the formation of the bicyclo[2.2.2]diazaoctane core common to the prenylated indole alkaloids discussed above was demonstrated in the biomimetic total synthesis of brevianamide B (10) (Scheme 5).14 Starting with epi-deoxybrevianamide E (40) formation of the lactim ether 41 was followed by oxidation to give the Diels-Alder precursor 42. Upon treatment with aqueous KOH, 42 tautomerized to the azadiene 43, which underwent intramolecular Diels-Alder to give a 2:1 mixture of diastereomers 44 and 45. The minor diastereomer was transformed to 10 by oxidation, pinacol, and lactim ether deprotection. This total synthesis was the first to confirm the Diels-Alder reaction proposed in the biosynthesis of 10.
Scheme 5.

Experimental confirmation of the proposed Diels-Alder reaction.
The biomimetic IMDA reaction was also applied toward the racemic synthesis of VM55599 (58) (Scheme 6).15 The authors were particularly interested in the effect that the methyl group on the proline ring would have on Diels-Alder diastereoselectivity. Coupling of the protected reverse prenylated tryptophan compound 47 with racemic β-methyl-β-hydroxyproline ethyl ester 48 gave the peptide 49. Cleavage of the Boc-group followed by cyclization provided the diketopiperazine 50, and dehydration with SOCl2 gave the enamide 51. Treatment with Meerwein’s salt gave the lactim ether 52, which underwent tautomerization and Diels-Alder reaction under basic conditions to give a mixture of all four possible diastereomers 54–57. Lactim ether deprotection and reduction of the tertiary amide gave VM55599 (58) in good yield.
Scheme 6.

Synthesis of VM55599.
In contrast to the above strategy for the generation azadienes under basic conditions, Liebscher and coworkers developed alternate, neutral conditions to prepare these azadienes in an attempt to determine the effect of the reaction conditions on the Diels-Alder diastereoselectivity (Scheme 7).16 Horner-Wadsworth-Emmons reaction of the indole aldehyde 59 with the phosphonate 60 provided the unsaturated compound 61. Treatment of 61 with neat acetyl chloride for 20 days gave the Diels-Alder cycloadduct 62 as a single distereomer. This result was particularly notable due to the exclusive diastereoselectivity, and the precedent established by this experiment was promising within the context of the diastereocontrolled synthesis of many prenylated indole alkaloids.
Scheme 7.

Liebscher’s Diels-Alder work.
The Diels-Alder precursor protocol developed by Liebscher soon proved to be of use in the asymmetric total synthesis of VM55599 (58) (Scheme 8).17 In order to prepare 58 in a stereocontrolled fashion, a new azadiene precursor was required, since the dehydro-β-methylproline precursor 52 used in the above racemic synthesis led to loss of the stereochemistry at the C-14 methyl group. However, Williams realized that a dehydrotryptophan could also be employed as a Diels-Alder precursor, and as a result, control of this key stereocenter could be achieved. The S-stereochemistry of the C-14 methyl group in 58 was derived from L-Ile, and Hoffman- Loeffler-Freytag conditions gave the protected methyl proline 63 in 45% overall yield. Saponification, amino acid coupling to glycine, Boc-deprotection, and ring closure led to the enantiopure diketopiperazine 65. Protection of the secondary amide and condensation with aldehyde 67 led to a mixture of epimers 68 in good overall yield, and a two-step sequence led to deprotection of the MTM-group and dehydration to give exclusively the Z-isomer 69.
Scheme 8.

Synthesis of the key diketopiperazine.
Treatment of 69 with acetyl chloride for two weeks gave a mixture of three diasteromeric cycloadducts, 72, 73, and 74 (Scheme 9). The authors suggest that 69 undergoes acylation to give the O-acyl lactim 70 that tautomerizes to furnish the azadiene 71, which then undergoes intramolecular Diels-Alder reaction from three of the four possible transition states followed by loss of acetate. Reduction of the major diastereomer 73 with excess DIBAL-H provided (−)-VM55599 (58), and allowed the absolute stereochemistry of this natural product to be rigorously assigned.
Scheme 9.

