

Summary of Recent Advances
Mast cells play as the major effector cells in immediate hypersensitivity through activation via the high-affinity IgE receptor, FcεRI, although many other functions have recently been discovered for this cell type. Given the broad array of proinflammatory mediators secreted from FcεRI-activated mast cells, as well as sensitization to allergens, IgE elevation, and increased mast cells in a majority of atopic dermatitis patients, mast cells are believed to be involved in the pathogenesis of atopic dermatitis. Numerous animal models have been used to study this epidemic disease. Here we review the recent progress to synthesize our current understanding of this disease and potential mechanisms for a mast cell's role in the disease.
Introduction
Mast cells are hematopoietic cells that originate from progenitor cells in the bone marrow. Mast cell progenitors enter the circulation and become mature mast cells after entering destination tissues under the influence of local microenvironment [1]. Mast cells have been long understood as the key effector cell type in IgE-mediated immediate hypersensitivity and allergic disorders, as well as in protective immune responses to certain parasites and bacteria [2,3]. However, recent progress in the field has broadened their roles in many immune responses [4], ranging from innate defense against venoms of bees and snakes [5] to multiple aspects of adaptive immune responses such as antigen presentation and leukocyte recruitment to draining lymph nodes [6] as well as downmodulation of immune responses [7]. Their pathogenic roles have been extended to include not only allergic diseases and helminth and bacterial infection, but also autoimmune diseases [8,9], allograft tolerance [10], angiogenesis in tissue repair [11], and carcinogenesis [12,13].
The high-affinity receptor for IgE (FcεRI) expressed on mast cells (and basophils) consists of four subunits (αβγ2): an IgE-binding α chain, a signal-amplifying, receptor-stabilizing β chain, and two disulfide-bonded γ chains that are the main signal transducers [14]. Upon encounter with multivalent antigen, IgE-bound FcεRI on mast cells become aggregated or crosslinked, leading to cell activation [15]. Activated mast cells secrete three classes of substances: (1) preformed chemical and protein mediators, such as histamine, serotonin, heparin and chondroitin sulfates, proteases, major basic protein, acid hydrolases, cathepsin, etc., (2) lipid mediators, such as prostaglandins, leukotrienes, and platelet-activating factor (PAF), and (3) preformed and/or de novo synthesized growth factors, cytokines, and chemokines, such as tumor necrosis factor (TNF)-α, TGF-β, MIP-1α, MCP-1, VEGF, IFN-α/β/γ, GM-CSF, IL-1α/β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, IL-15, IL-16, IL-18, IL-25, etc. (Fig. 1).
Figure 1.
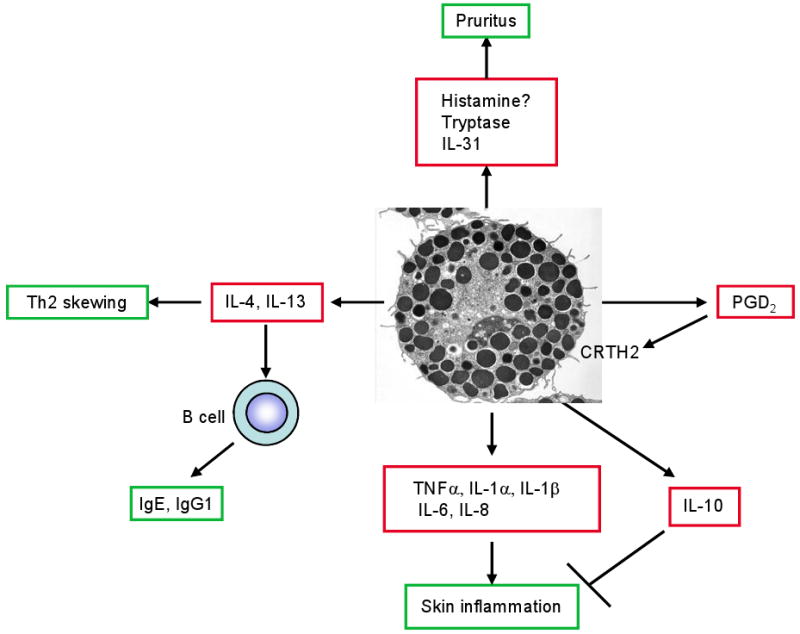
Mast cells secrete a wide array of preformed and de novo synthesized proinflammatory and immunomodulatory mediators upon stimulation by IgE and antigen or other stimuli. The mediators may contribute to various aspects of systemic and local immune responses in AD.
Anaphylactic responses to insect stings, injected medications, foods, and other agents are thought to be caused by IgE/antigen-dependent mast cell activation [16]. In addition to this classic pathway of systemic anaphylaxis [17,18], IgG antibodies can also induce anaphylaxis in a basophil-dependent manner in the mouse [19]. Histamine is primarily responsible for the development of shock in the classic pathway. In contrast, PAF is responsible in the latter alternative pathway. Despite the earlier controversy on the role of mast cells in asthma [20], recent studies clearly showed their importance [21,22]: mast cells can markedly enhance antigen-dependent airway hyperresponsiveness, airway eosinophil infiltration, and an increase in the number of proliferating cells in the airway epithelium that are induced in mouse models of asthma that either omit artificial adjuvants or use low doses of antigen challenge; mast cells' roles were also shown in a chronic asthma model [23]. In asthmatics, mast cells infiltrate airway smooth muscle and contribute to persistent inflammation in the airways [24-26]. The best evidence for the pivotal role of IgE and therefore that of FcεRI in human asthma came from clinical studies demonstrating that anti-IgE mAb Omalizumab decreases serum IgE levels and allergen-induced bronchoconstriction [27,28].
The most critical tool that has driven the high-paced findings is mast cell engraftment into mast cell-deficient animals, by which one can confirm that ‘defects’ or abnormal observations on mast cell-deficient animals can be reversed by providing exogenous mast cells [29]. Stem cell factor (SCF) is a critical growth and differentiation factor for mast cell development [30,31]. c-Kit is its receptor tyrosine kinase. Loss-of-function mutations in gene loci for this ligand (Sl)/receptor (W) pair lead to profound reductions of mast cells. Mast cell-deficient KitW/W-v mice have been a choice for mast cell engraftment for more than two decades. However, these mice have various other hematopoietic and non-hematopoietic abnormalities and are cumbersome to breed. In contrast, another strain of mast cell-deficient mice, KitW-sh/W-sh does not have anemia or neutropenia and are easy to breed, therefore KitW-sh/W-sh mice have become a favorite animal to study the mast cell function. However, because of multiple cellular abnormalities in KitW-sh/W-sh and KitW/W-v mice, it is essential to demonstrate that abnormalities in these mice can be rectified by ‘knock-in’ experiments before one can conclude that such abnormalities are due to mast cell deficiency [32].
Atopic dermatitis (AD)
AD, or eczema, is a chronic or chronically relapsing, pruritic inflammatory skin disease. The incidence of this disease has been increasing for the last three decades and affects at least 15% of children [33-35]. Clinical manifestations vary with age: eczematous lesions emerge on the cheeks and the scalp in infancy. Scratching causes crusted erosions. Later in the childhood, lesions involve flexures, the nape, and the dorsal skin of the limbs. In adolescence and adulthood, lichenification (thickening of the skin with accentuated skin markings) develops in flexures, head, and neck. Throughout these ages, persistent itch that often causes sleep deprivation and disfiguration of the skin substantially impairs the quality of a patient's life.
AD is characterized by skin inflammation, impaired skin barrier function, and IgE-mediated sensitization to food and environmental allergens. Histologically, acute eczematous lesions exhibit spongiosis (epidermal intercellular edema), hyperkeratosis (thickening of the stratum corneum), and parakeratosis (retention of nuclei in the stratum corneum), and chronic lesions are characterized by acanthosis (diffuse epidermal hyperplasia) and perivascular infiltration of lymphocytes and mast cells.
The etiology of this disease is incompletely understood, but it is multifactorial and the disease is manifested by complex interactions between genetic and environmental factors. Currently, there are two major schools of hypothesis to explain the pathogenesis of this probably heterogeneous disease: (I) one assumes that the primary defect is an immune dysregulation that causes Th2-predominant inflammation and IgE-mediated sensitization. In the other hypothesis (II), an intrinsic defect in skin barrier function underlies as a primary cause of the disease. In the latter scenario, even the uninvolved phase of the disease presents with cutaneous hypersensitivity of nonlesional skin, resulting from a defective (probably genetically predisposed) skin barrier that allows the penetration of allergens and microbial pathogens [36]. The acute phase characterized by eczematous skin lesions is facilitated by allergen/IgE-bound Langerhans cells and by an infiltration of skin-homing Th2 cells. Eczematous skin lesions develop as the result of complex immune and inflammatory responses driven by the release of proinflammatory cytokines and chemokines from various resident cell types. In the immune dysregulation hypothesis (I), on the contrary, defective skin barrier is interpreted as a consequence of inflammation. Regardless how to explain the disease progression, the chronic phase is characterized by lichenification of skin, an infiltration of Th1 cells, and tissue remodeling with increased collagen deposition and dermal thickening.
