

Abstract
The MET oncogene encodes the tyrosine kinase receptor for hepatocyte growth factor/scatter factor (HGF), known to stimulate invasive growth of epithelial cells. MET is overexpressed in a significant percentage of human cancers and is amplified during the transition between primary tumors and metastasis. To investigate whether this oncogene is directly responsible for the acquisition of the metastatic phenotype, we exploited a single-hit oncogenic version of MET, able to transform and to confer invasive and metastatic properties to nontumorigenic cells, both in vitro and in nude mice. We mutagenized the signal transducer docking site of Met (Y1349VHVX3Y1356VNV), which has the uncommon property of binding and activating multiple src homology region 2 (SH2)-containing intracellular effectors. Notably, a point mutation (H1351 → N) increased the transforming ability of the oncogene but abolished its metastatic potential. This mutation duplicates the Grb2 binding site, super-activating the Ras pathway and preventing the binding of the other intracellular transducers. Complementation in trans with another nonmetastatic mutant (N1358 → H), recruiting all the transducers downstream to Met except Grb2, rescued the invasive–metastatic phenotype. It is concluded that the metastatic potential of the MET oncogene relies on the properties of its multifunctional docking site, and that a single point mutation affecting signal transduction can dissociate neoplastic transformation from metastasis.
Metastasization is a complex multistep process, involving deregulated cell growth and acquisition of the ability to dissociate and to invade extracellular matrices (1). A number of oncogenes, including src and erbB2, impart tumorigenic and metastatic properties to transfected cells, and they are thought to play a role in the formation of naturally occurring metastasis in humans (2, 3). However, only the MET family oncogenes share the unique functional feature of mediating—other than growth—cell dissociation (“scattering”), motility, and invasion of extracellular matrices (4–6). The MET oncogene encodes the receptor for hepatocyte growth factor (7–9), otherwise known as “scatter factor”—a growth factor that also has angiogenic properties (10). MET is mutated in a small number of genetically inherited tumors (11) but is overexpressed at high frequency in sporadic cancers (5, 12) and is amplified during the transition between primary and metastatic cells (13). These findings suggest that MET provides neoplastic cells with a selective advantage for the acquisition of metastatic potential.
MATERIALS AND METHODS
Reagents and Cells.
All reagents, unless specified, were purchased from Sigma. Fisher rat fibroblasts were purchased from the American Type Culture Collection. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal calf serum (Flow Laboratories) in a 5% CO2/water-saturated atmosphere.
DNA Transfection and Focus-Forming Assay.
Cloning of TPR-MET cDNA and generation of the mutants by site-directed mutagenesis has been reported previously (14). Construction of the TPR-cSEA chimera has also been described (15). The TPR-vSEA construct was generated by ligating the 5′ portion of TPR-cSEA (PstI–ScaI) with the 3′ region of ENV-vSEA (ScaI-XbaI). Transfection of the constructs into Fisher rat fibroblasts was carried out by using the DNA/calcium phosphate coprecipitation procedure (CellPhect Transfection Kit, Pharmacia). For the focus-forming assay, after transfection cells were split at very low density and kept in DMEM/5% fetal calf serum. Formation of transformed foci was detected in 2–4 weeks.
Tumorigenicity and Experimental Metastasis Assay.
In vivo experiments were conducted in nude mice (6 in each group) injected with 106 transfected cells. For tumor growth evaluation, mice were injected s.c. and monitored for tumor growth. For experimental metastasis evaluation, mice were injected in the tail vein and examined after 3 weeks. The presence of multiple lung metastases was evaluated at necropsy and confirmed by histopathological observation of the lungs.
In Vitro Invasion Assay.
In this assay, 105 cells of each Fisher rat cell line were seeded on the upper side of a porous polycarbonate membrane (8.0 μm pore size) coated with the artificial basement membrane Matrigel (12.5 μg per filter; Collaborative Biomedical Products; Becton Dickinson Labware, Waltham, MA). After 48 hr of incubation the filters were removed and cells that invaded the Matrigel and attached to the lower chamber of the transwell were fixed with glutaraldehyde and stained with crystal violet. The assay was performed in the presence of 5% fetal calf serum.