Completion of the asymmetric total synthesis of (−)-VM55599.
Since many of these natural products only differ in the substitution on the indole ring, the preparation of the bicyclo[2.2.2]diazaoctane core separately would allow the rapid synthesis of a large number of these compounds in a highly convergent fashion. A model study aimed at studying the diastereoselectivity of the biomimetic Diels-Alder reaction led to a concise synthesis of just such a bicyclo[2.2.2]diazaoctane core 84, and the key bicyclic compound 84 could be used to rapidly prepare brevianamide B (10) (Scheme 10).18 Conjugate addtion of 1,3-dithiane-2-carboxylate 77 to the known ketone 76 gave the ester 78, and saponification and amino acid coupling provided 81 in good yield. Deprotection of the dithiane led to a mixture of the diketopiperazine 83 and the uncyclized amide 82, and the mixture was treated with excess AlCl3 to give the Diels-Alder cycloadduct 84. Interestingly, the stereochemistry of the cycloadduct 84 was exclusively the anti-configuration in contrast to the mixtures of syn- and anti-cycloadducts observed in the syntheses of VM55599 (58) and brevianamide B (10) presented earlier. Molecular modeling was used to justify this stereochemical preference. The Diels-Alder cycloadduct 84 is poised to undergo a Fischer indole synthesis, and treatment of 84 with phenyl hydrazine followed by ZnCl2 gave 85 in a good yield. Oxidative rearrangement of this known intermediate using previously reported conditions gave brevianamide B (10). Since only the anti-configuration is obtained in the Diels-Alder reaction, only natural products containing the anti-stereochemistry can be prepared by this IMDA/Fischer indole strategy. Also, the harsh conditions of the Fischer indole synthesis also limit the utility of this synthetic approach.
Scheme 10.

IMDA/Fischer indole strategy.
A biomimetic Diels-Alder reaction disconnection also proved to be useful in rendering the total synthesis of stephacidin A (3) more efficient and concise (Scheme 11).19 The reverse prenylated tryptophan derivative 86 was prepared in 11 steps from 6-hydroxyindole, and was coupled with cis-3-hydroxyproline ethyl ester hydrochloride 87 to give the peptide 88. Fmoc deprotection was accompanied by amide formation to give 89, which underwent Mitsunobu dehydration to give the enamide 90. Protection as the corresponding lactim ether 91 was followed by intramolecular Diels-Alder reaction to give a mixture of cycloadducts 92 and 93 with the syn-isomer 93 predominating. Deprotection of the major isomer 93 under acidic conditions gave stephacidin A (3) in only 17 steps and 5.4% overall yield.
Scheme 11.

Concise total synthesis of stephacidin A.
The Diels-Alder reactions described above all employed alkylated or acylated azadienes as synthetic intermediates, and the first example of a Diels-Alder reaction of a 5-hydroxypyrazin-2(1H)-one, the diene originally proposed as a biosynthetic intermediate in the preparation of the bicyclo[2.2.2]diazaoctane core in Nature in 1970, was not demonstrated in the laboratory until 2007!20 Williams found that starting with the previously reported, unprotected diketopiperazine 89, treatment with excess PBu3 and DEAD led to dehydration as well as tautomerization and Diels-Alder reaction to give 3 and its epimer (Scheme 12).
Scheme 12.

A more efficient total synthesis of stephacidin A.
Various control experiments showed that both PBu3 and DEAD were required in equal amounts in excess for the enamide to undergo Diels-Alder reaction. Control reactions showed neither PBu3 nor DEAD alone successfully mediated the Diels-Alder reaction. The authors suggest that the zwitterionic complex formed from attack of PBu3 on DEAD acts as a proton chaperone as shown in Figure 2.
Figure 2.
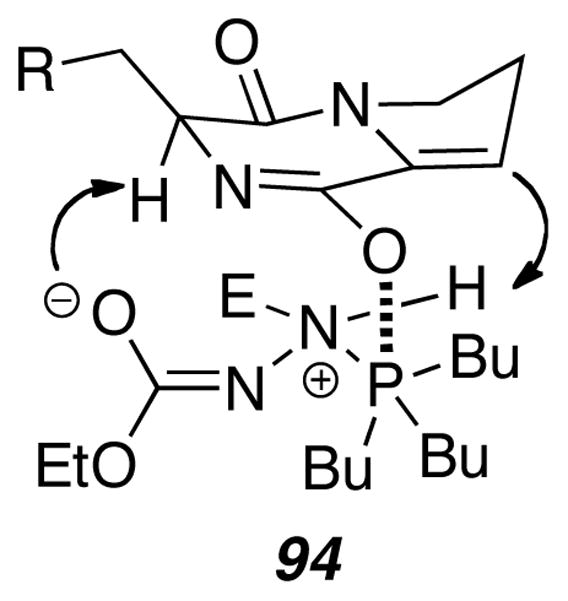
Proposed proton chaperone.
The synthetic intermediate 86 that was used in the above stephacidin A (3) synthesis was also employed in the synthesis of marcfortine C (103) by a biomimetic Diels-Alder reaction (Scheme 13).21 Coupling of 86 with the pipecolic acid derivative 95, and subsequent Fmoc-deprotection was accompanied by diketopiperazine formation to give 97. Heating in the presence of PBu3 and DEAD gave a mixture of cycloadducts favoring the syn-diastereomer 99. Amide reduction in the presence of excess DIBAL-H gave 100, and amine salt formation followed by indole oxidation and pinacol-type rearrangement gave marcfortine C (103).
Scheme 13.