Genetics of AD
Genome-wide association studies have identified several chromosomal loci as the possible locations of AD-associated genes, including 1q21, 3q21, 11q13, 16q, 17q25, 20p, and 3p26 [37,38]. These studies identified the AD susceptibility genes that support both (I) immune dysregulation and (II) impaired skin barrier function hypotheses: the involvement of IL4, IL13, IL18, and TIM1 genes supports the importance of CD4+ T cells and dysregulation of Th1 and Th2 genes in the pathophysiology of AD [39,40]. Recent studies also showed that thymic stromal lymphopoietin (TSLP), an IL-7-like cytokine, plays a crucial role in differentiation and maintenance of Th2 cells by activating myeloid dendritic cells (mDCs) [41]. TSLP-activated mDCs induce proliferation of CD4+ T cells, which differentiate into inflammatory Th2 cells that produce IL-4, IL-5, IL-13, and TNF-α, but little IL-10 [42]. TSLP is highly expressed in keratinocytes from AD patients [41], and transgenic (Tg) mice expressing TSLP in keratinocytes develop AD-like skin lesions [43,44]. On the other hand, impaired skin barrier function has been increasingly recognized as an underlying mechanism for the pathogenesis of AD [45,46]: SPINK5 (serine protease inhibitor, Kazal type 5) encodes a protease inhibitor LEKTI that is expressed in the uppermost epidermis and inhibits two serine proteases involved in desquamation [39,40]. Gene variants of SPINK5 [47-49] or filaggrin, a protein important for skin barrier and terminal differentiation of the epidermis, are linked to AD. Up to 15% of atopic dermatitis patients have mutations in the filaggrin gene [50-53]. The filaggrin gene is also mutated in the mouse model of AD, flaky tail [54].
Furthermore, involvement of the NOD1 and NOD2 genes, which encode cytosolic pathogen recognition receptors [39,40], and Toll-like receptors [39,40], suggest an important role for microbes in the pathogenesis of AD [55]. Indeed, AD patients often suffer from skin infections and a majority of the patients are colonized with Staphylococcus aureus, an infection thought to be critical for the pathogenesis and/or worsening of skin lesions [56,57]. Consistent with this, expression of antimicrobial peptides (LL-37 and HBD-2) in the epidermis is significantly decreased in AD patients, compared to psoriasis patients [17]. The combination of LL-37 and HBD-2 showed synergistic antimicrobial activity by effectively killing S. aureus. AD patients are also vulnerable to severe viral infections known as eczema vaccinatum and eczema herpeticum (EH) caused by vaccinia and herpes simplex viruses, respectively. A recent study showed that AD patients with a history of EH have a more severe Th2-polarized disease with greater allergen sensitization and more commonly have a history of food allergy, asthma, or both, compared to AD patients without EH [58]. AD patients with EH are more susceptible to cutaneous infections with S. aureus or molluscum contagiosum.
Mouse models of AD
Numerous mouse models of human AD have been developed since the first description of AD-like skin lesions in the dermatitis-prone NC/Nga mice in 1997 [59]. These models are essential for our current understanding of human AD, as they have been providing novel insights into the disease pathogenesis. There are two types of mouse models: (I) some mice develop skin lesions spontaneously; (II) in another type of model, skin lesions are induced by epicutaneous or intradermal challenge of immunized mice with a single allergen or a mixture of allergens. Another feature in the study of AD models is the use of genetically manipulated mice, but AD models of transgenic or gene-knockout mice either spontaneously develop skin lesions or are prone or resistant to skin lesion induction. As mouse AD models were reviewed in recent publications [60,61] and are reviewed in this issue, we discuss only major points below in the text and Table 1 (see our web site for a more comprehensive list: www.liai.org/pages/faculty-kawakami).
Table 1. Immunological features of mouse models of AD.
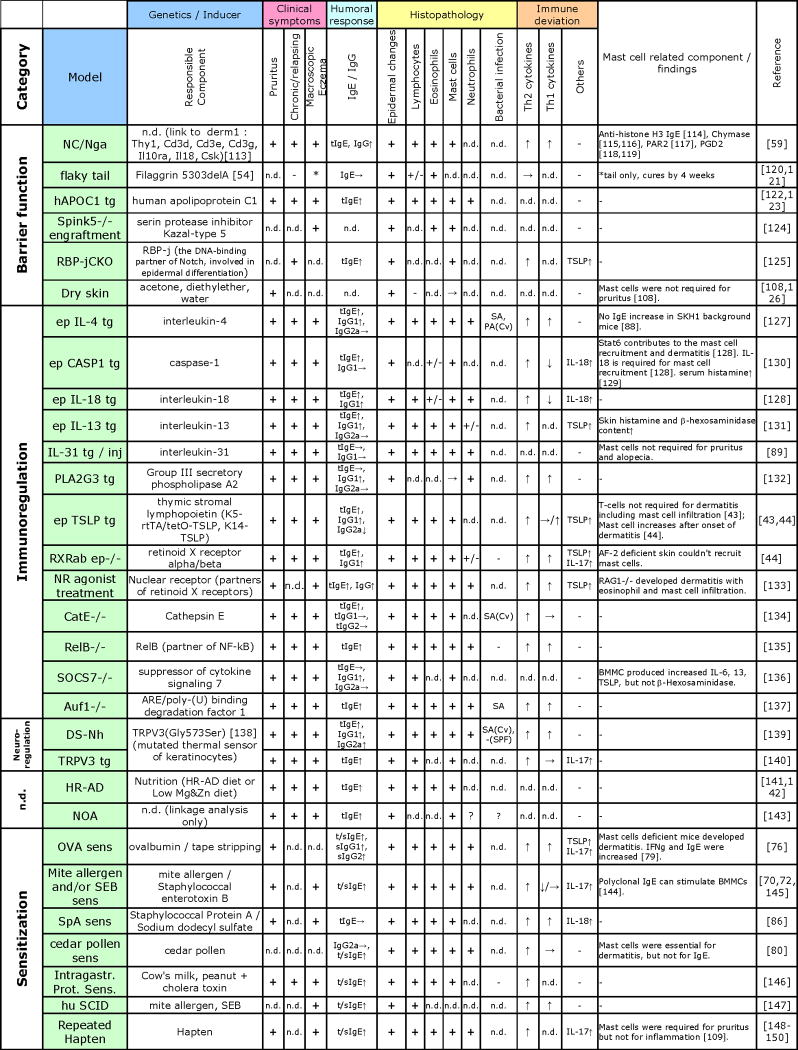 |
Abbreviations: tg, transgenic; inj, injection; tIgE, total IgE; sIgE, Ag-specific IgE; sens, sensitization; n.d., not determined or documented; Prot., protein; SA, Staphylococcus aureus; PA, Pseudomonas aeruginosa; Cv, conventional facility; SPF, specific pathogen free facility; ↑, increased; ↓, decreased; →, no change; BMMC, bone marrow derived mast cells
NC/Nga mouse model
NC/Nga mice spontaneously develop AD-like skin lesions under conventional (non-SPF [specific pathogen-free]) conditions [59]. Skin lesions in NC/Nga mice are characterized by infiltration of IL-4- and IL-5-producing CD4+ T cells, mast cells, and eosinophils, as well as high expression levels of Th2 chemokines (thymus and activation-regulation chemokine/CCL17 and monocyte-derived chemotactic cytokine/CCL22) and their receptor, CCR4 [62]. The elevation of serum IgE correlated with the onset of skin lesion development [59,62]. Constitutive activation of Jak3 may be involved in the hypersensitivity of B cells to IL-4 that leads to IgE elevation in these mice [63]. However, deficiency in Stat6, a critical signal transducer for IL-4 receptor, in NC/Nga mice did not inhibit skin lesion development [64]. Stat6-deficient NC/Nga mice had the skin microenvironment of IFN-γ production and an accumulation of IFN-γ-producing T cells in draining lymph nodes, suggesting that skin lesions in these mice are IgE- and Th2-independent. Under conventional conditions NC/Nga mice are infested with rodent mites [65,66], and the eradication of the mites with ivermectin (a broad-spectrum antiparasitic) leads to healing of skin lesions and reduced IgE levels [65]. However, the incidence of skin lesions in these mice under SPF conditions drastically varies from facility to facility (<5% in our facility). To overcome this problem, several groups including our own used mite extracts to induce dermatitis in NC/Nga mice [67-71]. By applying Dermatophagoides farinae (Der f; a common mite on humans) extracts and Staphylococcal enterotoxin B (SEB; a superantigen secreted by S. aureus) on the skin of NC/Nga and C57BL/6 (B6) mice, we established a highly efficient model to induce AD-like skin lesions with histologic and immunologic characteristics similar to those of spontaneously occurring skin lesions [72]. Using this dermatitis-inducible mice, we developed a mouse model of eczema vaccinatum [73]. Infection of eczematous skin with vaccinia virus induced severe erosive skin lesions, but not in the skin of healthy mice. Eczematous mice exhibited lower NK cell activity. The role of NK cells in controlling vaccinia virus-induced skin lesions was demonstrated by experiments depleting or transferring NK cells. IL-17 reduces NK cell activity in mice with preexisting dermatitis. Given low NK cell activities and increased IL-17 expression in AD patients, these results can explain the susceptibility of AD patients to eczema vaccinatum.