Soft Agar Colony Assay.
For the soft agar colony assay 2,000 cells were resuspended in 1 ml of DMEM/5% fetal calf serum and 0.33% Noble agar (Difco) and plated in a six-well plate onto the same medium containing 0.6% Noble agar. The number of colonies containing more than 30 cells was counted at 21 days.
Immunoprecipitation and Western Blotting.
Cells were lysed with a 1% Triton X-100 in Hepes buffer, extracts were clarified by centrifugation, and protein concentration was determined by using the BCA protein assay system (Pierce). The same amounts of total proteins were immunoprecipitated with anti-Met antibodies and Western blotted as previously described (14).
RESULTS AND DISCUSSION
The high incidence of MET overexpression in primary cancers (5, 12) and of gene amplification at later stages, in metastases (13), suggests that the signals transduced by the activated tyrosine kinase via its multifunctional docking site confer to neoplastic cells favorable growth properties at the primary site and selective advantage for the acquisition of metastatic potential. This conclusion is supported by the observation that cells transfected with an activated version of the MET gene acquire invasive and metastatic properties, both in vitro and in nude mice (16, 17).
Because MET encodes a transmembrane receptor, its role in invasion and metastasis should rely on its signal transduction properties. We have previously shown that the intracellular signals generated by the Met receptor are mediated by phosphorylation of two critical tyrosines located in the carboxyl-terminal tail (18). The tandemly repeated degenerate sequence Y1349VHVX3Y1356VNV represents a “multifunctional” docking site mediating high-affinity interactions with multiple src homology region 2 (SH2)-containing cytoplasmic effectors. We and others have shown that the catalytic transducers phosphatidylinositol 3-kinase (PI3K), phospholipase Cγ (PLCγ), and pp60c-src, as well as the noncatalytic signal amplifiers Shc and Gab1, bind either of the two phosphotyrosines (18–21). Conversely, Grb2—the adaptor protein that links Met to the Ras pathway—selectively binds Y1356, because it requires an asparagine (N) in the second position downstream of the phosphorylated tyrosine (22). We have previously shown (14) that loss of the link with Grb2 severely impairs transformation, without affecting the “scattering” response. This indicates that the level of signal reached by Grb2-independent routes is permissive for motility but not for transformation and that these two Met-mediated responses can be dissociated on the basis of their differential requirement for a direct link with Ras. Moreover, we have also shown (23) that disruption of the consensus for Grb2 binding in the mouse homologue of the MET gene severely impairs the targeted migration of the myoblasts from cervical myotomes to peripheral muscles.
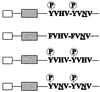
To identify the signaling pathways involved in metastasis we exploited an oncogenic version of MET (TPR-MET) able to transform cells in a single hit (16). Rat fibroblasts transformed by the rearranged oncogene acquired invasive–metastatic properties, in vitro and in vivo, as they (i) grew with anchorage-independent properties, (ii) invaded reconstituted basement membranes in vitro, and (iii) formed metastatic tumors in nude mice. Table 1 summarizes the phenotypes obtained after mutation of single amino acids of the Met “multifunctional” docking site. The two tyrosines (Y1349 and Y1356) are an absolute requirement, as shown by the fact that their replacement by phenylalanine (the “Double F” mutant) abrogated induction of both metastasis and foci of transformation. Asparagine (N1358) is also critical for malignant transformation, as shown by its mutation into histidine (H). This mutant (Grb2−), which still interacts with all the cytoplasmic signal transducers besides Grb2 (14), is severely impaired in its ability to activate the Ras pathway, because—as discussed above—an N in position +2 is required for recruiting the Grb2/SoS complex by means of the Y1356. The effect of enhancing Ras coupling at the expense of other signaling pathways was then studied by replacement of H1351 with an N. This results in the formation of a second high-affinity Grb2 binding site (YVNVX3YVNV). This mutant (2× Grb2) was highly transforming, but surprisingly, was unable to support the invasive–metastatic phenotype, both in vitro and in vivo. These results cannot be explained by different expression levels of the various Tpr-Met constructs, because the same amount of protein is found in cells transfected with the different mutants (Fig. 1). These findings show that the Ras pathway, although necessary, is not sufficient to induce metastasis, as other pathway(s) are required.