Total synthesis of marcfortine C.
The biomimetic Diels-Alder reaction of a 5-hydroxypyrazin-2(1H)-one was also used as the key reaction in the total synthesis of both malbrancheamide (110) and malbrancheamide B (111) (Scheme 14).22 The enamides 104 and 105 were individually treated with aqueous KOH in MeOH to give the syn-cylcoadducts 106 and 108, as well as the corresponding anti-epimers 107 and 109 respectively. From the syn-cycloadducts 106 and 108, treatment with excess DIBAL-H gave either malbrancheamide (110) or malbrancheamide B (111). These syntheses also allowed the preparation of 13C labeled malbrancheamide B and pre-malbrancheamide, and these biosynthetic precursors were employed in feeding studies.23
Scheme 14.

Diels-Alder reactions to synthesize the malbrancheamides.
The natural products (+)- and (−)-versicolamide B (8) are particularly intriguing from a biosynthetic perspective. Each of the enantiomeric natural products are produced by a different species within the same Aspergillus genus raising the possibility that each species has evolved enantiomerically distinct genes that direct the biosynthesis of either (+)- or (−)-versicolamide B (8). In order to gain support for a Diels-Alder biosynthesis, the total synthesis of both (+)- and (−)-versicolamide B (8) were undertaken (Scheme 15).24 Starting with the cis-diastereomer of 89, which was previously used in the stephacidin A (3) synthesis, indole oxidation accompanied by pinacol-type rearrangement gave a mixture (3:1) of the chromatographically separable oxindoles 112 and 113. Each was dehydrated under the previously reported Mitsunobu conditions to give the enamides 114 and 115.
Scheme 15.

Oxindole substrates.
Diels-Alder reaction under basic conditions furnished (+)-versicolamide B (8) from 114 and identical conditions gave (−)-versicolamide B (8) from 115 (Scheme 16). Each was accompanied by a minor diastereomeric cycloadduct 116, which like versicolamide B (8) also possesses an anti-configuration. The exclusive preference for the anti-cycloadducts is particularly striking considering that all previous cycloadditions utilizing indole-based azadienes display modest syn-selectivity (typically ~2.5:1; syn:anti). Ab initio calculations suggest that the exclusive anti-selectivity is due to a particularly stable transition state leading to the anti-cycloadduct when an oxindolic azadiene is used, whereas the corresponding indolic azadiene reacts through roughly equally stable syn- and anti-transition states.25
Scheme 16.

Biomimetic, asymmetric total synthesis of (+)- and (−)-versicolamide B.
4. Other Synthetic Strategies
Trost and coworkers devised a novel and concise route to the bicyclo[2.2.2]diazaoctane core of marcfortine B (103) via a key trimethylenemethane [3+2]-cycloaddition (TMM) and radical cyclization reaction (Scheme 17).26 Starting with the oxindole 117, synthesis of the TMM acceptor 118 was carried out in two steps. Clean cycloaddition and methylation of the resulting carboxylic acid provided the desired cyclopentane ring in 120. Epoxidation and elimination gave the allylic alcohol 121, and mesylation, displacement with the pipecolic acid derivative 122, and elimination gave the enamide 123. Michael addition of the amide nitrogen on the α,β-unsaturated ester gave the cyclized product 124 as a single diastereomer in quantitative yield. The authors speculate that the selectivity results from shielding of the re-face of the Michael acceptor by the aromatic portion of the molecule, and internal protonation of the resultant ester enolate by the amide hydrogen gave the observed stereochemistry. Reduction of the ester with DIBAL-H and installation of the xanthate ester 125 using standard conditions occurred without incident. Thus the stage was set for the key radical cyclization. Treatment of 125 with AIBN and Bu3SnH generated the corresponding primary radical, which attacked the enamide double bond to give 126. Subsequent trapping of the secondary radical with AIBN, C-H abstraction, and fragmentation are proposed to account for the newly formed double bond in 126. A number of subsequent steps including hydrogenation of the double bond and installation of the final seven-membered ring were required to complete the synthesis of 103.
Scheme 17.

