

Abstract
Abstract. Extracellular signal‐regulated kinase activity is essential for mediating cell cycle progression from G1 phase to S phase (DNA synthesis). In contrast, the role of extracellular signal‐regulated kinase during G2 phase and mitosis (M phase) is largely undefined. Previous studies have suggested that inhibition of basal extracellular signal‐regulated kinase activity delays G2‐ and M‐phase progression. In the current investigation, we have examined the consequence of activating the extracellular signal‐regulated kinase pathway during G2 phase on subsequent progression through mitosis. Using synchronized HeLa cells, we show that activation of the extracellular signal‐regulated kinase pathway with phorbol 12‐myristate 13‐acetate or epidermal growth factor during G2 phase causes a rapid cell cycle arrest in G2 as measured by flow cytometry, mitotic indices and cyclin B1 expression. This G2‐phase arrest was reversed by pre‐treatment with bisindolylmaleimide or U0126, which are selective inhibitors of protein kinase C proteins or the extracellular signal‐regulated kinase activators, MEK1/2, respectively. The extracellular signal‐regulated kinase‐mediated delay in M‐phase entry appeared to involve de novo synthesis of the cyclin‐dependent kinase inhibitor, p21 CIP1, during G2 through a p53‐independent mechanism. To establish a function for the increased expression of p21 CIP1 and delayed cell cycle progression, we show that extracellular signal‐regulated kinase activation in G2‐phase cells results in an increased number of cells containing chromosome aberrations characteristic of genomic instability. The presence of chromosome aberrations following extracellular signal‐regulated kinase activation during G2‐phase was further augmented in cells lacking p21 CIP1 . These findings suggest that p21 CIP1 mediated inhibition of cell cycle progression during G2/M phase protects against inappropriate activation of signalling pathways, which may cause excessive chromosome damage and be detrimental to cell survival.
INTRODUCTION
The extracellular signal‐regulated kinases 1 and 2 (ERK1/2) are members of the mitogen‐activated protein (MAP) kinase family and are involved in a variety of cellular functions including proliferation, differentiation, apoptosis and motility (Lewis et al. 1998; Pearson et al. 2001). ERK protein activation most often occurs sequentially through Ras G‐proteins, Raf kinases, and MAP/ERK kinases 1 and 2 (MEK1/2), which are the only known activators of ERK1/2 (Lewis et al. 1998). Upon activation, the ERK pathway promotes cell proliferation by indirect regulation of the activity of cyclin‐dependent kinases (Cdk) at specific times during the cell cycle. For example, Ras G‐protein activity couples growth factor receptor signalling to ERK proteins, which promote cyclin D1 expression, Cdk2 activity, and G1/S‐phase progression (Liu et al. 1995; Aktas et al. 1997). Ras also promotes progression through G1‐phase by increasing cyclin E/Cdk2 activity in cooperation with the ERK substrate, c‐Myc (Leone et al. 1997). Activated Cdk proteins phosphorylate the retinoblastoma (Rb) tumour‐suppressor protein, which acts as a cell cycle repressor of transcription during G1‐phase in its hypophosphorylated form (Hatakeyama & Weinberg 1995). Thus, Ras, indirectly through the activation of Cdk proteins, causes Rb phosphorylation and inhibition of Rb repression of genes required for entry into S phase (Mittnacht et al. 1997; Peeper et al. 1997). As expected, proteins downstream of Ras, including Raf, MEK, and ERK, have also been shown to play a central role in promoting entry into S‐phase progression by regulating the expression of cyclins and activation of Cdk proteins (Weber et al. 1997; Cheng et al. 1998; Lents et al. 2002).
The ERK pathway may also promote cell cycle progression by regulating the expression of Cdk inhibitors such as p21 CIP1 (Sewing et al. 1997; Kivinen & Laiho 1999; Coleman et al. 2003). For example, many growth factors that require Ras signalling through ERK to increase cell proliferation also induce p21 CIP1 expression (Leone et al. 1997; Kivinen & Laiho 1999). Similarly, pharmacological inhibition of MEK1/2 with PD98059 blocks growth factor‐mediated G1/S‐phase progression as well as p21 CIP1 expression (Kivinen & Laiho 1999). Although it may appear counter productive for the Ras/Raf/MEK/ERK pathway to promote the expression of both cyclin D proteins and the p21 CIP1 inhibitor, studies using cells that do not express the p21 CIP1 gene show a reduced ability to activate cyclin D/Cdk complexes and promote G1/S‐phase progression (Cheng et al. 1999). Given that increased p21 CIP1 expression is observed in more that 90% of breast cancer tissues that also over‐express cyclin D proteins (Russell et al. 1999), elevated p21 CIP1 levels have been proposed to function in delaying cell cycle progression in order to protect cancer cells against DNA damaging agents that would otherwise promote apoptosis (Gartel & Radhakrishnan 2005). Thus, tumour cells containing oncogenic Ras or Raf proteins may regulate the expression of cyclins and cell cycle inhibitors, which may simultaneously provide cells with a proliferative advantage and protection against damaging agents during cell cycle progression.
Despite its important role in regulating G1/S phase, the function of the ERK pathway in regulating somatic cell cycle progression through G2 phase and mitosis (M phase) is still under investigation. Inhibition of basal ERK pathway activity, with pharmacologic agents, RNA interference, or by over‐expression of dominant negative mutants of MEK1, causes a delay in G2 and M‐phase progression (Wright et al. 1999; Roberts et al. 2002; Liu et al. 2004). Several proteins of the ERK pathway have been shown to have increased catalytic activity during the mitotic phase. For example, a cytoplasmic form of Raf‐1 has been shown to be activated through a Ras‐independent mechanism and is uncoupled from MEK1/2 in total protein lysates isolated from cells arrested in mitosis with nocodazole (Laird et al. 1995; Ziogas et al. 1998; Laird et al. 1999). However, Raf‐1–dependent activation of MEK1 has been linked to the regulation of mitotic Golgi apparatus fragmentation (Colanzi et al. 2003). Thus, it is possible that localized Raf‐1/MEK/ERK signalling modules may be more relevant to cells progressing through G2 and M phases. Localized ERK pathway activation is supported by several studies demonstrating the presence of active forms of MEK and ERK proteins that are localized to the nucleus, chromosome kinetochores, centrosomes, microtubules, and the Golgi complex during G2 and M‐phase transitions (Acharya et al. 1998; Shapiro et al. 1998; Zecevic et al. 1998; Cha & Shapiro 2001; Willard & Crouch 2001). However, regulatory functions of the ERK pathway proteins and of the mitotic substrates at these intracellular locations remain largely unknown.
Extracellular signal‐regulated kinase pathway regulation during G2/M‐phase progression may involve protein kinase C (PKC) isoforms. Earlier studies have suggested that PKC proteins activate Raf‐1 through direct phosphorylation of serines 259 and 499 (Kolch et al. 1993). However, others have suggested that these phosphorylation sites are not required for Raf‐1 activation (Barnard et al. 1998; Schonwasser et al. 1998). Alternatively, PKC may indirectly activate Raf‐1 by phosphorylating and inactivating the Raf kinase inhibitory protein (RKIP) (Corbit et al. 2003). These studies proposed that phosphorylated RKIP dissociates from Raf‐1, which allows Raf‐1 activation to occur.
The classical PKC isoforms (α, βI, βII, and γ) are activated by diacylglycerol (DAG) and calcium, whereas activation of the novel PKC isoforms (δ, ɛ, η, and θ) only requires DAG (Ron & Kazanietz 1999). DAG analogues such as phorbol esters, are potent activators of the classical and novel PKC isoforms (Ron & Kazanietz 1999). Long‐term exposure (> 12 h) to phorbol 12‐myristate‐13‐acetate (PMA) has been reported to induce G2‐phase arrest through a mechanism involving phospholipid metabolites (Kaszkin et al. 1991) or G1‐phase arrest through inhibition of cyclin dependent kinases and activation of p21 CIP1 and p27 KIP1 expression (Hamada et al. 1996; Frey et al. 1997). Depending on the cell line, PKC isoforms may promote or inhibit G2/M‐phase progression. For example, PKC activity has been reported to decrease in mitotic glioma cells (Soma et al. 1994), but is elevated in promyelocytic leukaemia cells and has been involved in mitotic nuclear envelope breakdown (Goss et al. 1994). MCF‐7 breast cancer cells treated with PMA immediately after release from aphidicolin‐induced S‐phase arrest have shown increased p21 CIP1 expression and subsequent accumulation of cells in the G2 phase (Barboule et al. 1999). However, the signalling events that mediate the G2‐phase arrest in response to PMA have not been determined.