Tape-stripping/epicutaneous (EC) OVA sensitization model
EC exposure of protein antigens with or without disruption of the stratum corneum generally induces Th2 cytokines [74,75]. Raif Geha and colleagues developed an induction model in several strains including BALB/c and C57BL/6 mice by repeated EC sensitization of tape-stripped skin with ovalbumin (OVA) [76,77]. Dermatitis in this model mimics skin lesions of human AD in terms of infiltration of CD4+ T cells and eosinophils, and local expression of Th2 cytokines and chemokines. This model has been extensively used to study the cellular and molecular players in the development of skin lesions [61]. Interestingly, αβTCR (T cell receptor) T cells, but not γδTCR T, NKT, B, or mast cells, are required for the development in this model. Differential roles of IL-4, IL-5, and IFN-γ in skin lesion development and leukocyte infiltration were demonstrated using gene-manipulated mice, whereas IgE was not required for skin lesion development in this model [77]. In addition to these molecules, regulators that have been found to be positively involved in the development of skin lesions and inflammation in this model include TSLP, IL-10 (Th2 cytokine), CCR3 (chemokine receptor expressed in eosinophils), CCR4 (chemokine receptor expressed in skin-homing T cells), and the complement component C3. Negative regulators of skin inflammation in this model include C3a receptor and cyclooxygenase 2. In parallel to an increased realization of the importance of the impaired skin barrier in AD pathogenesis, e.g., filaggrin mutations, this group also emphasizes their findings that mechanical injury, i.e., tape stripping in their model, is critical for the development of skin lesions. Interestingly, they found that skin injury induces changes in the local environment that nurture Th2 and Th17 differentiation by increased expression of cytokines (IL-6, IL-23, IL-1, and IL-10), chemokines, and other genes [78].
Pathogenic roles of mast cells
As most studies showed increased numbers of mast cells in skin lesions in the AD models, it is generally assumed that mast cells contribute to skin inflammation. Unfortunately, however, few studies have directly addressed whether, to what extent, or by what mechanism mast cells play a role in spontaneous or induced development of AD-like skin lesions. An EC OVA sensitization study showed that skin inflammation is comparable in wild-type and KitW/W-v mice [79]. However, IFN-γ mRNA expression was increased in sensitized skin of KitW/W-v mice. In contrast, Der f/SEB induction experiments on KitW-sh/W-sh mice suggested a nonessential, but contributory role for mast cells to skin lesion development (T.A., M.K., and T.K., unpublished). Similarly, skin inflammation induced by EC sensitization with cedar pollen antigens was abolished in KitW/W-v and KitSl/Sl-d mice [80]. This cedar pollen dermatitis model was found to be independent of Stat6 and IgE, but dependent on CRTH2 (chemoattractant receptor homologous molecule expressed on Th2 cells), a prostaglandin D2 (PGD2) receptor. However, careful mast cell-reconstitution experiments have not been performed in either study. Interestingly, a recent study showed that FcεRI and FcRγ are involved in an EC OVA sensitization model [81]. However, what cell type is involved was not investigated in this study. Skin lesions were completely absent in FcRγ-deficient mice but partially inhibited in either FcεRIα- or FcγRIII/CD16-deficient mice. FcεRI controled both Th1 and Th2 skin responses, mast cell recruitment into draining lymph nodes, and IgE production. On the other hand, FcγRIII regulated only Th2 skin response, as well as T cell proliferation and IgG1 production. Considering the restricted expression of FcεRI in mast cells and basophils in mice and negative results on a mast cell's role in EC OVA sensitization experiments [79], these results collectively suggest that basophils might be involved in skin inflammation through the activation of FcεRI. However, a rigorous study needs to directly address this point using this induction protocol and to investigate apparent differences in the mast cell requirement between EC OVA sensitization- and Der f/SEB-induced dermatitis (or EC cedar pollen-induced dermatitis).
Although mast cells are increased in skin lesions of most AD cases and animal models, mastocytosis (an abnormal proliferation and accumulation of mast cells) is not associated with higher incidence of allergic diseases including AD [82]. Related to this issue, two kinds of keratinocyte-targeted K14-SCF Tg mice were generated: one produced both soluble and membrane-bound SCF, and the other produced only membrane-bound SCF [83]. The former (soluble/membrane) Tg mice caused cutaneous mastocytosis, but did not develop dermatitis.
Extrinsic vs. intrinsic AD – IgE in AD
A majority of AD patients have elevated serum IgE levels. In contrast with this so-called extrinsic AD, 20-30% of patients have low or normal levels of IgE. The latter, intrinsic AD, patients sometimes experience increases in serum IgE later in their life. Although AD might well be a syndrome caused by multiple etiologies (i.e., involving different combinations of multiple sets of cell types or multiple signaling pathways), it is also possible that the presence of intrinsic AD followed by a late-onset IgE elevation might suggests that IgE is not essential for the development of human AD, at least for the initiation of the disease. Several mouse studies support this notion: for example, Caspase 1 Tg mice, in which dermatitis development depends on IL-18, develop dermatitis even in the Stat6-deficient background. The aforementioned cedar pollen dermatitis does not require Stat6 or IgE [80], and skin lesions induced by Der f/SEB are normal in B cell-deficient mice (T.A., M.K., and T.K., unpublished). Furthermore, IL-18 has been increasingly recognized as an important player in AD pathogenesis. Clinically, IL-18 levels closely parallel disease severity [40,84]. Protein A (SpA), a virulence factor expressed on the surface of S. aureus, infection with which exacerbates AD, stimulates mouse keratinocytes to secrete IL-18 [85]. Disruption of the skin barrier with a subclinical dose of sodium dodecyl sufate, a detergent, plus daily EC application of SpA causes AD-like skin lesions in an IL-18-dependent manner [86]. This model does not induce elevation of serum IgE levels, thus similar to intrinsic AD. While treatment with detergent induces moderate Th1 cell response, additional SpA treatment is a prerequisite for differentiation of naive T cells toward unique Th1 cells, termed ‘super Th1 cells’, capable of producing both Th1 (IFN-γ) and Th2 (IL-13) cytokines and IL-3, as well as expressing CXCR3 (receptor for CXCL10 and CXCL11) and CCR5 (receptor for CCL3, CCL4, and CCL5). Induction of ‘super Th1 cells’ requires IL-18 stimulation. IL-18 was shown to be important for the development of infection-associated AD by induction of IL-3 from ‘super Th1 cells’. Cutaneous mastocytosis in this model is dependent on IL-18-dependent IL-3 production. Considering the increased Th1 cells in skin lesions of the chronic phase of AD and the prevalent infection with S. aureus, this model represent features of the chronic inflammation of human AD. Another potential player that counterbalance the Th2 predominance is IDECs (inflammatory dendritic epidermal cells), as these cells can produce IL-12 and IL-18 [87]. K14-IL-4-Tg/SKH1 mice, generated by crossing K14-IL-4-Tg/CByB6 mice with SKH1 (hairless, but apparently immunocompetent) mice, are another model of intrinsic AD [88]. The resulting hairless IL-4-Tg mice develop an inflammatory skin disease like that of the normally haired IL-4-Tg mice, accompanied by prominent skin infiltrations of T cells, mast cells, and eosinophils, as well as Th2 and Th1 cytokine up-regulation in chronic skin lesions. The inability of CD4+ T cells of the K14-IL-4-Tg/SKH1mice to up-regulate CD40L expression upon stimulation might account for their inability to up-regulate the IgE level. Another model of intrinsic AD is Tg mice overexpressing IL-31, a cytokine produced by activated T cells (Th2 cells express more IL-31 than Th1 cells) [89]. These models suggest that there can be multiple mechanisms for the development of intrinsic AD.
PGD2 and AD
Mast cells not only produce PGD2 abundantly upon FcεRI stimulation but also express a functional receptor CRTH2 for PGD2 [90]. PGD2 binds to two membrane receptors, D prostanoid receptor (DP)1 and DP2 (aka CRTH2). A DP2 agonist (13,14-dihydro-15-keto-PGD2) increases eosinophil recruitment at inflammatory sites and skin inflammation in an EC OVA sensitization model, whereas a DP1 agonist failed to induce eosinophil chemotaxis [91]. Administration of a CRTH2 antagonist, compound A, ameliorated skin inflammation caused by either EC OVA or FITC sensitization [92,93]. Compound A reduced total IgE, as well as antigen-specific IgE, IgG1, and IgG2a antibody levels. In compound A-treated mice, there were reduced activities of DCs to migrate to the draining lymph nodes and to stimulate naïve CD4+ T cells in response to FITC application to the skin. Ear-swelling responses induced by hapten-specific IgE and chronic contact hypersensitivity induced by repeated hapten application were less pronounced in CRTH2-deficient mice, compared to wild-type mice, whereas in vivo migration of Langerhans cells and DCs to regional lymph nodes was not impaired in CRTH2-deficient mice [94].
Pruritogenic role of mast cells in AD
Pruritus is one of the most prominent clinical features of AD and it disturbs the everyday life of AD patients. Itch-scratch vicious cycle also exacerbates the dermatitis by damaging the skin barrier and enhancing the itch [95]. This effect was also demonstrated in mouse models. Established dermatitis of NC/Nga mice was dramatically alleviated by clipping their toe-nails [96], while those with intact nails had sustained dermatitis. Interestingly, skin scratching was suggested to switch immune responses from Th2 to Th1 type in epicutaneously immunized mice [97]: mice sensitized epicutaneously with KLH showed Th2-biased immune response including expression of IgE and IL-13 in the local skin, whereas scratching on local abdominal skin using wire brush induced Th1 responses such as DTH reactions, increased IgG2a and IgG2b, and IFN-γ in the skin. This study suggests that scratching and its ensuing infection are important factor for the transition of the Th2-predominant acute phase to the chronic phase characterized by the presence of Th1 cells. Although mast cells respond to various AD-related stimuli, including IgE plus antigen, neuropeptides, bacterial components, and physical stimuli, the contribution of mast cells to the AD-related pruritus is not fully understood. Clinically, the major pruritogenic mediator from mast cells, i.e., histamine, turned out to be disappointing as a target of anti-itch therapeutics, because nonsedative antihistamines exhibited little effect on the eczema-related itch [98,99], while sedative antihistamines worked well both in human AD [98] and mouse AD models [99]. These results suggest the involvement of neurogenic components in the itch of AD. Since mast cells are closely located with the afferent neuron terminals in normal and inflamed tissues [100-103], functional interactions between mast cells and nerve fibers are implicated: tryptase is the primary protease produced by mast cells. Mast cell tryptase can cleave and activate its receptor, protease-activated receptor-2 (PAR-2), which is expressed on primary sensory nerves and keratinocytes [104-107]. The concentration and codein-induced release of tryptase are markedly increased in AD skin lesions, and induced itch in the atopic lesions was not suppressed by antihistamines [106]. Intracutaneously injected tryptase caused itch on the skin of healthy volunteers, which was alleviated by anti-PAR-2 neutralizing antibody and PAR-2 antagonist [107]. Moreover, while intradermal injection of a peptide that activates PAR-2 elicited pain followed by transient itch in healthy subjects, it provoked enhanced itch in AD lesions [106]. These results imply the involvement and enhancement of the tryptase/PAR2 pathway in AD skin. Mast cell-associated nerves in the skin are predominantly substance P positive [103]. Substance P can in turn activate mast cells through NK1 receptor, and released TNF-α can stimulate the neuron terminals through TNF receptors [95]. However, the roles of these interactive pathways in AD remain largely unknown. As mentioned above, IL-31 Tg mice developed AD-like skin lesions with severe pruritis. However, IL-31-induced pruritis could be observed in KitW/W-v mice [89].