Trost’s radical cyclization approach.
In the context of the synthesis of stephacidin A (3), stephacidin B (4), and avrainvillamide (39), Baran and coworkers developed an oxidative enolate coupling that proved useful as a key step for assembling the bicyclo[2.2.2]diazaoctane core of these natural products (Scheme 18). The Baran group explored various lower yielding approaches to stephacidin A (3)27,28 before ultimately reporting the most recent synthesis in 2006.29 The orthogonally protected tryptophan derivative 126 was prepared using a newly developed Heck-type coupling of an iodoaniline and a glutamate aldehyde. The protected amino acid was coupled to the proline derivative 127 under standard conditions to provide 128, which underwent carbamate deprotection, diketopiperazine formation, and subsequent protection of the secondary amide as the corresponding MOM-ether to give the key oxidative enolate coupling substrate 129. In order to optimize the intramolecular oxidative enolate coupling, the authors screened a number of oxidizing agents and found that Fe(acac)3 was the most effective. Thus, treatment of 129 with LDA and Fe(acac)3 led to efficient coupling and subsequent deprotection of the MOM-ether gave 130. Completion of the synthesis involved conversion of the methyl ester to the corresponding disubstituted alkene 131, which underwent thermal Boc-deprotection and cyclization to provide stephacidin A (3) in modest and variable yield. The authors speculate that this cyclization involves initial Boc-deprotection followed by a formal ene reaction to give a spirocyclic intermediate, which undergoes a 1,2-shift to provide the natural product. From stephacidin A (3), the authors also prepared avrainvillamide (39) and stephacidin B (4).
Scheme 18.

Baran’s total synthesis of stephacidin A.
Myers’ approach to avrainvillamide (39) and stephacidin B (4) involved a key aminoacyl radical cyclization to assemble the bicyclo[2.2.2]diazaoctane core and subsequent transition metal coupling and nitrone formation to install the last three rings (Scheme 19).30 The enantioenriched cyclohexanone 132 was readily available in a few steps from commercially available 1,4-cyclohexanedione monoketal. Alkylation of the ketone enolate with the isopropylsulfonate 133 led to a single diastereomer 134. Interestingly, alkylation using the corresponding methanesulfonate led to diminished yields, and the authors suggest that competitive proton transfer from the methanesulfonate group could account for such a result. Next, a Strecker-like addition of hydrogen cyanide gave the amino nitrile 135 in 65% yield as well as the distereomeric amino nitrile in 16% yield. The major isomer 135 underwent epimerization of the α-carbon of the ketone to establish the stereochemistry in avrainvillamide (39) and stephacidin B (4), and subsequent conversion of the nitrile to the corresponding primary amide under platinum catalysis gave 137 in good yield. The primary amide 137 was then treated with thiophenol under basic conditions resulting in conjugate addition of thiophenol and cyclic hemiaminal formation to afford 138. Cleavage of the N-Boc group was accompanied by dehydration of the cyclic aminal, and acylation of the free amine with 1-methyl-2,5-cyclohexadiene-1-carbonyl chloride to give 140. The specific acylating agent 139 was chosen due to the fact that it had been reported as an efficient acyl radical precursor.31 In fact, when 140 was heated with tert-amyl peroxybenzoate, the aminoacyl radical 141 is presumably formed, which then attacks the more-substituted carbon of the enamide C-C double bond and expels the phenlthiyl radical to give the bicyclo[2.2.2]diazaoctane core structure 142. Thus, the authors had completed the most synthetically challenging tetracyclic half of avrainvillamide (39), and completion of the synthesis would require building the second half. In order to achieve this goal, 142 underwent a three-step sequence to remove the silyl protecting group, oxidize the free alcohol to the ketone, and introduce the α-iodoenone to give coupling partner 143. Ullman coupling of 143 with the readily available aryl iodide 144 provided 145 in good yield, and installation of the unsaturated nitrone found in avrainvillamide (39) was carried out simply by reduction of the nitro-group of 145 with activated zinc to give 39. The authors also showed that avrainvillamide (39) readily dimerizes to stephacidin B (4) under a variety mild conditions firmly establishing their biosynthetic relationship.
Scheme 19.