The goal of the current study was to examine the consequence of ERK activation in regulating G2/M‐phase progression. Our findings demonstrate that ERK activation during the G2 phase delays cell cycle progression into mitosis through a mechanism involving de novo synthesis of p21 CIP1 . In addition, ERK activation during G2 phase causes an increase in the hallmarks of chromosome instability, a phenomenon that is exacerbated in cells lacking p21 CIP1 . These data suggest that ERK‐mediated induction of p21 CIP1 during G2 phase and cell cycle arrest may protect cells from excessive DNA damage that might occur during mitotic transitions and result in compromised cell survival.
MATERIALS AND METHODS
Cell culture and reagents
HeLa cells (#CCL‐2, ATCC, Manassas, VA), human retina epithelial cells that stably express human telomerase reverse transcriptase (hTERT‐RPE cells, #CRL‐4000; ATCC, Manassas, VA), HCT116 parental, or HCT116 p21−/– cells (kindly provided by Dr Bert Vogelstein, Johns Hopkins University) were cultured in a complete medium consisting of Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum and antibiotics (penicillin, 100 U/ml; streptomycin, 100 µg/ml) from Invitrogen (Carlsbad, CA). Epidermal growth factor (EGF) and PMA were purchased from Sigma (St. Louis, MO) and used at final concentrations of 0.01–100 ng/ml and 0.01–0.1 µm, respectively. The MEK1/2 inhibitor, U0126, and the PKC inhibitor, bisindolylmaleimide (Bis) I, were purchased from Calbiochem (La Jolla, CA) and used at final concentrations of 10 µm and 1 µm, respectively. The proteasome inhibitor, MG115, and protein synthesis inhibitor, cyclohexamide, were purchased from Calbiochem and used at final concentrations of 10 µg/ml and 10 µm, respectively.
Antibodies specific for cyclin B1 (sc‐245) and p21 CIP1 (sc‐397) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific for phosphorylated ERK1/2 (M‐8159) and α‐tubulin (T‐6557) were purchased from Sigma. Immunoblotting analysis was performed as previously described (Dangi et al. 2003).
Cells were synchronized at the G1/S‐phase boundary using a double thymidine block as previously described (Dangi et al. 2003). Briefly, cells (approximately 50% confluent) were treated with 2 mm thymidine in complete medium for 16 h. The cells were released back into the cell cycle by washing with Hanks buffered saline solution (HBSS, Invitrogen) and were incubated for an additional 8 h in complete medium in the absence of thymidine. Cells were treated a second time with 2 mm thymidine in complete medium for 16 h, which resulted in an average of 85–90% of them synchronized at the G1/S‐phase boundary of the cell cycle (Dangi et al. 2003). Synchronized cells were washed with HBSS, were released back into the cell cycle and were harvested at various times after release with or without the indicated treatments. Protein lysates were collected from synchronized cells after two washes with cold phosphate buffered saline (PBS, pH 7.2; Invitrogen) by scraping with 300 µl of cold tissue lysis buffer (20 mm Tris‐base, pH 7.4, 137 mm NaCl, 2 mm ethylenediamenetetraacetic acid (EDTA), 1% Triton X‐100, 25 mmβ‐glycerophosphate, 2 mm sodium pyrophosphate, 10% glycerol, 1 mm sodium orthovanadate, 1 mm phenylmethylsulphonyl fluoride (PMSF), 1 mm benzamidine). The lysates were centrifuged at 20 000 (Xg) to remove insoluble material, were diluted with an equal volume of 2X sodium dodecyl sulphate (SDS)‐sample buffer, and the proteins were separated on SDS‐polyacrylamide gel electrophoresis for immunoblot analysis.
Immunofluorescence and mitotic index assays
HeLa or HCT116 cells were grown on round coverslips (No. 1, 18 mm; VWR, West Chester, PA) in 6‐cm tissue culture plates and then were synchronized at the G1/S boundary as described in the previous section. Cells were released back into the cell cycle for 7 h, which corresponded to G2 phase, and were treated with PMA or EGF. In some cases, 20–30 min prior to PMA or EGF treatment, cells were pre‐treated in the presence or absence of Bis or U0126. Immunofluorescence was performed as previously described (Cha & Shapiro 2001). Briefly, at varying times after release, coverslips were fixed with 4% paraformaldehyde diluted in PBS, from a 16% stock (Electron Microscopy Sciences, Hatfield, PA) for 5 min and were then permeabilized with 0.1% triton X‐100 for 3 min. Alternatively, cells were fixed in cold methanol (−20 °C) for 10 min prior to staining. Following blocking with bovine serum albumin, cells were incubated with antibodies against γ‐tubulin or p62 nucleoporin followed by fluoroscein or Texas red‐conjugated secondary antibodies. Cellular DNA was stained with 4′, 6‐diamidino‐2‐phenylindole (DAPI, 0.2 µg/ml in PBS) for 5 min. Mitotic chromosomes were identified, based on their characteristic condensed structure, using a Nikon E800 fluorescence microscope, and photographs were taken using a Hamamatsu CCD camera (ORCA‐ER‐285; Biovision Technologies, Exton, PA, USA). Images of mitotic cells, which included cells in prophase, pro‐metaphase, metaphase, anaphase and telophase, were processed using iplab software (Scanalytics, Inc. Fairfax, VA). Mitotic cells were expressed as a fraction of total cells counted to determine the mitotic index (MI). Cells containing multiple nuclei (two or more) were counted and expressed as a percentage of the total cell number. Centrosomes were identified by γ‐tubulin staining and cells containing greater that two centrosomes were expressed as a percentage of the total cell number. Approximately 250–300 cells were counted for each condition.
Flow cytometry
Synchronized cells were trypsinized, washed with cold PBS, fixed with cold (−20 °C) 70% ethanol and stored at 4 °C overnight. Cells were then incubated with 100 µg/mL propidium iodide (Sigma) dissolved in 0.2 M Tris pH 7.5, 20 mm EDTA, 1 mg/ml RNase A (Sigma) for 1 h at room temperature and were then diluted with an equal volume of PBS. DNA content was measured by flow cytometry (FACScan Analyser; Becton Dickinson, Franklin Lakes, NJ) and was analysed using the Sync Wizard Model, ModFit LT software (Becton Dickinson). Fifteen thousand cells were counted under each condition. To determine G1, S, and G2/M‐phase cell populations, the settings for 2 N and 4 N DNA content peaks were obtained within each experiment using the G1/S arrested cells (2N) as the reference and applied to all samples within a given experiment.
RESULTS
PKC mediates PMA‐induced G2 arrest
Protein kinase C proteins are linked to the regulation of the ERK pathway and cell cycle progression. Using PMA as a potent activator of PKC, we first addressed the requirement for PKC and ERK signalling in mediating PMA‐induced cell cycle delay during G2 phase. HeLa cells synchronized at the G1/S phase boundary were released back into the cell cycle, pre‐treated in the absence or presence of the PKC inhibitor, Bis during G2 phase, and were then stimulated with or without PMA. Cells harvested at various times after release were analysed by flow cytometry for cell‐cycle progression and ERK activation. Fluorescence‐activated cell sorter (FACS) analysis of DNA content showed that untreated cells enter G2/M phase after 7 h release from G1/S, exit mitosis, and return to G1 phase 13 h after release from the initial G1/S‐phase block (Fig. 1a). In contrast, PMA‐treated cells remain primarily in G2/M phase up to 13 h after release from G1/S block (Fig. 1a). The PMA‐induced arrest in G2/M phase was reversed by the PKC inhibitor, Bis I (Fig. 1a). Delays in G2/M‐phase progression were also observed in non‐transformed telomerase immortalized human retinal pigment epithelial (hTERT‐RPE) cells (Fig. 1b) or HEK293 cells (data not shown) treated with PMA during G2 phase.