A dry skin mouse model, induced by acetone and diethylether treatment followed by water application, showed increased scratching with increased transepidermal water loss (TEWL, an indicator of skin barrier function) and decreased capacitance (an indicator of stratum corneum hydration). In this model, both total and degranulating mast cell numbers were unchanged after 5 days of dry skin induction. Mast cell-deficient KitW/W-v mice showed no differences in scratching behavior than wild type littermates. Scratching was suppressed by opioid receptor antagonists [108]. On the other hand, in a repeated hapten treatment model of AD, mast cell-deficient KitSl/Sl-d mice showed no increase in scratching behavior than vehicle treated mice, although the histopathological findings revealed the epidermal thickening, severe inflammatory infiltrate, and IgE elevation in both wild type and KitSl/Sl-d mice [109]. These observations collectively suggest both mast cell-mediated and mast cell-independent mechanisms underlie the AD-related itch. Extensive explorations are needed to elucidate the role of mast cells and their interaction with the nervous system in AD.
Conclusions and future perspectives
An emerging scenario supported by clinical observations and EC OVA sensitization models (Fig. 2) is that impairment in skin barrier function will allow access of allergens and microbes to antigen-presenting cells such as Langerhans cells and dermal DCs and endow the microenvironment with a Th2 (and Th17) cell-nurturing capacity. These APCs will mature (lose the ability to uptake antigen and acquire antigen-presenting ability) during the migration to draining lymph nodes. Naïve T cells stimulated by the APCs will differentiate to Th2 cells. Th2 effector cells migrate back to inflammatory skin sites to cause damage and to recruit other immune cells such as eosinophils and mast cells. Inflammation will further damage skin barrier function. Infection with bacteria and virus will also worsen AD by various mechanisms. In contrast, other models using crude allergen extracts suggest that activation of multiple (both immune and non-immune) cell types is involved from the initial stage of disease. For instance, house dust mite extracts affect T, B, and mast cells as well as epithelial cells [110]. Human and mouse studies also showed the importance of keratinocytes as the major player not only of the skin barrier but also as a part of the immune system (keratinocytes are the major source of TSLP and IL-18).
Figure 2.
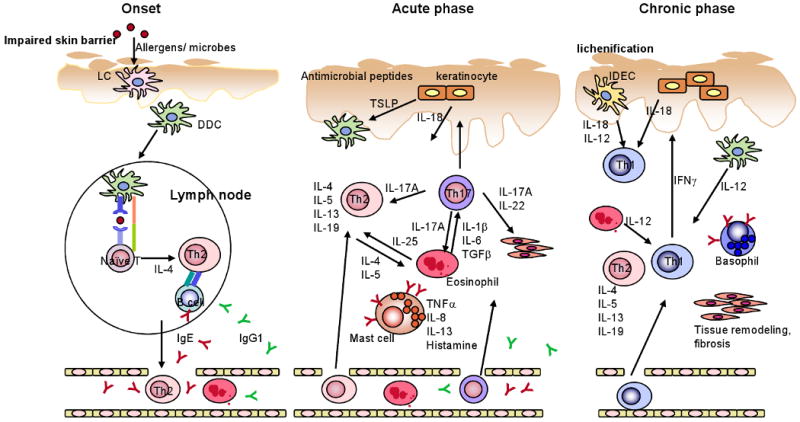
Schematic showing some major cellular and molecular components that are involved in AD pathogenesis. Adapted by permission from Macmillan Publishers Ltd: Journal of Investigative Dermatology, Di Cesare et al. [112], copyright (2008).↑
The current situation surrounding the role of mast cells in the AD pathogenesis has resemblances to that of mast cells in asthma/airway inflammation in 1990s. Some early experiments showed that mast cells play an essential role in allergen-induced airway inflammation, whereas others showed that mast cells play no significant role. Several experiments later resolved this controversy by demonstrating that mast cells play a critical role when mice were immunized with low doses of allergen, without adjuvant, or with low frequency, while they are dispensable when immunized with high doses of allergen or together with adjuvant [21,22,111]. Thus, mast cells seem to amplify the inflammatory processes in asthma/airway inflammation. However, the role of IgE in AD is less clear, as several models have convincingly demonstrated the lack of an increase in IgE levels and/or any significant role of IgE in causation of skin lesions. This issue is also similar to that of mast cells. To resolve these related issues, experiments mimicking human AD should be performed, e.g., using real-world allergenic environments, in addition to clean experiments based on a single allegen that have an advantage of many useful (immunological and genetic) reagents and tend to provide clean-cut results. For example, use of multiple vs. single allergens and adjuvants of purified microbial agents vs. crude extracts of allergenic organisms might reveal subtle effects of IgE and mast cells on skin lesion development. Such a study might also shed novel insights into the hygiene hypothesis.
The current research focuses mainly on impaired skin barrier function and Th2/Th1 (and Th17) imbalance as the underlying mechanisms for AD. Numerous studies have revealed roles of mast cells as effector cells of allergic reactions, which might also be true in the development of skin lesions in AD. However, little is known in their potential role in modulating the course of AD disease progression. Another important point is that a large number of genes are being shown to be associated with AD through genome-wide association studies. However, validation of such candidate genes as those truly involved in mouse models has just begun. This will probably be the most rewarding area of research, which will disclose novel cellular and molecular mechanisms of AD pathogenesis and eventually lead to novel efficacious therapeutic modalities in the next decade or so.
Acknowledgments
We acknowledge support from National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases contract NO1 AI40030 to T.K. This publication is no. 1163 from the La Jolla Institute for Allergy and Immunology.