Myers’ radical cyclization and nitrone synthesis.
Drawing on work originally developed on model systems,32,33 Simpkins and coworkers recently completed an elegant asymmetric total synthesis of (−)-malbrancheamide B (111) using a cationic cascade sequence (Scheme 20). Beginning with the easily accessible aldehyde 146, aldol reaction with the ester 147 gave the alcohol 148, and dehydration followed by saponification delivered the acid 149. Standard amino acid coupling with the prenylated proline derivative 150 gave the peptide 151 as a mixture of E- and Z-isomers, and acid mediated cyclization delivered the diketopiperazine 152. The authors employed a key cationic cascade strategy to prepare the hexacyclic intermediate 153. TMS-OTf simultaneously deprotected the indole nucleus, promoted the cyclization of indole on the prenyl group, and promoted the cyclization of the prenyl group on the diketopiperazine. From 153, reductive cleavage of the N-OBn bond was achieved with LiCl and SmI2, and reduction of 154 with DIBAL-H gave (−)-malbrancheamide B ((−)-111). While this synthesis is less efficient overall than Williams’ racemic synthesis of 111, the Simpkins group completed the synthesis in less synthetic steps and delivered enantiomerically pure 111.
Scheme 20.

Simpkins’ cationic cascade toward (−)-malbrancheamide B.
5. Miscellaneous
A number of research groups have also described novel methodologies for the preparation of bicyclo[2.2.2]diazaoctane ring systems that are notable even though these specific methods have not been employed in the preparation of natural products.
Sammes was the first to propose that a Diels-Alder reaction could be involved in the biosynthesis of the brevianamides (Scheme 1).6 In an effort to secure experimental support for such a Diels-Alder biosynthetic hypothesis, Sammes and coworkers prepared a pyrazine derivative 155, and treated this compound with a number of dienophiles (Scheme 21).34 Reaction of 155 with dimethylacetylenedicarboxylate gave the cycloadduct 156 in good yield. While this cycloaddition was promising, the proposed biosynthetic Diels-Alder occurs with a pyrazine and an unactivated dienophile. Thus, the reaction of 155 with an unactivated dienophile, norbornadiene, was undertaken providing the cycloadduct 157 in modest yield. This work was important in establishing the feasibility of the Diels-Alder biosynthetic construction in the laboratory, and forms the basis on which a number of natural product syntheses were later predicated.
Scheme 21.

Early work by Sammes.
In the course of some work on 1,3-dipolar cycloadditions of proline derivatives, Fabre and coworkers noted a side-reaction that afforded bicyclo[2.2.2]diazaoctane products.35 When an acylated proline contains a pyridine ring, such as 158, then treatment with acetic anhydride generates an azadiene 159 that undergoes a formal Diels-Alder reaction (although the authors designated this as a dipolar cycloaddition reaction) to give a mixture of cycloadducts 160 and 161. This interesting system can be viewed as an adventitious progenitor of total syntheses incorporating the Diels-Alder reactions of acylated pyrazines that followed publication of this report.16,17
A prelude to Williams’ intramolecular SN2′ cyclization involved a similar intramolecular ring opening of an epoxide to assemble the bicyclo[2.2.2]diazaoctane ring system common to the paraherquamides, brevianamides, and stephacidins.36 Starting with racemic protected homoserine 162, amide formation under standard conditions provided 164, which underwent reductive deprotection and cyclization to give the diketopiperazine 165. Protecting group manipulations and alcohol oxidation led to the aldehyde 167, and Corey–Chaykovsky conditions afforded the key epoxide substrate 168. Treatment of 168 with LiHMDS led to a mixture of 169 and 170 in a 2.8:1 ratio. Interestingly, a mixture of the six- and seven-membered products, 169 and 170 respectively, was obtained favoring the desired six-membered bicyclo[2.2.2]diazaoctane 169. While the epoxide-opening strategy did not prove optimal in the context of natural product synthesis, this work inspired the successful intramolecular SN2′ cyclization chemistry used in a number of notable syntheses discussed above.
6. Conclusion
The total synthesis of prenylated indole alkaloids has proven to be a fertile area for the development of a number of powerful synthetic methodologies. The brevianamides, paraherquamides, stephacidins, notoamides and malbrancheamides all present daunting synthetic challenges, and the solutions encompassed in the total syntheses described above have often proven widely useful. In many cases, these total syntheses have disclosed important information about the reactivity of proposed biosynthetic intermediates allowing the refinement of various biosynthetic proposals, and the synthetic technology developed has also informed the preparation of isotopically labeled proposed biosynthetic precursors that have been employed with success in incorporation studies. As the number of highly biologically active prenylated indole alkaloids isolated from Nature continues to grow, one can expect that the synthesis of these compounds will continue to be an active area of research.
Scheme 22.