Figure 1.
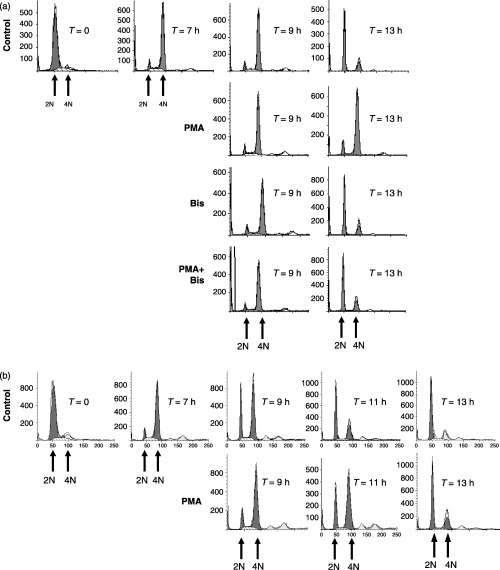
Fluorescence‐activated cell sorting (FACS) analysis of PMA‐mediated G2‐phase arrest. HeLa cells were synchronized at G1/S boundary and released back into the cell cycle. (a) Cells were left untreated (control) or treated with PMA (0.1 µm) at 7 h after G1/S release, with the bisindolylmaleimide (Bis, 1 µm) at 6.5 h after G1/S release, or with PMA and Bis together. (b) FACS analysis of synchronized hTERT cells treated in the absence (control) or presence of PMA at 7 h after G1/S‐phase release. Cells were harvested at the times indicated after release from G1/S and the number of cells (Y axis) containing 2N or 4N DNA content (X axis) were determined by FACS.
In agreement with previous studies (Roberts et al. 2002), basal ERK activity was elevated in G2/M phase and PMA treatment caused sustained ERK activation for up to 6 h (Fig. 2a). Bis had little effect on basal ERK activity but it did inhibit PMA‐induced ERK activity (Fig. 2a). In control cells, cyclin B1 expression showed a typical transient expression as cells progressed through G2 phase (7 and 7.3 h after G1/S release), mitosis (9 h), and back into G1 phase (11 and 13 h). In contrast, cyclin B1 expression in PMA‐treated cells was sustained for up to 13 h and supported the FACS data, indicating that these cells were arrested in the G2 or M phase (Fig. 2a). However, pre‐treatment with Bis before PMA restored the transient expression of cyclin B1 indicate that PKC activity is required for maintaining PMA‐induced G2/M‐phase arrest (Fig. 2a).
Figure 2.
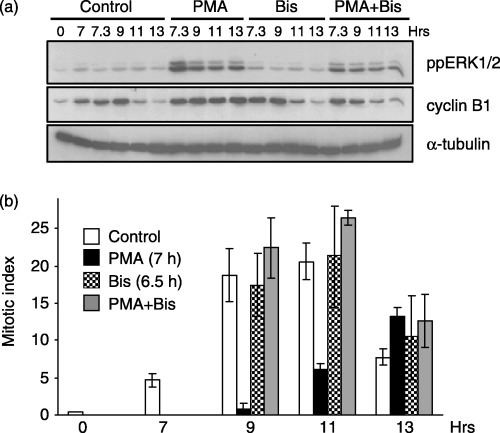
PMA‐induced G2‐phase arrest requires PKC activity. Cells synchronized at the G1/S boundary were released back into the cell cycle for 6.5 h and pre‐treated with or without Bis (1 µm). At 7 h post‐G1/S release, cells were treated in the absence or presence of PMA (0.1 µm) and harvested at the times indicated. (a) Immunoblot analysis of active ERK1/2 (ppERK1/2, top panel) and cyclin B1 (middle panel) as the marker of G2/M progression. The expression of α‐tubulin (lower panel) is shown for a protein loading control. (b) The MI at various times after G1/S release in controls (open bars), PMA‐treated (black bars), Bis‐treated (checkered bars), or Bis‐ and PMA‐treated (grey bars) cells. Data represent the mean and standard error from three independent experiments.
To determine whether the PMA‐induced cell cycle delay was occurring in the G2 or the M phase, the MI was evaluated under various conditions. As shown in Fig. 2b, the MI of PMA‐treated cells at 9 and 11 h after G1/S release was greatly reduced compared to untreated control cells. By 13 h after release, the MI of PMA‐treated cells began to increase, indicating a more than 4 h delay in mitotic entry (Fig. 2b). The delay in mitotic entry was reversed by pre‐treatment with Bis (Fig. 2b), supporting the requirement for PKC in mediating PMA‐induced G2‐phase arrest.
ERK mediates PMA‐induced G2‐arrest
We next examined the requirement for ERK signalling in mediating PMA‐induced G2‐phase arrest. G2‐phase cells generated as described in Fig. 1 were pre‐treated in the absence or presence of the MEK1/2 inhibitor, U0126, followed by PMA stimulation. Similar to inhibition of PKC, inhibition of ERK activation by U0126 also reversed most of the PMA‐induced G2‐arrest (Fig. 3). As expected, U0126 blocked PMA‐induced ERK activation (Fig. 4a) and reversed the PMA‐induced G2‐phase arrest as indicated by MI measurements (Fig. 4b). In addition, U0126 treatment also enhanced the MI at 13 h post‐G1/S‐phase release (Fig. 4b) and did not reverse elevated cyclin B1 expression (Fig. 4a). These findings indicated a delay in mitotic progression and are consistent with our previous studies that have demonstrated a requirement for ERK during metaphase to anaphase transitions (Roberts et al. 2002). Thus, these data indicate that PMA‐induced G2‐phase arrest requires PKC activation of the ERK pathway.
Figure 3.
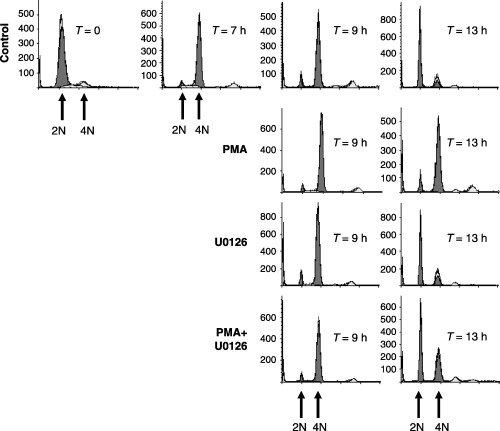
FACS analysis of ERK dependency in mediating PMA‐induced G2‐phase arrest. HeLa cells were synchronized at the G1/S boundary and released back into the cell cycle. Cells were left untreated (control), treated with PMA (0.1 µm) at 7 h after release, treated with U0126 (10 µm) at 6.5 h after G1/S release or treated with PMA and U0126 together. Cells were harvested at the times indicated after release from G1/S and the number of cells (Y axis) containing 2N or 4N DNA content (X axis) were determined by FACS.
Figure 4.
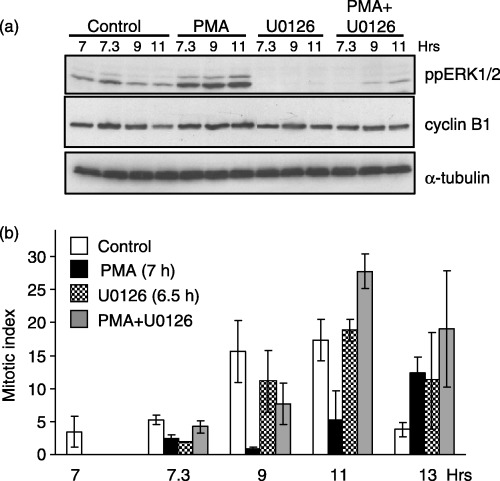
PMA‐induced G2‐phase arrest requires ERK activity. Cells synchronized at the G1/S boundary were released back into the cell cycle and treated in the presence of absence of PMA (0.1 µm, 7 h after G1/S release), U0126 (10 µm, 6.5 h after G1/S release), or PMA and U0126 together. (a) Immunoblot analysis of ppERK1/2 (top panel), cyclin B1 (middle panel), and α‐tubulin (lower panel) as a protein loading control. (b) The MI at varying times after G1/S release in untreated (white bars), PMA‐treated (black bars), U0126‐treated (checkered bars), or PMA‐ and U0126‐treated cells (grey bars). Data represent the mean and standard error from three independent experiments.