Footnotes
Conflict of interest statement: No authors have any conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the past two years, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Kitamura Y, Ito A. Mast cell-committed progenitors. Proc Natl Acad Sci U S A. 2005;102:11129–11130. doi: 10.1073/pnas.0505073102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galli SJ. Mast cells and basophils. Curr Opin Hematol. 2000;7:32–39. doi: 10.1097/00062752-200001000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Galli SJ, Maurer M, Lantz CS. Mast cells as sentinels of innate immunity. Curr Opin Immunol. 1999;11:53–59. doi: 10.1016/s0952-7915(99)80010-7. [DOI] [PubMed] [Google Scholar]
- 4.Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol. 2008;9:1215–1223. doi: 10.1038/ni.f.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metz M, Piliponsky AM, Chen CC, Lammel V, Abrink M, Pejler G, Tsai M, Galli SJ. Mast cells can enhance resistance to snake and honeybee venoms. Science. 2006;313:526–530. doi: 10.1126/science.1128877. [DOI] [PubMed] [Google Scholar]
- 6.McLachlan JB, Hart JP, Pizzo SV, Shelburne CP, Staats HF, Gunn MD, Abraham SN. Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat Immunol. 2003;4:1199–1205. doi: 10.1038/ni1005. [DOI] [PubMed] [Google Scholar]
- 7.Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 2007;8:1095–1104. doi: 10.1038/ni1503. [DOI] [PubMed] [Google Scholar]
- 8.Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191:813–822. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 10.Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- 11.Heissig B, Rafii S, Akiyama H, Ohki Y, Sato Y, Rafael T, Zhu Z, Hicklin DJ, Okumura K, Ogawa H, et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J Exp Med. 2005;202:739–750. doi: 10.1084/jem.20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007;13:1211–1218. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 14.Kinet JP. The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu Rev Immunol. 1999;17:931–972. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- 15.Metzger H. The receptor with high affinity for IgE. Immunol Rev. 1992;125:37–48. doi: 10.1111/j.1600-065x.1992.tb00624.x. [DOI] [PubMed] [Google Scholar]
- 16.Finkelman FD. Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol. 2007;120:506–515. doi: 10.1016/j.jaci.2007.07.033. quiz 516-507. [DOI] [PubMed] [Google Scholar]
- 17.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 18.Strait RT, Morris SC, Yang M, Qu XW, Finkelman FD. Pathways of anaphylaxis in the mouse. J Allergy Clin Immunol. 2002;109:658–668. doi: 10.1067/mai.2002.123302. [DOI] [PubMed] [Google Scholar]
- 19.Tsujimura Y, Obata K, Mukai K, Shindou H, Yoshida M, Nishikado H, Kawano Y, Minegishi Y, Shimizu T, Karasuyama H. Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity. 2008;28:581–589. doi: 10.1016/j.immuni.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 20.Galli SJ. Complexity and redundancy in the pathogenesis of asthma: reassessing the roles of mast cells and T cells. J Exp Med. 1997;186:343–347. doi: 10.1084/jem.186.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi T, Miura T, Haba T, Sato M, Serizawa I, Nagai H, Ishizaka K. An essential role of mast cells in the development of airway hyperresponsiveness in a murine asthma model. J Immunol. 2000;164:3855–3861. doi: 10.4049/jimmunol.164.7.3855. [DOI] [PubMed] [Google Scholar]
- 22.Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med. 2000;192:455–462. doi: 10.1084/jem.192.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest. 2006;116:1633–1641. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradding P. The role of the mast cell in asthma: a reassessment. Curr Opin Allergy Clin Immunol. 2003;3:45–50. doi: 10.1097/00130832-200302000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Robinson DS. The role of the mast cell in asthma: induction of airway hyperresponsiveness by interaction with smooth muscle? J Allergy Clin Immunol. 2004;114:58–65. doi: 10.1016/j.jaci.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 26.Tunon-de-Lara JM, Berger P, Begueret H. Mast cells in airway smooth muscle. N Engl J Med. 2002;347:1040–1041. doi: 10.1056/NEJM200209263471318. author reply 1040-1041. [DOI] [PubMed] [Google Scholar]
- 27.D'Amato G, Liccardi G, Noschese P, Salzillo A, D'Amato M, Cazzola M. Anti-IgE monoclonal antibody (omalizumab) in the treatment of atopic asthma and allergic respiratory diseases. Curr Drug Targets Inflamm Allergy. 2004;3:227–229. doi: 10.2174/1568010043343615. [DOI] [PubMed] [Google Scholar]
- 28.Spector S. Omalizumab: efficacy in allergic disease. Panminerva Med. 2004;46:141–148. [PubMed] [Google Scholar]
- 29.Nakano T, Sonoda T, Hayashi C, Yamatodani A, Kanayama Y, Yamamura T, Asai H, Yonezawa T, Kitamura Y, Galli SJ. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J Exp Med. 1985;162:1025–1043. doi: 10.1084/jem.162.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galli SJ, Zsebo KM, Geissler EN. The kit ligand, stem cell factor. Adv Immunol. 1994;55:1–96. doi: 10.1016/s0065-2776(08)60508-8. [DOI] [PubMed] [Google Scholar]
- 31.Okayama Y, Kawakami T. Development, migration, and survival of mast cells. Immunol Res. 2006;34:97–115. doi: 10.1385/IR:34:2:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metz M, Grimbaldeston MA, Nakae S, Piliponsky AM, Tsai M, Galli SJ. Mast cells in the promotion and limitation of chronic inflammation. Immunol Rev. 2007;217:304–328. doi: 10.1111/j.1600-065X.2007.00520.x. [DOI] [PubMed] [Google Scholar]
- 33.Leung DY, Bieber T. Atopic dermatitis. Lancet. 2003;361:151–160. doi: 10.1016/S0140-6736(03)12193-9. [DOI] [PubMed] [Google Scholar]
- 34.Geha RS. Allergy and hypersensitivity. Nature versus nurture in allergy and hypersensitivity. Curr Opin Immunol. 2003;15:603–608. doi: 10.1016/j.coi.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 35.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 36.Leung DYM. New insights into the complex gene-environment interactions evolving into atopic dermatitis. J Allergy Clin Immunol. 2006;118:37–39. [Google Scholar]
- 37.Cookson W. The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat Rev Immunol. 2004;4:978–988. doi: 10.1038/nri1500. [DOI] [PubMed] [Google Scholar]
- 38.Esparza-Gordillo J, Weidinger S, Folster-Holst R, Bauerfeind A, Ruschendorf F, Patone G, Rohde K, Marenholz I, Schulz F, Kerscher T, et al. A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat Genet. 2009 doi: 10.1038/ng.347. [DOI] [PubMed] [Google Scholar]
- 39.Graves PE, Siroux V, Guerra S, Klimecki WT, Martinez FD. Association of atopy and eczema with polymorphisms in T-cell immunoglobulin domain and mucin domain-IL-2-inducible T-cell kinase gene cluster in chromosome 5 q 33. J Allergy Clin Immunol. 2005;116:650–656. doi: 10.1016/j.jaci.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 40.Novak N, Kruse S, Potreck J, Maintz L, Jenneck C, Weidinger S, Fimmers R, Bieber T. Single nucleotide polymorphisms of the IL18 gene are associated with atopic eczema. J Allergy Clin Immunol. 2005;115:828–833. doi: 10.1016/j.jaci.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 41.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 42.Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203:269–273. doi: 10.1084/jem.20051745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, Comeau MR, Campbell DJ, Ziegler SF. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. 2005;202:541–549. doi: 10.1084/jem.20041503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M, Messaddeq N, Teletin M, Pasquali JL, Metzger D, Chambon P. Retinoid X receptor ablation in adult mouse keratinocytes generates an atopic dermatitis triggered by thymic stromal lymphopoietin. Proc Natl Acad Sci U S A. 2005;102:14795–14800. doi: 10.1073/pnas.0507385102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elias PM, Hatano Y, Williams ML. Basis for the barrier abnormality in atopic dermatitis: outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol. 2008;121:1337–1343. doi: 10.1016/j.jaci.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elias PM, Steinhoff M. “Outside-to-inside” (and now back to “outside”) pathogenic mechanisms in atopic dermatitis. J Invest Dermatol. 2008;128:1067–1070. doi: 10.1038/jid.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato A, Fukai K, Oiso N, Hosomi N, Murakami T, Ishii M. Association of SPINK5 gene polymorphisms with atopic dermatitis in the Japanese population. Br J Dermatol. 2003;148:665–669. doi: 10.1046/j.1365-2133.2003.05243.x. [DOI] [PubMed] [Google Scholar]
- 48.Nishio Y, Noguchi E, Shibasaki M, Kamioka M, Ichikawa E, Ichikawa K, Umebayashi Y, Otsuka F, Arinami T. Association between polymorphisms in the SPINK5 gene and atopic dermatitis in the Japanese. Genes Immun. 2003;4:515–517. doi: 10.1038/sj.gene.6363889. [DOI] [PubMed] [Google Scholar]
- 49.Walley AJ, Chavanas S, Moffatt MF, Esnouf RM, Ubhi B, Lawrence R, Wong K, Abecasis GR, Jones EY, Harper JI, et al. Gene polymorphism in Netherton and common atopic disease. Nat Genet. 2001;29:175–178. doi: 10.1038/ng728. [DOI] [PubMed] [Google Scholar]
- ••50.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]; This study demonstrated that loss-of-function mutations in the filaggrin gene are very strong predisposing factors for atopic dermatitis, as these variants are carried by approximately 9% of people of European origin.
- 51.Morar N, Willis-Owen SA, Moffatt MF, Cookson WO. The genetics of atopic dermatitis. J Allergy Clin Immunol. 2006;118:24–34. doi: 10.1016/j.jaci.2006.03.037. quiz 35-26. [DOI] [PubMed] [Google Scholar]
- 52.Sandilands A, Terron-Kwiatkowski A, Hull PR, O'Regan GM, Clayton TH, Watson RM, Carrick T, Evans AT, Liao H, Zhao Y, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–654. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- 53.Nomura T, Sandilands A, Akiyama M, Liao H, Evans AT, Sakai K, Ota M, Sugiura H, Yamamoto K, Sato H, et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol. 2007;119:434–440. doi: 10.1016/j.jaci.2006.12.646. [DOI] [PubMed] [Google Scholar]
- •54.Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, Callanan JJ, Kawasaki H, Shiohama A, Kubo A, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41:602–608. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports a 1-bp deletion mutation, 5303delA, within the filaggrin gene in the spontaneous mouse mutant flaky tail (ft).
- 55.Tosi MF. Innate immune responses to infection. J Allergy Clin Immunol. 2005;116:241–249. doi: 10.1016/j.jaci.2005.05.036. quiz 250. [DOI] [PubMed] [Google Scholar]
- 56.Strange P, Skov L, Lisby S, Nielsen PL, Baadsgaard O. Staphylococcal enterotoxin B applied on intact normal and intact atopic skin induces dermatitis. Arch Dermatol. 1996;132:27–33. [PubMed] [Google Scholar]
- 57.Jappe U. Superantigens and their association with dermatological inflammatory diseases: facts and hypotheses. Acta Derm Venereol. 2000;80:321–328. doi: 10.1080/000155500459231. [DOI] [PubMed] [Google Scholar]
- 58.Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, Gallo R, Paller AS, Lieff S, Reese J, Zaccaro D, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009 doi: 10.1016/j.jaci.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuda H, Watanabe N, Geba GP, Sperl J, Tsudzuki M, Hiroi J, Matsumoto M, Ushio H, Saito S, Askenase PW, et al. Development of atopic dermatitis-like skin lesion with IgE hyperproduction in NC/Nga mice. Int Immunol. 1997;9:461–466. doi: 10.1093/intimm/9.3.461. [DOI] [PubMed] [Google Scholar]
- 60.Gutermuth J, Ollert M, Ring J, Behrendt H, Jakob T. Mouse models of atopic eczema critically evaluated. Int Arch Allergy Immunol. 2004;135:262–276. doi: 10.1159/000082099. [DOI] [PubMed] [Google Scholar]
- •61.Jin H, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol. 2009;129:31–40. doi: 10.1038/jid.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a superb review of animal models of AD.