Table 1.
Biological Properties of TPR-MET mutants
| Tpr-Met protein* | Transformation,† % | Tumorigenicity‡ | Anchorage-independent growth§ | Invasion¶ | Metastasis‖ |
|---|---|---|---|---|---|
| |||||
| Wild type | 100 | 6/6 | 15 ± 3 | 130 ± 30 | 12/12 |
| Double F | 0 | ND | ND | ND | ND |
| Grb2− | 11 | 6/6 | 0 | <10 | 0/12 |
| 2× Grb2 | 150 | 6/6 | 0 | <10 | 0/12 |
Schematic representation of Tpr-Met wild type and of the Tpr-Met proteins bearing mutations in the “multifunctional” docking site. White boxes represent the Tpr moiety; gray boxes represent the Met tyrosine kinase domain. YVHV-YVNV indicates the sequence of the multifunctional docking site. The N, critical for Grb2 binding, is underlined.
Ability to induce foci of transformation, expressed as relative transforming activity (percent of TPR-MET).
Number of mice developing tumors 3 weeks after subcutaneous injection of 106 cells. ND, not done.
Ability to form colonies in soft agar, evaluated as number of colonies (>30 cells) grown after 21 days.
Ability to invade reconstituted basement membranes “in vitro,” evaluated as number of cells per microscopic field migrated into the transwell lower chamber after 48 hr.
Number of mice dead because of lung metastasis, 3 weeks after injection of 106 cells into the tail vein.
Figure 1.

Expression of Tpr-Met mutants in Fisher rat fibroblasts. The same amounts of Tpr-Met proteins (arrow) were immunoprecipitated by using Met-specific antibodies. Immunoprecipitated proteins were separated on SDS/10% PAGE, blotted, and probed with anti-Met antibodies.
Interestingly, a natural variant of the MET multifunctional docking site, containing a double Grb2 binding motif (YVNLX3YVNL) is found in the homologous avian protooncogene cSEA. On studying the biological properties of the recombinant oncogene TPR-SEA we observed that the avian oncogene is endowed with transforming activity (15), but is devoid of metastatic potential. The behavior of TPR-cSEA was comparable to that of the 2× Grb2 mutant of MET (Fig. 2). We interpret this result to mean that the presence of two contiguous high-affinity Grb2 binding sites prevents the binding of other (known or still unidentified) signal transducers that are critical for metastasis.
Figure 2.

(Left) Schematic representation of Tpr-Met and Tpr-Sea proteins. White boxes represent the Tpr moiety; gray boxes represent the Met tyrosine kinase domain; the black box represents the Sea tyrosine kinase domain. YVHV-YVNV indicates the sequence of the Met multifunctional docking site; YVNL-YVNL indicates the sequence of the Sea multifunctional docking site. The N, critical for Grb2 binding, is underlined. (Right) Transformation refers to ability to induce foci of transformation, expressed as relative transforming activity (percent of Tpr-Met wild type). Metastasis indicates percentage of mice dead because of lung metastases, 3 weeks after injection of 106 cells into the tail vein.
Thus, the invasive–metastatic phenotype induced by Met is based on the ability of its “multifunctional” docking site to concomitantly activate multiple signaling pathways. To prove this assumption we then tested whether the wild-type phenotype could be rescued by complementation in trans. Fig. 3 shows that cells cotransfected with the two inactive mutants (Grb2− and 2× Grb2) resumed the ability to invade in vitro reconstituted basement membrane, a feature known to have excellent concordance with the invasive–metastatic potential in vivo (24). Interestingly, we found that complementation in cis naturally occurs in the viral counterpart of cSEA (25), the oncogene vSEA carried by the S13 virus (26). In the viral protein, replacement of a leucine with a methionine in the multifunctional docking site (YVNLX3YVNM) generates an optimal, high-affinity, phosphatidylinositol 3-kinase binding site (22). Cells transfected with a TPR-vSEA construct displayed an invasive phenotype “in vitro” (Fig. 4).
Figure 3.