Activation of ERK during G2 phase by EGF delays M‐phase progression
We next tested whether activating the ERK pathway by EGF during G2 also affected G2/M progression. Cells synchronized in the G2 phase were pre‐treated with or without U0126 and then were treated in the presence or absence of EGF. FACS analysis indicated that EGF mediated an approximately 2 h delay in G2/M progression compared to controls (Fig. 5, 11 h time point) and that this delay was reversed by pre‐treatment with U0126 (Fig. 5). As expected, EGF activation of ERK was inhibited by U0126 (Fig. 6a). Cyclin B1 degradation and mitotic entry as determined by MI were also delayed by approximately 2 h in cells treated with EGF in the G2 phase and this was reversed by the presence of U0126 (Fig. 6a,b). As in Fig. 5b, U0126‐treated cells showed an elevated MI at the 13‐h time point (Fig. 6b), indicative of a delay in metaphase to anaphase transitions (Roberts et al. 2002). These data provide further support that the level of ERK activity during G2 phase regulates mitotic entry and progression.
Figure 5.
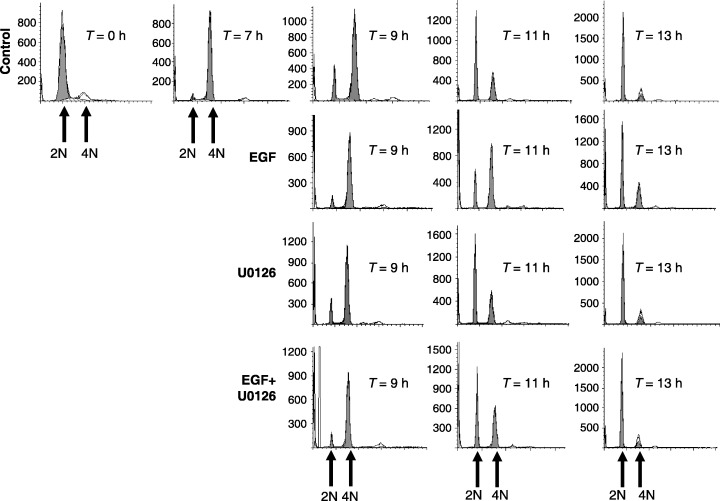
FACS analysis of ERK dependency in mediating EGF‐induced delay in G2/M progression. HeLa cells were synchronized at the G1/S boundary and released back into the cell cycle. Cells were left untreated (control), treated with EGF (100 ng/mL) at 7 h after G1/S release, treated with U0126 (10 µm) at 6.5 h after G1/S release, or treated with EGF and U0126 together. Cells were harvested at the times indicated after release from G1/S and the number of cells (Y axis) containing 2N or 4N DNA content (X axis) were determined by FACS.
Figure 6.
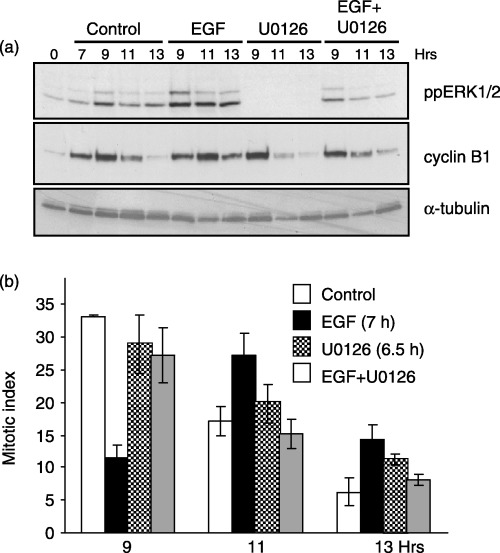
EGF‐induced G2‐phase arrest requires ERK activity. Cells synchronized at the G1/S boundary were released back into the cell cycle and treated with EGF (100 ng/ml, 7 h after G1/S release), U0126 (10 µm, 6.5 h after G1/S release), or both EGF and U0126. (a) Immunoblots show the levels of ppERK1/2 (top panel), cyclin B1 (middle panel) and α‐tubulin (lower panel) as a protein loading control. (b) The MI at varying times after G1/S release in untreated (white bars), EGF‐treated (black bars), U0126‐treated (checkered bars), or EGF‐ and U0126‐treated cells (grey bars). Data represent the mean and standard error from three independent experiments.
An ERK activation threshold determines M‐phase progression
Our data suggest that PMA or EGF‐mediated activation of the ERK pathway during the G2 phase delays mitotic entry. We next determined whether an ERK activity threshold was needed to be reached before a delay in G2/M‐phase progression could be observed. To test this, cells synchronized at the G1/S boundary were released into G2 phase and were treated with varying doses of EGF or PMA. Treatment with 0.1 ng/ml of EGF caused a transient activation of the ERK pathway but had little effect on cyclin B1 expression (Fig. 7a). Increasing the EGF dose further enhanced the magnitude and duration of ERK activity and the expression of cyclin B1 at 11 h after G1/S release as compared to untreated controls (Fig. 7a). As another measure of cell cycle progression, the expression of the Cdk inhibitor p21 CIP1 was measured. Whereas the lowest dose of EGF that caused ERK activation had little effect on p21 CIP1, higher EGF doses increased p21 CIP1 expression in a manner that correlated with ERK activity and cyclin B1 expression (Fig. 7a).
Figure 7.
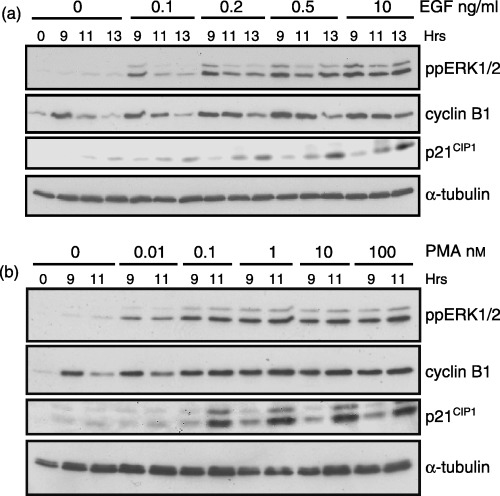
Cyclin B1 and p21CIP1 expression during G2/M‐phase correlate with ERK activity. Cells synchronized in G1/S were released back into the cell cycle and treated with the indicated dose of EGF (a) or PMA (b). Immunoblot analysis shows the level of ppERK1/2 (top panel), cyclin B1, and p21 CIP1 (middle panels) expression. The expression of α‐tubulin (lower panel) is shown for a protein loading control.
Similarly, the low doses of PMA (0.01 nm) activated ERK but had little effect on cyclin B1 expression as compared to controls (Fig. 7b). When PMA concentrations were increased by 10‐fold or more, maximal ERK activation was observed and a corresponding increase in cyclin B1 and p21 CIP1 expression consistent with G2‐phase arrest (Fig. 7b). Thus, the level of ERK activity during G2 phase appears to regulate cell cycle progression through regulation of p21 CIP1 expression.
ERK regulates p21 CIP1 expression during G2 phase
Several studies support a role for the CIP/KIP family of Cdk inhibitors in regulating cell cycle progression during G2 phase in response to various stress conditions (Niculescu et al. 1998; Coqueret 2003; Di Gennaro et al. 2003). Thus, the requirement for PKC and ERK proteins in regulating the expression of p21 CIP1 was examined in G2‐phase cells treated with PMA. Increased p21 CIP1 expression was observed within 2 h of PMA treatment (9 h after G1/S release) and continued to increase after 4 and 6 h treatment with PMA (11 and 13 h after G1/S release, respectively) (Fig. 8a,b). The increased p21 CIP1 expression corresponded to the accumulation of G2/M‐phase cells analysed previously by FACS analysis (1, 3) and the accumulation of cyclin B1 protein (2, 4). Inhibition of PKC or ERK activation during G2 phase with Bis or U0126, respectively, inhibited PMA‐induced p21 CIP1 expression (Fig. 8a,b). These data suggest that elevated PKC and ERK activities delay G2/M progression by increasing the expression of p21 CIP1 during G2 phase.