- 62.Vestergaard C, Yoneyama H, Murai M, Nakamura K, Tamaki K, Terashima Y, Imai T, Yoshie O, Irimura T, Mizutani H, et al. Overproduction of Th2-specific chemokines in NC/Nga mice exhibiting atopic dermatitis-like lesions. J Clin Invest. 1999;104:1097–1105. doi: 10.1172/JCI7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsumoto M, Ra C, Kawamoto K, Sato H, Itakura A, Sawada J, Ushio H, Suto H, Mitsuishi K, Hikasa Y, et al. IgE hyperproduction through enhanced tyrosine phosphorylation of Janus kinase 3 in NC/Nga mice, a model for human atopic dermatitis. J Immunol. 1999;162:1056–1063. [PubMed] [Google Scholar]
- 64.Yagi R, Nagai H, Iigo Y, Akimoto T, Arai T, Kubo M. Development of atopic dermatitis-like skin lesions in STAT6-deficient NC/Nga mice. J Immunol. 2002;168:2020–2027. doi: 10.4049/jimmunol.168.4.2020. [DOI] [PubMed] [Google Scholar]
- 65.Iijima OT, Takeda H, Komatsu Y, Matsumiya T, Takahashi H. Atopic dermatitis in NC/Jic mice associated with Myobia musculi infestation. Comp Med. 2000;50:225–228. [PubMed] [Google Scholar]
- 66.Morita E, Kaneko S, Hiragun T, Shindo H, Tanaka T, Furukawa T, Nobukiyo A, Yamamoto S. Fur mites induce dermatitis associated with IgE hyperproduction in an inbred strain of mice, NC/Kuj. J Dermatol Sci. 1999;19:37–43. doi: 10.1016/s0923-1811(98)00047-4. [DOI] [PubMed] [Google Scholar]
- 67.Gao XK, Nakamura N, Fuseda K, Tanaka H, Inagaki N, Nagai H. Establishment of allergic dermatitis in NC/Nga mice as a model for severe atopic dermatitis. Biol Pharm Bull. 2004;27:1376–1381. doi: 10.1248/bpb.27.1376. [DOI] [PubMed] [Google Scholar]
- 68.Heishi M, Imai Y, Katayama H, Hashida R, Ito M, Shinagawa A, Sugita Y. Gene expression analysis of atopic dermatitis-like skin lesions induced in NC/Nga mice by mite antigen stimulation under specific pathogen-free conditions. Int Arch Allergy Immunol. 2003;132:355–363. doi: 10.1159/000074903. [DOI] [PubMed] [Google Scholar]
- 69.Kang JS, Lee K, Han SB, Ahn JM, Lee H, Han MH, Yoon YD, Yoon WK, Park SK, Kim HM. Induction of atopic eczema/dermatitis syndrome-like skin lesions by repeated topical application of a crude extract of Dermatophagoides pteronyssinus in NC/Nga mice. Int Immunopharmacol. 2006;6:1616–1622. doi: 10.1016/j.intimp.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 70.Matsuoka H, Maki N, Yoshida S, Arai M, Wang J, Oikawa Y, Ikeda T, Hirota N, Nakagawa H, Ishii A. A mouse model of the atopic eczema/dermatitis syndrome by repeated application of a crude extract of house-dust mite Dermatophagoides farinae. Allergy. 2003;58:139–145. doi: 10.1034/j.1398-9995.2003.23790.x. [DOI] [PubMed] [Google Scholar]
- 71.Unno T, Suto H, Yoshiike T, Ogawa H, Ra C. Induction of atopic dermatitis-like skin lesion in NC/Nga mice--the influence of the skin barrier destroying solution to the induction of dermatitis. Arerugi. 2001;50:1152–1162. [PubMed] [Google Scholar]
- 72.Kawakami Y, Yumoto K, Kawakami T. An improved mouse model of atopic dermatitis and suppression of skin lesions by an inhibitor of tec family kinases. Allergol Int. 2007;56:403–409. doi: 10.2332/allergolint.O-07-486. [DOI] [PubMed] [Google Scholar]
- 73.Kawakami Y, Tomimori Y, Yumoto K, Hasegawa S, Ando T, Tagaya Y, Crotty S, Kawakami T. Inhibition of NK cell activity by IL-17 allows vaccinia virus to induce severe skin lesions in a mouse model of eczema vaccinatum. J Exp Med. 2009;206:1219–1225. doi: 10.1084/jem.20082835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang LF, Lin JY, Hsieh KH, Lin RH. Epicutaneous exposure of protein antigen induces a predominant Th2-like response with high IgE production in mice. J Immunol. 1996;156:4077–4082. [PubMed] [Google Scholar]
- 75.Strid J, Hourihane J, Kimber I, Callard R, Strobel S. Disruption of the stratum corneum allows potent epicutaneous immunization with protein antigens resulting in a dominant systemic Th2 response. Eur J Immunol. 2004;34:2100–2109. doi: 10.1002/eji.200425196. [DOI] [PubMed] [Google Scholar]
- 76.Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest. 1998;101:1614–1622. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spergel JM, Mizoguchi E, Oettgen H, Bhan AK, Geha RS. Roles of TH1 and TH2 cytokines in a murine model of allergic dermatitis. J Clin Invest. 1999;103:1103–1111. doi: 10.1172/JCI5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •78.He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci U S A. 2007;104:15817–15822. doi: 10.1073/pnas.0706942104. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that EC immunization with OVA drive IL-17 expression in the skin by IL-23 and TGF-β-dependent manner.
- 79.Alenius H, Laouini D, Woodward A, Mizoguchi E, Bhan AK, Castigli E, Oettgen HC, Geha RS. Mast cells regulate IFN-gamma expression in the skin and circulating IgE levels in allergen-induced skin inflammation. J Allergy Clin Immunol. 2002;109:106–113. doi: 10.1067/mai.2002.120553. [DOI] [PubMed] [Google Scholar]
- 80.Oiwa M, Satoh T, Watanabe M, Niwa H, Hirai H, Nakamura M, Yokozeki H. CRTH2-dependent, STAT6-independent induction of cedar pollen dermatitis. Clin Exp Allergy. 2008;38:1357–1366. doi: 10.1111/j.1365-2222.2008.03007.x. [DOI] [PubMed] [Google Scholar]
- •81.Abboud G, Staumont-Salle D, Kanda A, Roumier T, Deruytter N, Lavogiez C, Fleury S, Remy P, Papin JP, Capron M, et al. FcεRI and FcγRIII/CD16 differentially regulate atopic dermatitis in mice. J Immunol. 2009;182:6517–6526. doi: 10.4049/jimmunol.0801055. [DOI] [PubMed] [Google Scholar]; Using an EC OVA sensitization model, this study demonstrated the contribution of FcεRI and FcγRIII/CD16 to the AD pathology.
- 82.Gonzalez de Olano D, de la Hoz Caballer B, Nunez Lopez R, Sanchez Munoz L, Cuevas Agustin M, Dieguez MC, Alvarez Twose I, Castells MC, Escribano Mora L. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA) Clin Exp Allergy. 2007;37:1547–1555. doi: 10.1111/j.1365-2222.2007.02804.x. [DOI] [PubMed] [Google Scholar]
- 83.Kunisada T, Lu SZ, Yoshida H, Nishikawa S, Mizoguchi M, Hayashi S, Tyrrell L, Williams DA, Wang X, Longley BJ. Murine cutaneous mastocytosis and epidermal melanocytosis induced by keratinocyte expression of transgenic stem cell factor. J Exp Med. 1998;187:1565–1573. doi: 10.1084/jem.187.10.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tanaka T, Tsutsui H, Yoshimoto T, Kotani M, Matsumoto M, Fujita A, Wang W, Higa S, Koshimoto T, Nakanishi K, et al. Interleukin-18 is elevated in the sera from patients with atopic dermatitis and from atopic dermatitis model mice, NC/Nga. Int Arch Allergy Immunol. 2001;125:236–240. doi: 10.1159/000053821. [DOI] [PubMed] [Google Scholar]
- 85.Nakano H, Tsutsui H, Terada M, Yasuda K, Matsui K, Yumikura-Futatsugi S, Yamanaka K, Mizutani H, Yamamura T, Nakanishi K. Persistent secretion of IL-18 in the skin contributes to IgE response in mice. Int Immunol. 2003;15:611–621. doi: 10.1093/intimm/dxg062. [DOI] [PubMed] [Google Scholar]
- •86.Terada M, Tsutsui H, Imai Y, Yasuda K, Mizutani H, Yamanishi K, Kubo M, Matsui K, Sano H, Nakanishi K. Contribution of IL-18 to atopic-dermatitis-like skin inflammation induced by Staphylococcus aureus product in mice. Proc Natl Acad Sci U S A. 2006;103:8816–8821. doi: 10.1073/pnas.0602900103. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed the importance of IL-18 for the development of infection-associated AD by induction of IL-3 from super Th1 cells.