Rescue of the invasive phenotype by reconstitution in trans of Met multifunctional docking site. Cells expressing the wild-type Tpr-Met invaded the in vitro reconstituted basement membrane and migrated to the Transwell lower chamber. Invasion was severely impaired in cells expressing Tpr-Met mutants affecting the multifunctional docking site (Grb2− and 2× Grb2, see text). Coexpression of the two inactive mutants restored the ability to invade the basement membrane. Cells migrating into the lower chamber were fixed with glutaraldehyde and stained with crystal violet. (×4.)
Figure 4.

Cells expressing TPR-vSEA invaded the “in vitro” reconstituted basement membrane and migrated into the Transwell lower chamber. Invasion was severely impaired in cells expressing TPR-cSEA. Cells migrating into the lower chamber were fixed with glutaraldehyde and stained with crystal violet. (×4.)
The result of the complementation experiments leaves two possibilities for interpretation. (i) The genetic program leading to metastasis (i.e., cell “scattering,” matrix invasion, and growth) is initiated by MET by activation of multiple signaling pathways, triggered independently and concomitantly by multiple transducers, binding to either the phosphorylated tyrosine-1349 (YVHV) or tyrosine-1356 (YVNV). (ii) Alternatively, the metastasis program is triggered by MET by a single specific pathway sustained by an effector (still to be identified) that requires simultaneous binding to both sequences Y1349VHV and Y1356VNV. In cells expressing the two mutants these sequences are brought to close proximity by heterodimerization of the mutated proteins. The signal transduction “amplifier” Gab1, which binds either tyrosine-1349 or tyrosine-1356 (21), is a likely candidate; this molecule, however, is also phosphorylated in response to other tyrosine kinase receptors, such as the epidermal growth factor receptor (27), which is unable to trigger the invasive phenotype (4). Interestingly, the interaction of the isolated Met-binding-domain of Gab1 with Tpr-Met wild type and with the 2× Grb2 mutant was found to be comparable (data not shown). This finding suggests that the ability of Tpr-Met to associate with Gab1 does not seem sufficient, per se, for Met-mediated invasion and metastasis.
In either case, this paper demonstrates that the metastatic potential of the MET oncogene relies on its two-tyrosine docking site, and it identifies a single point mutation that dissociates the transforming properties of an oncogene from its ability to elicit the invasive–metastatic phenotype.
Acknowledgments
We thank Dr. G. Gaudino and Dr. M. Santoro for providing the TPR-SEA construct. The excellent technical assistance of R. Callipo, L. Palmas, and G. Petruccelli is gratefully acknowledged. We thank Antonella Cignetto for secretarial help and Elaine Wright for editing the manuscript. This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) to P.M.C. and from the Consiglio Nazionale delle Ricerche Applicazioni Cliniche della Ricerca Oncologica 95.00340.PF39 to P.M.C.
References
- 1.Liotta L A, Stetler-Stevenson W G. In: Cancer Principles and Practice of Oncology. 4th Ed. De Vita V Jr, Hellman S, Rosenberg S A, editors. Philadelphia: Lippincott; 1993. pp. 134–149. [Google Scholar]
- 2.Egan S E, Wright J A, Jarolim L, Yanagihara K, Bassin R H, Greenberg A H. Science. 1987;238:202–205. doi: 10.1126/science.3659911. [DOI] [PubMed] [Google Scholar]
- 3.Yu D, Wang S, Dulski K M, Tsai C M, Nicolson G L, Hung M C. Cancer Res. 1994;54:3260–3266. [PubMed] [Google Scholar]
- 4.Medico E, Huff J, Jelinek M-A, Follenzi A, Gaudino G, Parsons T, Comoglio P M. Mol Biol Cell. 1996;3:495–504. doi: 10.1091/mbc.7.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comoglio P M, Boccaccio C. Genes to Cells. 1996;1:347–354. doi: 10.1046/j.1365-2443.1996.37037.x. [DOI] [PubMed] [Google Scholar]