Figure 8.
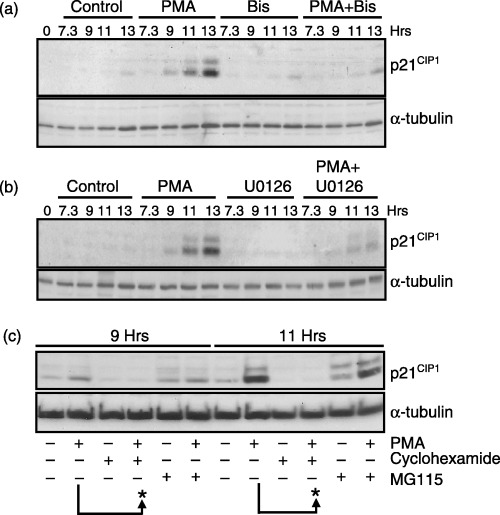
De novo synthesis of p21CIP1 during G2‐phase requires PKC and ERK activity. HeLa cells were synchronized at the G1/S boundary, released, and harvested at the times indicated. (a) Immunoblot analysis of p21 CIP1 expression in control cells or cells treated with PMA, Bis, or PMA plus Bis as described in 1, 2. (b) Immunoblot of p21 CIP1 in control cells or cells treated with PMA, U0126, or PMA plus U0126 as described in 3, 4. (c) Immunoblot analysis of p21 CIP1 in untreated or cells treated with PMA in the presence or absence of cyclohexamide (10 µm) or MG115 (10 µg/mL) at 7 h after G1/S‐phase release. The arrows and asterisks highlight the expression of p21 CIP1 at 9 or 11 h after G1/S release in cells treated with PMA minus or plus cyclohexamide or MG115. Τhe expression of α‐tubulin (lower panel) is shown for a protein loading control in a, b, and c.
It was next investigated whether PMA‐induced expression of p21 CIP1 during G2 phase was the result of increased protein synthesis or to chromosome stability. Synchronized cells were treated with cyclohexamide at 7.5 h after G1/S release to block protein synthesis. After 30 min, cells were treated with PMA (8 h after G1/S release) and harvested after an additional 1 or 3 h incubation (9 and 11 h post‐G1/S‐phase release). As shown, cyclohexamide blocked basal and PMA‐induced p21 CIP1 expression at 9 and 11 h post‐G1/S release (Fig. 8c). In contrast, inhibition of the proteasome and protein degradation with MG115 had little effect on PMA‐induced p21 CIP1 expression (Fig. 8c). MG115 treatment in the absence of PMA caused a small increase in p21 CIP1 expression, suggesting a role for the proteasome in p21 CIP1 turnover during G2 and M phase (Fig. 8c). These data indicate that one mechanism for PMA‐mediated G2‐phase arrest is through ERK‐mediated de novo synthesis of p21 CIP1 .
p21 CIP1 is required for PMA‐induced G2‐phase arrest
To demonstrate that p21 CIP1 expression is required for PMA‐induced G2‐phase arrest, cell cycle progression was monitored in HCT116 colorectal carcinoma cells lacking p21 CIP1 . FACS analysis showed that greater than 85% of both parental and p21 CIP1 knockout cells synchronized at the G1/S‐phase boundary with 2N DNA content using excess thymidine (data not shown). Treatment with PMA at 6 h after release, when most cells have entered G2/M‐phase, caused a delay in mitotic progression as measured by FACS in the parental but not the cells lacking p21 CIP1 (Fig. 9a,b). Examination of mitotic indices demonstrated that PMA induced a 2‐h delay in mitotic entry in parental cells but had no effect on mitotic entry in cells lacking p21 CIP1 (Fig. 9c,d). Although the data also show that cells lacking p21 CIP1 progress through G2/M‐phase at different rates than the parental cells, these findings nevertheless indicate that p21 CIP1 is required for PMA‐induced G2‐phase arrest.
Figure 9.
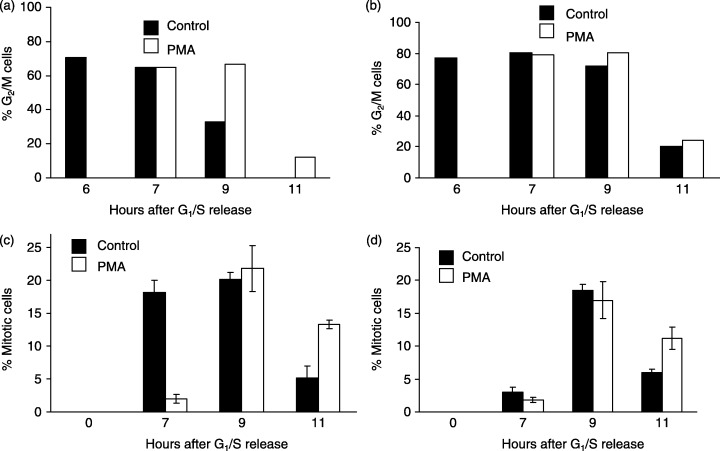
Expression of p21CIP1 is required for PMA‐induced G2‐phase arrest. HCT116 parental (a and c) or p21 CIP1 knockout cells (b and d) were synchronized at the G1/S‐phase boundary, treated in the absence (closed bars) or presence (open bars) of 0.1 µm PMA at 6 h after release, and harvested at the times indicated after G1/S release for FACS analysis of G2/M‐phase cells (a and b) or mitotic indices (c and d).
A role for ERK‐mediated p21 CIP1 expression during G2 phase in regulating chromosome stability
The last set of experiments described here examined the consequence of ERK activation during G2 phase and p21 CIP1 expression, on hallmark characteristics of chromosome stability. Defects in mitosis and cytokinesis are manifested by cells that are binucleate and/or contain multiple centrosomes; both of which are indicators of chromosome instability (Saavedra et al. 1999; , Fenech 2002), among the cells described in these experiments. HeLa cells synchronized in G2 were treated with EGF in the presence or absence of U0126, and then were incubated for 17 h to allow completion of one cell cycle, and then were processed for immunofluorescence. Approximately 15% of the cells treated with EGF were bi‐nucleate compared to approximately 3% in untreated cells (Fig. 10a,b). The increase in bi‐nucleate cells caused by EGF could be prevented with U0126 treatment (Fig. 10a). This response was unique to ERK activation during the G2 phase, as asynchronous cells treated with EGF showed only a slight increase in bi‐nucleate cells as compared to G2‐phase synchronized cells treated with EGF (Fig. 10c). Similarly, G2‐phase cells treated with EGF also exhibited an increase in the number of cells containing greater than two centrosomes, which also was partially prevented with U0126 (Fig. 10d). These findings suggest that ERK activation during G2 phase promotes cellular changes that are consistent with chromosome instability.
Figure 10.