- 87.Novak N, Valenta R, Bohle B, Laffer S, Haberstok J, Kraft S, Bieber T. FcepsilonRI engagement of Langerhans cell-like dendritic cells and inflammatory dendritic epidermal cell-like dendritic cells induces chemotactic signals and different T-cell phenotypes in vitro. J Allergy Clin Immunol. 2004;113:949–957. doi: 10.1016/j.jaci.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 88.Chen L, Overbergh L, Mathieu C, Chan LS. The development of atopic dermatitis is independent of Immunoglobulin E up-regulation in the K14-IL-4 SKH1 transgenic mouse model. Clin Exp Allergy. 2008;38:1367–1380. doi: 10.1111/j.1365-2222.2008.02987.x. [DOI] [PubMed] [Google Scholar]
- 89.Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, Haugen HS, Maurer M, Harder B, Johnston J, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–760. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- 90.Boehme SA, Franz-Bacon K, Chen EP, Ly TW, Kawakami Y, Bacon KB. Murine bone marrow-derived mast cells express chemoattractant receptor-homologous molecule expressed on T-helper class 2 cells (CRTh2) Int Immunol. 2009;21:621–632. doi: 10.1093/intimm/dxp031. [DOI] [PubMed] [Google Scholar]
- 91.Spik I, Brenuchon C, Angeli V, Staumont D, Fleury S, Capron M, Trottein F, Dombrowicz D. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174:3703–3708. doi: 10.4049/jimmunol.174.6.3703. [DOI] [PubMed] [Google Scholar]
- 92.Boehme SA, Chen EP, Franz-Bacon K, Sasik R, Sprague LJ, Ly TW, Hardiman G, Bacon KB. Antagonism of CRTH2 ameliorates chronic epicutaneous sensitization-induced inflammation by multiple mechanisms. Int Immunol. 2009;21:1–17. doi: 10.1093/intimm/dxn118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boehme SA, Franz-Bacon K, Chen EP, Sasik R, Sprague LJ, Ly TW, Hardiman G, Bacon KB. A small molecule CRTH2 antagonist inhibits FITC-induced allergic cutaneous inflammation. Int Immunol. 2009;21:81–93. doi: 10.1093/intimm/dxn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Satoh T, Moroi R, Aritake K, Urade Y, Kanai Y, Sumi K, Yokozeki H, Hirai H, Nagata K, Hara T, et al. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177:2621–2629. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- 95.Yosipovitch G, Greaves MW, Schmelz M. Itch. Lancet. 2003;361:690–694. doi: 10.1016/S0140-6736(03)12570-6. [DOI] [PubMed] [Google Scholar]
- 96.Yamamoto M, Haruna T, Ueda C, Asano Y, Takahashi H, Iduhara M, Takaki S, Yasui K, Matsuo Y, Arimura A. Contribution of itch-associated scratch behavior to the development of skin lesions in Dermatophagoides farinae-induced dermatitis model in NC/Nga mice. Arch Dermatol Res. 2008 doi: 10.1007/s00403-008-0912-8. [DOI] [PubMed] [Google Scholar]
- 97.Matsushima H, Hayashi S, Shimada S. Skin scratching switches immune responses from Th2 to Th1 type in epicutaneously immunized mice. J Dermatol Sci. 2003;32:223–230. doi: 10.1016/s0923-1811(03)00106-3. [DOI] [PubMed] [Google Scholar]
- 98.Krause L, Shuster S. Mechanism of action of antipruritic drugs. Br Med J (Clin Res Ed) 1983;287:1199–1200. doi: 10.1136/bmj.287.6400.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fujii M, Nabe T, Tomozawa J, Kohno S. Involvement of skin barrier dysfunction in itch-related scratching in special diet-fed hairless mice. Eur J Pharmacol. 2006;530:152–156. doi: 10.1016/j.ejphar.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 100.Naukkarinen A, Harvima IT, Aalto ML, Horsmanheimo M. Mast cell tryptase and chymase are potential regulators of neurogenic inflammation in psoriatic skin. Int J Dermatol. 1994;33:361–366. doi: 10.1111/j.1365-4362.1994.tb01069.x. [DOI] [PubMed] [Google Scholar]
- 101.Stead RH, Tomioka M, Quinonez G, Simon GT, Felten SY, Bienenstock J. Intestinal mucosal mast cells in normal and nematode-infected rat intestines are in intimate contact with peptidergic nerves. Proc Natl Acad Sci U S A. 1987;84:2975–2979. doi: 10.1073/pnas.84.9.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 103.Naukkarinen A, Harvima I, Paukkonen K, Aalto ML, Horsmanheimo M. Immunohistochemical analysis of sensory nerves and neuropeptides, and their contacts with mast cells in developing and mature psoriatic lesions. Arch Dermatol Res. 1993;285:341–346. doi: 10.1007/BF00371834. [DOI] [PubMed] [Google Scholar]
- 104.Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 105.Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA, Schechter N, Woolkalis M, Brass LF. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- 106.Steinhoff M, Neisius U, Ikoma A, Fartasch M, Heyer G, Skov PS, Luger TA, Schmelz M. Proteinase-activated receptor-2 mediates itch: a novel pathway for pruritus in human skin. J Neurosci. 2003;23:6176–6180. doi: 10.1523/JNEUROSCI.23-15-06176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ui H, Andoh T, Lee JB, Nojima H, Kuraishi Y. Potent pruritogenic action of tryptase mediated by PAR-2 receptor and its involvement in anti-pruritic effect of nafamostat mesilate in mice. Eur J Pharmacol. 2006;530:172–178. doi: 10.1016/j.ejphar.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 108.Miyamoto T, Nojima H, Shinkado T, Nakahashi T, Kuraishi Y. Itch-associated response induced by experimental dry skin in mice. Jpn J Pharmacol. 2002;88:285–292. doi: 10.1254/jjp.88.285. [DOI] [PubMed] [Google Scholar]
- 109.Yamashita H, Michibata Y, Mizukami H, Ogihara Y, Morita A, Nose M. Dermal mast cells play a central role in the incidence of scratching behavior in mice induced by multiple application of the hapten, 2,4,6-trinitrochlorobenzene. Exp Dermatol. 2005;14:438–444. doi: 10.1111/j.0906-6705.2005.00304.x. [DOI] [PubMed] [Google Scholar]
- 110.Yu CK, Chen CL. Activation of mast cells is essential for development of house dust mite Dermatophagoides farinae-induced allergic airway inflammation in mice. J Immunol. 2003;171:3808–3815. doi: 10.4049/jimmunol.171.7.3808. [DOI] [PubMed] [Google Scholar]
- 111.Mayr SI, Zuberi RI, Zhang M, de Sousa-Hitzler J, Ngo K, Kuwabara Y, Yu L, Fung-Leung WP, Liu FT. IgE-dependent mast cell activation potentiates airway responses in murine asthma models. J Immunol. 2002;169:2061–2068. doi: 10.4049/jimmunol.169.4.2061. [DOI] [PubMed] [Google Scholar]
- 112.Di Cesare A, Di Meglio P, Nestle FO. A role for Th17 cells in the immunopathogenesis of atopic dermatitis? J Invest Dermatol. 2008;128:2569–2571. doi: 10.1038/jid.2008.283. [DOI] [PubMed] [Google Scholar]
- 113.Kohara Y, Tanabe K, Matsuoka K, Kanda N, Matsuda H, Karasuyama H, Yonekawa H. A major determinant quantitative-trait locus responsible for atopic dermatitis-like skin lesions in NC/Nga mice is located on Chromosome 9. Immunogenetics. 2001;53:15–21. doi: 10.1007/s002510000286. [DOI] [PubMed] [Google Scholar]
- 114.Matsubara T, Aoki N, Hino S, Okajima T, Nadano D, Matsuda T. Serum and monoclonal immunoglobulin E antibodies from NC/Nga mice with severe atopic-like dermatitis recognize an auto-antigen, histone H3. Clin Exp Allergy. 2009;39:579–590. doi: 10.1111/j.1365-2222.2008.03174.x. [DOI] [PubMed] [Google Scholar]
- 115.Terakawa M, Fujieda Y, Tomimori Y, Muto T, Tanaka T, Maruoka H, Nagahira K, Ogata A, Nakatsuka T, Fukuda Y. Oral chymase inhibitor SUN13834 ameliorates skin inflammation as well as pruritus in mouse model for atopic dermatitis. Eur J Pharmacol. 2008;601:186–191. doi: 10.1016/j.ejphar.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 116.Watanabe N, Tomimori Y, Terakawa M, Ishiwata K, Wada A, Muto T, Tanaka T, Maruoka H, Nagahira K, Nakatsuka T, et al. Oral administration of chymase inhibitor improves dermatitis in NC/Nga mice. J Invest Dermatol. 2007;127:971–973. doi: 10.1038/sj.jid.5700708. [DOI] [PubMed] [Google Scholar]
- 117.Tsujii K, Andoh T, Ui H, Lee JB, Kuraishi Y. Involvement of Tryptase and Proteinase-Activated Receptor-2 in Spontaneous Itch-Associated Response in Mice With Atopy-like Dermatitis. J Pharmacol Sci. 2009;109:388–395. doi: 10.1254/jphs.08332fp. [DOI] [PubMed] [Google Scholar]
- 118.Inoue T, Sugimoto M, Sakurai T, Saito R, Futaki N, Hashimoto Y, Honma Y, Arai I, Nakaike S. Modulation of scratching behavior by silencing an endogenous cyclooxygenase-1 gene in the skin through the administration of siRNA. J Gene Med. 2007;9:994–1001. doi: 10.1002/jgm.1091. [DOI] [PubMed] [Google Scholar]
- 119.Sugimoto M, Arai I, Futaki N, Hashimoto Y, Sakurai T, Honma Y, Nakaike S. Time course changes of scratching counts, dermatitis symptoms, and levels of cutaneous prostaglandins in NC/Nga mice. Exp Dermatol. 2006;15:875–882. doi: 10.1111/j.1600-0625.2006.00480.x. [DOI] [PubMed] [Google Scholar]