Fabre’s intermolecular Diels-Alder reaction
Scheme 23.

Epoxide opening.
Acknowledgments
R.M.W. is grateful to the National Institutes of Health (CA70375) for providing financial support for the work reported from this laboratory on the brevianamides, paraherquamides, stephacidins, notoamides and malbrancheamides.
Biographies

Robert M. Williams was born in New York in 1953 and attended Syracuse University where he received the B.A. degree in Chemistry in 1975. He obtained the Ph.D. degree in 1979 at MIT (W.H. Rastetter) and was a postdoctoral fellow at Harvard (1979–1980; R.B. Woodward/Yoshito Kishi). He joined Colorado State University in 1980 and was named a University Distinguished Professor in 2002. His interdisciplinary research program (over 250 publications) at the chemistry-biology interface is focused on the total synthesis of biomedically significant natural products, biosynthesis of secondary metabolites, studies on antitumor drug-DNA interactions, HDAC inhibitors, amino acids and peptides.

Kenneth Aaron Miller (Pittsburgh, 1979) graduated from the University of Georgia in 2002 with a B.S. in Chemistry. He received is Ph.D. from the University of Texas - Austin in 2007 under the supervision of Prof. Stephen F. Martin, and continued his studies as a postdoctoral fellow at Colorado State University with Prof. Robert M. Williams. He joined the Chemistry Department at Fort Lewis College in 2009 where he is currently an Assistant Professor. His research interests include bioactive natural product synthesis and the development of green chemical methods for the synthesis of common heterocyclic scaffolds.
References
- 1.Williams RM, Cox RJ. Acc Chem Res. 2003;36:127–139. doi: 10.1021/ar020229e. [DOI] [PubMed] [Google Scholar]
- 2.Williams RM. Chem Pharm Bull. 2002;50:711–740. doi: 10.1248/cpb.50.711. [DOI] [PubMed] [Google Scholar]
- 3.Williams RM, Stocking EM, Sanz-Cevera JF. Topics Curr Chem. 2000;209:97–173. [Google Scholar]
- 4.Qian-Cutrone J, Huang S, Shu YZ, Vyas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q. J Am Chem Soc. 2002;124:14556–14557. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Luis S, Rodriguez R, Acevedo L, Gonzalez MC, Lira-Rocha A, Mata R. Tetrahedron. 2006;62:1817–1822. [Google Scholar]
- 6.Porter AEA, Sammes PG. J Chem Soc Chem Commun. 1970:1103. [Google Scholar]
- 7.Baldas J, Birch AJ, Russell RA. J Chem Soc Perkin Trans I. 1974:50. [Google Scholar]
- 8.Williams RM, Glinka T, Kwast E. J Am Chem Soc. 1988;110:5927–5929. [Google Scholar]
- 9.Cushing TD, Sanz-Cervera JF, Williams RM. J Am Chem Soc. 1993;115:9323–9324. [Google Scholar]
- 10.Trost BM, Fortunak MD. Organometallics. 1982;1:7–13. [Google Scholar]
- 11.Williams RM, Cao J, Tsujishima H, Cox RJ. J Am Chem Soc. 2003;125:12172–12178. doi: 10.1021/ja036713+. [DOI] [PubMed] [Google Scholar]