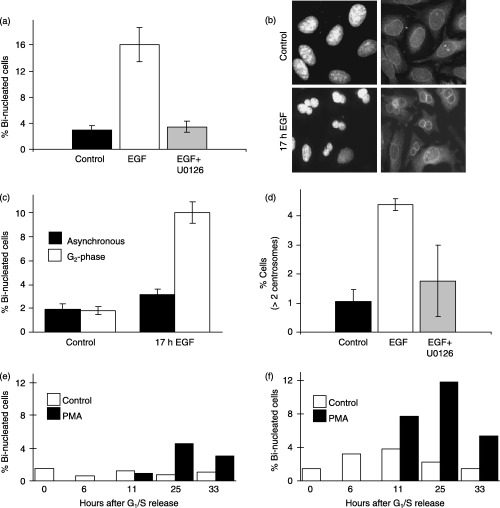
ERK activation during G2‐phase promotes hallmarks of chromosome instability; protective function of p21CIP1 expression. (a) Cells synchronized at the G1/S‐phase boundary were released for 7 h and treated with or without EGF (10 ng/mL) in the absence or presence of U0126 (10 µm). After 17 h, cells were immunostained for p62 nucleoporin as a marker of the nuclear envelope and counter‐stained with DAPI. The graph shows the percentage of bi‐nucleated cells under each condition. (b) Representative image of DAPI and p62 nucleoporin staining in control and EGF treated cells showing bi‐nucleated cells. (c) Percentage of bi‐nucleated cells in asynchronous or G2‐phase synchronized cells treated for 17 h in the absence or presence of EGF. (d) Percentage of cells containing greater than two centrosomes in control, EGF or EGF plus U0126 treated cells as described for (a). HCT116 parental (e) or p21 CIP1 knockout cells (f) were synchronized at G1/S phase, treated in the absence or presence of PMA at 6 h after release, and the percentage of bi‐nucleated cells were determined at the times indicated.
Finally, a function for p21 CIP1 expression during G2 phase in regulating the generation of bi‐nucleate cells was examined in HCT116 parental and p21 CIP1 knockout cells. Synchronized cells were treated during G2 phase in the absence or presence of PMA. At times after PMA treatment, cells were fixed and stained with DAPI for nuclear analysis. Similar to EGF, PMA treatment increased the number of bi‐nucleate cells in both parental and p21 CIP1 knockout cells (Fig. 10e,f). However, in the absence of p21 CIP1, there were substantially more bi‐nucleate cells in untreated or PMA‐treated conditions, as compared to the parental cells (Fig. 10e,f). The ratio of PMA to unstimulated cell was also expressed using the basal levels of bi‐nucleate cells in parental or p21 CIP1 knockout cells. When five time points for the unstimulated conditions were averaged, the p21 CIP1 knockout cells showed approximately 2.5‐fold more bi‐nucleate cells compared to the parental cells in the absence of PMA treatment. When this difference is taken into account, PMA still induced approximately threefold more bi‐nucleate cells in p21 CIP1 knockout cells compared to the parental cells at the 11‐h time point (data not shown). However, at the 25‐ or 33‐h time points, this analysis showed no difference in the PMA‐induced bi‐nucleate cells between the p21 CIP1 knockout cells and the parental cells (data not shown). Thus, although the magnitude in number of bi‐nucleate cells in p21 CIP1 knockout cells treated with or without PMA are greater than the parental cells (Fig. 10e,f), the fold increase as a result of PMA is only observed during the first cell cycle. Nevertheless, these data support a role for p21 CIP1 expression during G2 phase in providing protection against a phenotype characteristic of chromosome instability.
DISCUSSION
Using synchronized cells, these studies demonstrate that activation of the ERK pathway during G2 phase with PMA or EGF will initiate a rapid delay in G2 to M‐phase progression. The ERK‐mediated delay during the G2 phase appears to occur, in part, through the de novo synthesis of the cell‐cycle inhibitor p21 CIP1 in p53 defective cells. PMA or EGF dose–response assays indicated that some ERK activity above basal levels is tolerated during G2 phase. However, once an ERK activity threshold was reached, then the delay in G2‐phase progression could be observed. A potential consequence of activated ERK during G2 phase is an increased number of bi‐nucleate cells that, in this case, are an indicator of defects in mitosis and cytokinesis and a characteristic of chromosome instability. Although a role for the ERK pathway during PMA or EGF‐induced G1 or G2‐phase arrest was implied previously (Kinzel et al. 1990; Kaszkin et al. 1991; Kosaka et al. 1996; Barboule et al. 1999), the current studies are to our knowledge, the first to show a link between ERK activity and the regulation of p21 CIP1 expression during G2 phase.
The function of the ERK pathway in modulating G2 and M‐phase transitions remains largely unknown. Recent studies using pharmacological inhibitors, catalytically inactive mutants of ERK pathway proteins, and RNA interference suggests that a basal ERK activity is important for G2 and M‐phase progression in several transformed and non‐transformed cell lines (Abbott & Holt 1999; Wright et al. 1999; Hayne et al. 2000; Roberts et al. 2002; Liu et al. 2004). In addition, activation of the ERK pathway caused by over‐expression of constitutively active Ras, Raf‐1, or MEK1 mutants can induce a p53‐dependent cell cycle arrest and senescence in non‐transformed cells (Lin et al. 1998; Zhu et al. 1998; Ferbeyre et al. 2002). Moreover, over‐expression of the Mos kinase and subsequent ERK activation caused a p53‐dependent cell‐cycle arrest characterized by an increased number of bi‐nucleate cells (Fukasawa & Vande Woude 1997). Thus, although some basal level of ERK activity is needed during G2/M phase, ERK activation above basal levels may also be detrimental at this time of the cell cycle.
Our studies used primarily p53 defective cells, which have suggested that G2‐phase arrest mediated by ERK activation and p21 CIP1 induction is regulated through p53‐independent mechanisms. This is in agreement with others that used conditional activation of Raf kinase and ERK to cause a G1‐phase arrest in mouse fibroblasts through p53‐independent induction of p21 CIP1 (Woods et al. 1997). Thus, p53‐independent mechanisms for cell cycle control as previously suggested (Taylor & Stark 2001) and p53‐dependent mechanisms appear to function in ERK‐mediated G1 or G2‐phase arrest.
One role for G2‐phase arrest as a result of ERK activation may be to protect cells from signals that may interfere with mitotic transitions. A major function for the ERK pathway is to mediate transcription events in response to extracellular signals. However, the structural constraints imposed by condensed chromosomes result in transcription inhibition in mitotic cells. Thus, elevated ERK activity throughout the cell during G2 phase might be sensed as an inappropriate activation of transcription events that would detract from the high energy requirements needed for the dynamic structural changes that occur during mitosis. To compensate, our findings suggest that ERK may negatively regulate the cell cycle by imposing a G2‐phase checkpoint through induction of p21 CIP1 until ERK activity returns to levels that are compatible with proper mitotic transitions.
Increased expression of p21 CIP1, which is found in some cancer cells, may act to protect cells against apoptosis‐inducing agents (Gartel & Radhakrishnan 2005). Similarly, activation of the ERK pathway in cancer cells may be involved in the development of drug resistance (Davis et al. 2003). ERK‐induced G2‐phase arrest following treatment with genotoxic agents, such as doxorubicin, or microtubule‐interfering drugs, such as paclitaxel, may provide a protective mechanism that prevents cells with extensive DNA damage from entering mitosis and generating additional damage that would initiate an apoptotic response. Thus, G2‐phase arrest may allow the cells to repair DNA damage induced by chemotherapeutic drugs. As such, efforts towards simultaneous inhibition of ERK or other signalling pathways may be a more effective approach to treating drug‐resistant cancer cells. For example, simultaneous inhibition of the PKC, Chk1, and the ERK pathway with UCN‐01 and MEK1/2 inhibitors has been shown to be effective in killing leukaemia cells that harbour the BCR‐Abl fusion protein and are resistant to the anticancer drug ST1571, also known as Gleevec (Yu et al. 2002).
The role of other MAP kinase signalling pathways in regulating cell cycle progression through G2 phase and mitosis may also play a role during genotoxic or other stress conditions. Activation of p38 MAP kinase was reported to mediate G2‐phase arrest in response to genotoxic or osmotic stress (Bulavin et al. 2001; Dmitrieva et al. 2002; Hirose et al. 2003). Similarly, conditional activation of MEKK3, a kinase that mediates inflammatory responses, induces G2‐phase arrest through a mechanism that is partially dependent on p38 MAP kinase activity (Ellinger‐Ziegelbauer et al. 1999; Garner et al. 2002). Several MAP kinases may mediate cell cycle arrest in the context of breast cancer cell lines treated with the chemotherapeutic drug paclitaxel, which causes G2‐phase arrest and apoptosis in a manner that requires ERK and p38 MAP kinase activities but not a functional p53 protein (Bacus et al. 2001). However, in our studies, we have found no evidence that p38 MAP kinase is activated in G2 or M‐phase cells treated with or without PMA (data not shown). This agrees with others who report that PMA is a potent activator of ERK but not p38 MAP kinase (Morton et al. 2004).