- 6.Gherardi E, Stoker M. Cancer Cells. 1991;3:227–232. [PubMed] [Google Scholar]
- 7.Giordano S, Ponzetto C, Di Renzo M F, Cooper C S, Comoglio P M. Nature (London) 1989;339:155–156. doi: 10.1038/339155a0. [DOI] [PubMed] [Google Scholar]
- 8.Bottaro D P, Rubin J S, Faletto D L, Chan A M, Kmiecik T E, Vande Woude G F, Aaronson S A. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 9.Naldini L, Tamagnone L, Vigna E, Sachs M, Hartmann G, Birchmeier W, Daikuhara Y, Tsubouchi H, Blasi F, Comoglio P M. EMBO J. 1992;10:2867–2878. doi: 10.1002/j.1460-2075.1992.tb05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bussolino F, Di Renzo M F, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio P M. J Cell Biol. 1992;119:629–641. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt L, Duh F M, Chen F, Kishida T, Glenn G, et al. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 12.Di Renzo M F, Narsimhan R P, Olivero M, Bretti S, Giordano S, Medico E, Gaglia P, Zara P, Comoglio P M. Oncogene. 1991;6:1997–2003. [PubMed] [Google Scholar]
- 13.Di Renzo M F, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S, Plebani M, Gespach C, Comoglio P C. Clin Cancer Res. 1995;1:147–154. [PubMed] [Google Scholar]
- 14.Ponzetto C, Zhu Z, Audero E, Maina F, Bardelli A, Basile M L, Giordano S, Narsimhan R, Comoglio P M. J Biol Chem. 1996;271:14119–14123. doi: 10.1074/jbc.271.24.14119. [DOI] [PubMed] [Google Scholar]
- 15.Santoro M, Collesi C, Grisendi S, Gaudino G, Comoglio P M. Mol Cell Biol. 1996;16:7072–7083. doi: 10.1128/mcb.16.12.7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper C S, Park M, Blair D, Tainsky M A, Huebner K, Croce C M, Vande Woude G F. Nature (London) 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 17.Rong S, Segal S, Anver M, Resau J H, Vande Woude G. Proc Natl Acad Sci USA. 1994;91:4731–4735. doi: 10.1073/pnas.91.11.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, Graziani A, Panayotou G, Comoglio P M. Cell. 1994;77:261–271. doi: 10.1016/0092-8674(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 19.Pelicci G, Giordano S, Zhen Z, Salcini A E, Lanfrancone L, Bardelli A, Panayotou G, Waterfield M D, Ponzetto C, Pelicci P G, Comoglio P M. Oncogene. 1995;10:1631–1638. [PubMed] [Google Scholar]
- 20.Fixman E D, Fournier T M, Kakimura D M, Naujokas M A, Park M. J Biol Chem. 1996;271:13116–13122. doi: 10.1074/jbc.271.22.13116. [DOI] [PubMed] [Google Scholar]
- 21.Weidner M, Di Cesare S, Sachs M, Brinkmann V, Behrens J, Birchmeier W. Nature (London) 1996;384:173–176. doi: 10.1038/384173a0. [DOI] [PubMed] [Google Scholar]
- 22.Songyang Z, Shoelson S, Chaudhuri M, Gish G, Pawson T, Haser W G, King F, Roberts T, Ratnofski S, Lechleider R J, Neel B G, Birge R B, Fajardo J E, Chou M M, Hanafusa H, Schaffhausen B, Cantley L C. Cell. 1993;72:1–20. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 23.Maina F, Casagranda F, Audero E, Simeone A, Comoglio P M, Klein R, Ponzetto C. Cell. 1996;87:531–542. doi: 10.1016/s0092-8674(00)81372-0. [DOI] [PubMed] [Google Scholar]
- 24.Albini A, Iwamoto Y, Kleinman H K, Martin G R, Aaronson S A, Kozlowsky J M, McEwan R N. Cancer Res. 1989;47:3239–3245. [PubMed] [Google Scholar]
- 25.Huff J L, Jelinek M A, Borgman C A, Lansing T J, Parsons T J. Proc Natl Acad Sci USA. 1993;90:6140–6144. doi: 10.1073/pnas.90.13.6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith D R, Vogt P K, Hayman M J. Proc Natl Acad Sci USA. 1989;86:5291–5295. doi: 10.1073/pnas.86.14.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holgado-Madruga M, Emlet D R, Moscatello D K, Godwin A K, Wrong A J. Nature (London) 1996;379:560–562. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]