- 12.Artman GD, Grubbs AW, Williams RM. J Am Chem Soc. 2007;129:6336–6342. doi: 10.1021/ja070259i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- 14.Williams RM, Sanz-Cervera JF, Sancenon F, Marco JA, Halligan K. J Am Chem Soc. 1998;120:1090–1091. doi: 10.1016/s0968-0896(98)00102-3. [DOI] [PubMed] [Google Scholar]
- 15.Stocking EM, Sanz-Cervera JF, Williams RM. J Am Chem Soc. 2000;122:1675–1683. [Google Scholar]
- 16.Jin S, Wessig P, Liebscher J. J Org Chem. 2001;66:3984–3997. doi: 10.1021/jo0100897. [DOI] [PubMed] [Google Scholar]
- 17.Sanz-Cervera JF, Williams RM. J Am Chem Soc. 2002;124:2556–2559. doi: 10.1021/ja017425l. [DOI] [PubMed] [Google Scholar]
- 18.Adams LA, Valente MWN, Williams RM. Tetrahedron. 2006;62:5195–5200. [Google Scholar]
- 19.Greshock TG, Grubbs AW, Tsukamoto S, Williams RM. Angew Chem Int Ed. 2007;46:2262–2265. doi: 10.1002/anie.200604378. [DOI] [PubMed] [Google Scholar]
- 20.Greshock TJ, Williams RM. Org Lett. 2007;9:4255–4258. doi: 10.1021/ol701845t. [DOI] [PubMed] [Google Scholar]
- 21.Greshock TJ, Grubbs AW, Williams RM. Tetrahedron. 2007;63:6124–6130. doi: 10.1016/j.tet.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller KA, Welch TR, Greshock TJ, Ding Y, Sherman DH, Williams RM. J Org Chem. 2008;73:3116–3119. doi: 10.1021/jo800116y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding Y, Greshock TG, Miller KA, Sherman DH, Williams RM. Org Lett. 2008;10:4893–4866. doi: 10.1021/ol8019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller KA, Williams RM. Nature Chem. 2009;1:XXX–XXX. doi: 10.1038/nchem.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domingo LR, Zaragoza RJ, Williams RM. J Org Chem. 2003;68:2895–2902. doi: 10.1021/jo020564g. [DOI] [PubMed] [Google Scholar]
- 26.Trost BM, Cramer N, Bernsmann H. J Am Chem Soc. 2007;129:3086–3087. doi: 10.1021/ja070142u. [DOI] [PubMed] [Google Scholar]
- 27.Baran PS, Guerrero CA, Ambhaikar NB, Hafensteiner BD. Angew Chem Int Ed. 2005;44:606–609. doi: 10.1002/anie.200461864. [DOI] [PubMed] [Google Scholar]
- 28.Baran PS, Guerrero CA, Hafensteiner BD, Ambhaikar NB. Angew Chem Int Ed. 2005;44:3892–3895. doi: 10.1002/anie.200500655. [DOI] [PubMed] [Google Scholar]
- 29.Baran PS, Hafensteiner BD, Ambhaikar NB, Guerrero CA, Gallagher JD. J Am Chem Soc. 2006;128:8678–8693. doi: 10.1021/ja061660s. [DOI] [PubMed] [Google Scholar]
- 30.Herzon SB, Myers AG. J Am Chem Soc. 2005;127:5342–5344. doi: 10.1021/ja0510616. [DOI] [PubMed] [Google Scholar]
- 31.Jackson LV, Walton JC. Chem Commun. 2000:2327. [Google Scholar]
- 32.Pichowicz M, Simpkins NS, Blake AJ, Wilson C. Tetrahedron. 2008;64:3713–3735. [Google Scholar]
- 33.Frebault F, Simpkins NS, Fenwick A. J Am Chem Soc. 2009;131:4214–4215. doi: 10.1021/ja900688y. [DOI] [PubMed] [Google Scholar]
- 34.Machin PJ, Porter AEA, Sammes PG. J Chem Soc Perkin Trans. 1973;14:404–409. [Google Scholar]
- 35.Fabre JL, Farge D, James C, Lave D. Tetrahedron Lett. 1985;26:5447–5450. [Google Scholar]
- 36.Williams RM, Maruyama LK. J Org Chem. 1987;52:4044–4047. [Google Scholar]