During DNA damage or microtubule disruption, the c‐Jun N‐terminal kinase (JNK) pathway may also promote G2‐phase arrest and susceptibility to undergo apoptosis through a mechanism involving inhibition of cdc2 and the anti‐apoptotic protein Bcl‐2 (Yamamoto et al. 1999; Goss et al. 2003). Similar to ERK, a basal level of JNK activity may regulate proper progression through G2 and M phase of the cell cycle. Recent studies suggest that inhibition of JNK activity with relatively non‐specific chemical inhibitors or by genetic knockout of MKK7, a JNK activator, causes a delay in G2 to M‐phase progression (Mingo‐Sion et al. 2004; Wada et al. 2004). Although we have found no evidence that JNK proteins are activated in untreated or PMA‐treated synchronized HeLa cells during G2 phase (data not shown), the three major MAP kinase family members may co‐ordinate cell cycle progression during G2 phase in a manner that may be dependent on cell type and extracellular signal.
ACKNOWLEDGEMENTS
This work was supported in part by a grant from the National Institutes of Health‐National Cancer Institute (CA‐105299). We thank Dr Bert Vogelstein (Johns Hopkins University) for providing the HCT116 p21 CIP1 parental and knockout cell lines.
REFERENCES
- Abbott DW, Holt JT (1999) Mitogen‐activated protein kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J. Biol. Chem. 274, 2732–2742. [DOI] [PubMed] [Google Scholar]
- Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V (1998) Signaling via mitogen‐activated protein kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell 92, 183–192. [DOI] [PubMed] [Google Scholar]
- Aktas H, Cai H, Cooper GM (1997) Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1 . Mol. Cell. Biol. 17, 3850–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, Keyomarsi K, Yarden Y, Seger R (2001) Taxol‐induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 20, 147–155. [DOI] [PubMed] [Google Scholar]
- Barnard D, Diaz B, Clawson D, Marshall M (1998) Oncogenes, growth factors and phorbol esters regulate Raf‐1 through common mechanisms. Oncogene 17, 1539–1547. [DOI] [PubMed] [Google Scholar]
- Barboule N, Lafon C, Chadebech P, Vidal S, Valette A (1999) Involvement of p21 in the PKC‐induced regulation of the G2/M cell cycle transition. FEBS Lett. 444, 32–37. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ Jr (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107. [DOI] [PubMed] [Google Scholar]
- Cha H, Shapiro P (2001) Tyrosine‐phosphorylated extracellular signal‐regulated kinase associates with the Golgi complex during G2/M phase of the cell cycle. Evidence for regulation of Golgi structure. J. Cell Biol. 153, 1355–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF (1998) Assembly of cyclin D‐dependent kinase and titration of p27Kip1 regulated by mitogen‐activated protein kinase kinase (MEK1). Proc. Natl. Acad. Sci. USA 95, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ (1999) The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin d‐dependent kinases in murine fibroblasts. EMBO J. 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Sutterlin C, Malhotra V (2003) RAF1‐activated MEK1 is found on the Golgi apparatus in late prophase and is required for Golgi complex fragmentation in mitosis. J. Cell Biol. 161, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Marshall CJ, Olson MF (2003) Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1‐imposed block in proteasome‐mediated degradation. EMBO J. 22, 2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coqueret O (2003) New roles for p21 and p27 cell‐cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 13, 65–70. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR (2003) Activation of Raf‐1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J. Biol. Chem. 278, 13061–13068. [DOI] [PubMed] [Google Scholar]
- Yu C, Dai Y, Dent P, Grant S (2002) Coadministration of UCN‐01 with MEK1/2 inhibitors potently induces apoptosis in BCR/ABL+ leukemia cells sensitive and resistant to ST1571. Cancer Biol. Ther. 1, 674–682. [DOI] [PubMed] [Google Scholar]
- Dangi S, Cha H, Shapiro P (2003) Requirement for phosphatidylinositol‐3 kinase activity during progression through S‐phase and entry into mitosis. Cell Signal 15, 667–675. [DOI] [PubMed] [Google Scholar]
- Davis JM, Navolanic PM, Weinstein‐Oppenheimer CR, Steelman LS, Hu W, Konopleva M, Blagosklonny MV, McCubrey JA (2003) Raf‐1 and Bcl‐2 induce distinct and common pathways that contribute to breast cancer drug resistance. Clin. Cancer Res. 9, 1161–1170. [PubMed] [Google Scholar]
- Di Gennaro E, Barbarino M, Bruzzese F, De Lorenzo S, Caraglia M, Abbruzzese A, Avallone A, Comella P, Caponigro F, Pepe S, Budillon A (2003) Critical role of both p27KIP1 and p21CIP1/WAF1 in the antiproliferative effect of ZD1839 (‘Iressa’), an epidermal growth factor receptor tyrosine kinase inhibitor, in head and neck squamous carcinoma cells, J. Cell Physiol. 195, 139–150. [DOI] [PubMed] [Google Scholar]
- Dmitrieva NI, Bulavin DV, Fornace AJ Jr, Burg MB (2002) Rapid activation of G2/M checkpoint after hypertonic stress in renal inner medullary epithelial (IME) cells is protective and requires p38 kinase. Proc. Natl. Acad. Sci. USA 99, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinger‐Ziegelbauer H, Kelly K, Siebenlist U (1999) Cell cycle arrest and reversion of Ras‐induced transformation by a conditionally activated form of mitogen‐activated protein kinase kinase kinase 3. Mol. Cell. Biol. 19, 3857–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M (2002) Chromosomal biomarkers of genomic instability relevant to cancer. Drug Discov. Today 7, 1128–1137. [DOI] [PubMed] [Google Scholar]
- Ferbeyre G, De Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW (2002) Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell. Biol. 22, 3497–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey MR, Saxon ML, Zhao X, Rollins A, Evans SS, Black JD (1997) Protein kinase C isozyme‐mediated cell cycle arrest involves induction of p21(waf1/cip1) and p27(kip1) and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J. Biol. Chem. 272, 9424–9435. [DOI] [PubMed] [Google Scholar]
- Fukasawa K, Vande Woude GF (1997) Synergy between the mos/mitogen‐activated protein kinase pathway and loss of p53 function in transformation and chromosome instability. Mol. Cell. Biol. 17, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AP, Weston CR, Todd DE, Balmanno K, Cook SJ (2002) Delta MEKK3:ER* activation induces a p38 alpha/beta 2‐dependent cell cycle arrest at the G2 checkpoint. Oncogene 21, 8089–8104. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Radhakrishnan SK (2005) Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 65, 3980–3985. [DOI] [PubMed] [Google Scholar]
- Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, Fields AP (1994) Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J. Biol. Chem. 269, 19074–19080. [PubMed] [Google Scholar]
- Goss VL, Cross JV, Ma K, Qian Y, Mola PW, Templeton DJ (2003) SAPK/JNK regulates cdc2/cyclin B kinase through phosphorylation and inhibition of cdc25c. Cell. Signal. 15, 709–718. [DOI] [PubMed] [Google Scholar]
- Hamada K, Takuwa N, Zhou W, Kumada M, Takuwa Y (1996) Protein kinase C inhibits the CAK‐CDK2 cyclin‐dependent kinase cascade and G1/S cell cycle progression in human diploid fibroblasts. Biochim. Biophys. Acta. 1310, 149–156. [DOI] [PubMed] [Google Scholar]
- Hatakeyama M, Weinberg RA (1995) The role of RB in cell cycle control. Prog. Cell Cycle Res. 1, 9–19. [DOI] [PubMed] [Google Scholar]
- Hayne C, Tzivion G, Luo Z (2000) Raf‐1/MEK/MAPK pathway is necessary for the G2/M transition induced by nocodazole. J. Biol. Chem. 275, 31876–31882. [DOI] [PubMed] [Google Scholar]