- 120.Lane PW. Two new mutations in linkage group XVI of the house mouse. Flaky tail and varitint-waddler-J. J Hered. 1972;63:135–140. doi: 10.1093/oxfordjournals.jhered.a108252. [DOI] [PubMed] [Google Scholar]
- 121.Presland RB, Boggess D, Lewis SP, Hull C, Fleckman P, Sundberg JP. Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris. J Invest Dermatol. 2000;115:1072–1081. doi: 10.1046/j.1523-1747.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- 122.Jong MC, Gijbels MJ, Dahlmans VE, Gorp PJ, Koopman SJ, Ponec M, Hofker MH, Havekes LM. Hyperlipidemia and cutaneous abnormalities in transgenic mice overexpressing human apolipoprotein C1. J Clin Invest. 1998;101:145–152. doi: 10.1172/JCI791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nagelkerken L, Verzaal P, Lagerweij T, Persoon-Deen C, Berbee JF, Prens EP, Havekes LM, Oranje AP. Development of atopic dermatitis in mice transgenic for human apolipoprotein C1. J Invest Dermatol. 2008;128:1165–1172. doi: 10.1038/sj.jid.5701182. [DOI] [PubMed] [Google Scholar]
- 124.Descargues P, Deraison C, Bonnart C, Kreft M, Kishibe M, Ishida-Yamamoto A, Elias P, Barrandon Y, Zambruno G, Sonnenberg A, et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat Genet. 2005;37:56–65. doi: 10.1038/ng1493. [DOI] [PubMed] [Google Scholar]
- 125.Demehri S, Morimoto M, Holtzman MJ, Kopan R. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 2009;7:e1000067. doi: 10.1371/journal.pbio.1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grubauer G, Feingold KR, Harris RM, Elias PM. Lipid content and lipid type as determinants of the epidermal permeability barrier. J Lipid Res. 1989;30:89–96. [PubMed] [Google Scholar]
- 127.Chan LS, Robinson N, Xu L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: an experimental animal model to study atopic dermatitis. J Invest Dermatol. 2001;117:977–983. doi: 10.1046/j.0022-202x.2001.01484.x. [DOI] [PubMed] [Google Scholar]
- 128.Konishi H, Tsutsui H, Murakami T, Yumikura-Futatsugi S, Yamanaka K, Tanaka M, Iwakura Y, Suzuki N, Takeda K, Akira S, et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/stat6 under specific pathogen-free conditions. Proc Natl Acad Sci U S A. 2002;99:11340–11345. doi: 10.1073/pnas.152337799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murakami T, Yamanaka K, Tokime K, Kurokawa I, Tsutsui H, Nakanishi K, Mizutani H. Topical suplatast tosilate (IPD) ameliorates Th2 cytokine-mediated dermatitis in caspase-1 transgenic mice by downregulating interleukin-4 and interleukin-5. Br J Dermatol. 2006;155:27–32. doi: 10.1111/j.1365-2133.2006.07241.x. [DOI] [PubMed] [Google Scholar]
- 130.Yamanaka K, Tanaka M, Tsutsui H, Kupper TS, Asahi K, Okamura H, Nakanishi K, Suzuki M, Kayagaki N, Black RA, et al. Skin-specific caspase-1-transgenic mice show cutaneous apoptosis and pre-endotoxin shock condition with a high serum level of IL-18. J Immunol. 2000;165:997–1003. doi: 10.4049/jimmunol.165.2.997. [DOI] [PubMed] [Google Scholar]
- 131.Zheng T, Oh MH, Oh SY, Schroeder JT, Glick AB, Zhu Z. Transgenic expression of interleukin-13 in the skin induces a pruritic dermatitis and skin remodeling. J Invest Dermatol. 2009;129:742–751. doi: 10.1038/jid.2008.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sato H, Taketomi Y, Isogai Y, Masuda S, Kobayashi T, Yamamoto K, Murakami M. Group III secreted phospholipase A2 transgenic mice spontaneously develop inflammation. Biochem J. 2009;421:17–27. doi: 10.1042/BJ20082429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li M, Hener P, Zhang Z, Kato S, Metzger D, Chambon P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A. 2006;103:11736–11741. doi: 10.1073/pnas.0604575103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsukuba T, Okamoto K, Okamoto Y, Yanagawa M, Kohmura K, Yasuda Y, Uchi H, Nakahara T, Furue M, Nakayama K, et al. Association of cathepsin E deficiency with development of atopic dermatitis. J Biochem (Tokyo) 2003;134:893–902. doi: 10.1093/jb/mvg216. [DOI] [PubMed] [Google Scholar]
- 135.Barton D, HogenEsch H, Weih F. Mice lacking the transcription factor RelB develop T cell-dependent skin lesions similar to human atopic dermatitis. Eur J Immunol. 2000;30:2323–2332. doi: 10.1002/1521-4141(2000)30:8<2323::AID-IMMU2323>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 136.Knisz J, Banks A, McKeag L, Metcalfe DD, Rothman PB, Brown JM. Loss of SOCS7 in mice results in severe cutaneous disease and increased mast cell activation. Clin Immunol. 2009 doi: 10.1016/j.clim.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 137.Sadri N, Schneider RJ. Auf1/Hnrnpd-deficient mice develop pruritic inflammatory skin disease. J Invest Dermatol. 2009;129:657–670. doi: 10.1038/jid.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Asakawa M, Yoshioka T, Matsutani T, Hikita I, Suzuki M, Oshima I, Tsukahara K, Arimura A, Horikawa T, Hirasawa T, et al. Association of a mutation in TRPV3 with defective hair growth in rodents. J Invest Dermatol. 2006;126:2664–2672. doi: 10.1038/sj.jid.5700468. [DOI] [PubMed] [Google Scholar]
- 139.Haraguchi M, Hino M, Tanaka H, Maru M. Naturally occurring dermatitis associated with Staphylococcus aureus in DS-Nh mice. Exp Anim. 1997;46:225–229. doi: 10.1538/expanim.46.225. [DOI] [PubMed] [Google Scholar]
- 140.Yoshioka T, Imura K, Asakawa M, Suzuki M, Oshima I, Hirasawa T, Sakata T, Horikawa T, Arimura A. Impact of the Gly573Ser substitution in TRPV3 on the development of allergic and pruritic dermatitis in mice. J Invest Dermatol. 2009;129:714–722. doi: 10.1038/jid.2008.245. [DOI] [PubMed] [Google Scholar]
- 141.Fujii M, Tomozawa J, Mizutani N, Nabe T, Danno K, Kohno S. Atopic dermatitis-like pruritic skin inflammation caused by feeding a special diet to HR-1 hairless mice. Exp Dermatol. 2005;14:460–468. doi: 10.1111/j.0906-6705.2005.00313.x. [DOI] [PubMed] [Google Scholar]
- 142.Makiura M, Akamatsu H, Akita H, Yagami A, Shimizu Y, Eiro H, Kuramoto M, Suzuki K, Matsunaga K. Atopic dermatitis-like symptoms in HR-1 hairless mice fed a diet low in magnesium and zinc. J Int Med Res. 2004;32:392–399. doi: 10.1177/147323000403200407. [DOI] [PubMed] [Google Scholar]
- 143.Watanabe O, Natori K, Tamari M, Shiomoto Y, Kubo S, Nakamura Y. Significantly elevated expression of PF4 (platelet factor 4) and eotaxin in the NOA mouse, a model for atopic dermatitis. J Hum Genet. 1999;44:173–176. doi: 10.1007/s100380050136. [DOI] [PubMed] [Google Scholar]
- 144.Kashiwakura JI, Kawakami Y, Yuki K, Zajonc DM, Hasegawa S, Tomimori Y, Caplan B, Saito H, Furue M, Oettgen HC, et al. Polyclonal IgE Induces Mast Cell Survival and Cytokine Production. Allergol Int. 2009;58 doi: 10.2332/allergolint.08-OA-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Laouini D, Kawamoto S, Yalcindag A, Bryce P, Mizoguchi E, Oettgen H, Geha RS. Epicutaneous sensitization with superantigen induces allergic skin inflammation. J Allergy Clin Immunol. 2003;112:981–987. doi: 10.1016/j.jaci.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 146.Li XM, Kleiner G, Huang CK, Lee SY, Schofield B, Soter NA, Sampson HA. Murine model of atopic dermatitis associated with food hypersensitivity. J Allergy Clin Immunol. 2001;107:693–702. doi: 10.1067/mai.2001.114110. [DOI] [PubMed] [Google Scholar]
- 147.Herz U, Schnoy N, Borelli S, Weigl L, Kasbohrer U, Daser A, Wahn U, Kottgen E, Renz H. A human-SCID mouse model for allergic immune response bacterial superantigen enhances skin inflammation and suppresses IgE production. J Invest Dermatol. 1998;110:224–231. doi: 10.1046/j.1523-1747.1998.00119.x. [DOI] [PubMed] [Google Scholar]
- 148.Kitagaki H, Fujisawa S, Watanabe K, Hayakawa K, Shiohara T. Immediate-type hypersensitivity response followed by a late reaction is induced by repeated epicutaneous application of contact sensitizing agents in mice. J Invest Dermatol. 1995;105:749–755. doi: 10.1111/1523-1747.ep12325538. [DOI] [PubMed] [Google Scholar]
- 149.Nagai H, Matsuo A, Hiyama H, Inagaki N, Kawada K. Immunoglobulin E production in mice by means of contact sensitization with a simple chemical, hapten. J Allergy Clin Immunol. 1997;100:S39–44. doi: 10.1016/s0091-6749(97)70003-4. [DOI] [PubMed] [Google Scholar]
- 150.Webb EF, Tzimas MN, Newsholme SJ, Griswold DE. Intralesional cytokines in chronic oxazolone-induced contact sensitivity suggest roles for tumor necrosis factor alpha and interleukin-4. J Invest Dermatol. 1998;111:86–92. doi: 10.1046/j.1523-1747.1998.00239.x. [DOI] [PubMed] [Google Scholar]