- Hirose Y, Katayama M, Stokoe D, Haas‐Kogan DA, Berger MS, Pieper RO (2003) The p38 mitogen‐activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA‐methylating agents. Mol. Cell. Biol. 23, 8306–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaszkin M, Furstenberger G, Richards J, Seidler L, Kinzel V (1991) A proposed common mechanism by which phorbol esters and epidermal growth factor delay the progression from G2 phase to mitosis of HeLa cells through phospholipid metabolites. Cancer Res. 51, 4328–4335. [PubMed] [Google Scholar]
- Kinzel V, Kaszkin M, Blume A, Richards J (1990) Epidermal growth factor inhibits transiently the progression from G2‐phase to mitosis: a receptor‐mediated phenomenon in various cells. Cancer Res. 50, 7932–7936. [PubMed] [Google Scholar]
- Kivinen L, Laiho M (1999) Ras‐ and mitogen‐activated protein kinase kinase‐dependent and independent pathways in p21Cip1/Waf1 induction by fibroblast growth factor‐2, platelet‐derived growth factor, and transforming growth factor‐beta1, Cell Growth Differ. 10, 621–628. [PubMed] [Google Scholar]
- Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR (1993) Protein kinase C alpha activates RAF‐1 by direct phosphorylation. Nature 364, 249–252. [DOI] [PubMed] [Google Scholar]
- Kosaka C, Sasaguri T, Ishida A, Ogata J (1996) Cell cycle arrest in the G2 phase induced by phorbol ester and diacylglycerol in vascular endothelial cells. Am. J. Physiol. 270, C170–C178. [DOI] [PubMed] [Google Scholar]
- Laird AD, Taylor SJ, Oberst M, Shalloway D (1995) Raf‐1 is activated during mitosis. J. Biol. Chem. 270, 26742–26745. [DOI] [PubMed] [Google Scholar]
- Laird AD, Morrison DK, Shalloway D (1999) Characterization of Raf‐1 activation in mitosis. J. Biol. Chem. 274, 4430–4439. [DOI] [PubMed] [Google Scholar]
- Lents NH, Keenan SM, Bellone C, Baldassare JJ (2002) Stimulation of the Raf/MEK/ERK cascade is necessary and sufficient for activation and Thr‐160 phosphorylation of a nuclear‐targeted CDK2. J. Biol. Chem. 277, 47469–47475. [DOI] [PubMed] [Google Scholar]
- Leone G, Degregori J, Sears R, Jakoi L, Nevins JR (1997) Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 387, 422–426. [DOI] [PubMed] [Google Scholar]
- Lewis TS, Shapiro PS, Ahn NG (1998) Signal tranduction through MAP kinase cascades. Adv. Cancer Res. 74, 49–139. [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, Van Aelst L, Serrano M, Lowe SW (1998) Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 12, 3008–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Chao JR, Jiang MC, Ng SY, Yen JJ, Yang‐Yen HF (1995) Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3T3 cells. Mol. Cell. Biol. 15, 3654–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yan S, Zhou T, Terada Y, Erikson RL (2004) The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene 23, 763–776. [DOI] [PubMed] [Google Scholar]
- Mingo‐Sion AM, Marietta PM, Koller E, Wolf DM, Van Den Berg CL (2004) Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene 23, 596–604. [DOI] [PubMed] [Google Scholar]
- Mittnacht S, Paterson H, Olson MF, Marshall CJ (1997) Ras signalling is required for inactivation of the tumour suppressor pRb cell‐cycle control protein. Curr. Biol. 7, 219–221. [DOI] [PubMed] [Google Scholar]
- Morton S, Davis RJ, Cohen P (2004) Signalling pathways involved in multisite phosphorylation of the transcription factor ATF‐2. FEBS Lett. 572, 177–183. [DOI] [PubMed] [Google Scholar]
- Niculescu AB 3rd Chen X, Smeets M, Hengst L, Prives C, Reed SI (1998) Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 18, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH (2001) Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Peeper DS, Upton TM, Ladha MH, Neuman E, Zalvide J, Bernards R, Decaprio JA, Ewen ME (1997) Ras signalling linked to the cell‐cycle machinery by the retinoblastoma protein. Nature 386, 177–181. [DOI] [PubMed] [Google Scholar]
- Roberts EC, Shapiro PS, Nahreini TS, Pages G, Pouyssegur J, Ahn NG (2002) Distinct cell cycle timing requirements for extracellular signal‐regulated kinase and phosphoinositide 3‐kinase signaling pathways in somatic cell mitosis. Mol. Cell. Biol. 22, 7226–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Kazanietz MG (1999) New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 13, 1658–1676. [PubMed] [Google Scholar]
- Russell A, Hendley J, Germain D (1999) Inhibitory effect of p21 in MCF‐7 cells is overcome by its coordinated stabilization with d‐type cyclins. Oncogene 18, 6454–6459. [DOI] [PubMed] [Google Scholar]
- Saavedra HI, Fukasawa K, Conn CW, Stambrook PJ (1999) MAPK mediates RAS‐induced chromosome instability. J. Biol. Chem. 274, 38083–38090. [DOI] [PubMed] [Google Scholar]
- Schonwasser DC, Marais RM, Marshall CJ, Parker PJ (1998) Activation of the mitogen‐activated protein kinase/extracellular signal‐regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 18, 790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewing A, Wiseman B, Lloyd AC, Land H (1997) High‐intensity Raf signal causes cell cycle arrest mediated by p21Cip1 . Mol. Cell. Biol. 17, 5588–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro PS, Vaisberg E, Hunt AJ, Tolwinski NS, Whalen AM, McIntosh JR, Ahn NG (1998) Activation of the MKK/ERK pathway during somatic cell mitosis: direct interactions of active ERK with kinetochores and regulation of the mitotic 3F3/2 phosphoantigen. J. Cell Biol. 142, 1533–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma MR, Baetta R, Bergamaschi S, De Renzis MR, Davegna C, Battaini F, Fumagalli R, Govoni S (1994) PKC activity in rat C6 glioma cells: changes associated with cell cycle and simvastatin treatment. Biochem. Biophys. Res. Commun. 200, 1143–1149. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Stark GR (2001) Regulation of the G2/M transition by p53. Oncogene 20, 1803–1815. [DOI] [PubMed] [Google Scholar]
- Wada T, Joza N, Cheng HY, Sasaki T, Kozieradzki I, Bachmaier K, Katada T, Schreiber M, Wagner EF, Nishina H, Penninger JM (2004) MKK7 couples stress signalling to G2/M cell‐cycle progression and cellular senescence. Nat. Cell Biol. 6, 215–226. [DOI] [PubMed] [Google Scholar]
- Weber JD, Raben DM, Phillips PJ, Baldassare JJ (1997) Sustained activation of extracellular‐signal‐regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem. J. 326 (1), 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard FS, Crouch MF (2001) MEK, ERK, and p90RSK are present on mitotic tubulin in Swiss 3T3 cells: a role for the MAP kinase pathway in regulating mitotic exit. Cell. Signal. 13, 653–664. [DOI] [PubMed] [Google Scholar]
- Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M (1997) Raf‐induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1 . Mol. Cell. Biol. 17, 5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JH, Munar E, Jameson DR, Andreassen PR, Margolis RL, Seger R, Krebs EG (1999) Mitogen‐activated protein kinase activity is required for the G(2)/M transition of the cell cycle in mammalian fibroblasts. Proc. Natl. Acad. Sci. USA 96, 11335–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ichijo H, Korsmeyer SJ (1999) BCL‐2 is phosphorylated and inactivated by an ASK1/Jun N‐terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 19, 8469–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic M, Catling AD, Eblen ST, Renzi L, Hittle JC, Yen TJ, Gorbsky GJ, Weber MJ (1998) Active MAP kinase in mitosis: localization at kinetochores and association with the motor protein CENP‐E. J. Cell Biol. 142, 1547–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Woods D, McMahon M, Bishop JM (1998) Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 12, 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziogas A, Lorenz IC, Moelling K, Radziwill G (1998) Mitotic Raf‐1 is stimulated independently of Ras and is active in the cytoplasm. J. Biol. Chem. 273, 24108–24114. [DOI] [PubMed] [Google Scholar]