

Abstract
Multiple endocrine neoplasia type 1 (MEN1), an inherited tumor syndrome affecting endocrine organs including pancreatic islets, results from mutation of the tumor suppressor gene Men1 that encodes protein menin. Although menin is known to be involved in regulating cell proliferation in vitro, it is not clear how menin regulates cell cycle and whether mutation of Men1 acutely promotes pancreatic islet cell proliferation in vivo. Here we show that excision of the floxed Men1 in mouse embryonic fibroblasts (MEFs) accelerates G0/G1 to S phase entry. This accelerated S-phase entry is accompanied by increased cyclin-dependent kinase 2 (CDK2) activity as well as decreased expression of CDK inhibitors p18Ink4c and p27Kip1. Moreover, Men1 excision results in decreased expression of p18Ink4c and p27Kip1 in the pancreas. Furthermore, complementation of menin-null cells with wild-type menin represses S phase entry. To extend the role of menin in repressing cell cycle in cultured cells to in vivo pancreatic islets, we generated a system in which floxed Men1 alleles can be excised in a temporally controllable manner. As early as seven days following Men1 excision, pancreatic islet cells display increased proliferation, leading to detectable enlargement of pancreatic islets fourteen days after Men1 excision. These observations are consistent with the notion that an acute effect of Men1 mutation is accelerated S phase entry and enhanced cell proliferation in pancreatic islets. Together, these results suggest a molecular mechanism whereby menin suppresses MEN1 tumorigenesis at least partly through repressing G0/G1 to S transition.
Keywords: menin, tumor suppressor, MEN1, cell proliferation, islet cells, cell cycle
INTRODUCTION
Multiple endocrine neoplasia type 1 (MEN1) is a dominantly inherited tumor syndrome that results from the mutation of the tumor suppressor gene Men1, which encodes menin (1, 2). Menin interacts with multiple proteins that play critical roles in the regulation of cell proliferation, including JunD (3), Smad 3 (4) and Activator of S-Phase Kinase (ASK) (5). ASK is the crucial regulatory factor for protein kinase cdc7 that is required for initiation of DNA replication (6, 7), and menin functionally represses the activity of ASK (5). In addition, menin interacts with a protein complex containing the Mixed Lineage Leukemia protein (MLL) (8, 9), and upregulates transcription of various target genes including the cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p18Ink4c in transformed fibroblasts (10) and insulinoma cells (11). While these observations provide a potential mechanistic link between menin and cell cycle regulation, a direct link between menin’s function and cell cycle progression has not been established. An obstacle to answering this question has been a lack of synchronizable cells in which Men1 can be conditionally inactivated in vitro that the effect of Men1 deletion on the cell cycle progression can be examined.
Mouse models have greatly increased our understanding of molecular pathology of the MEN1 syndrome. Tumors derived from mice heterozygous for Men1 display loss of heterozygosity (LOH) (12, 13), confirming the role of menin as a bona fide tumor suppressor. Tumors arise in the parathyroid (14), pituitary (15), and pancreatic islet cells (15–17) from the mice in which Men1 is conditionally inactivated in these respective organs, establishing an important role for menin in suppressing tumor development in endocrine organs. However, because the excision of Men1 is not under temporal control in these mice, it is challenging to study the acute effects of deletion of Men1 on proliferation of pancreatic islet cells. Thus, although tumor cells in insulinomas of the mice display enhanced cell proliferation as shown by 5′-Bromo-2′-Deoxyuridine-5′-Triphosphate (BrdU) uptake (17), it is difficult to determine how soon after Men1 deletion increased islet cell proliferation occurs. If increased islet proliferation is an acute consequence of Men1 deletion, then this would suggest that loss of menin-mediated repression of cell proliferation is at least in part responsible for the early events of MEN1 tumorigenesis. A mouse model in which Men1 can be deleted in a temporally controllable manner will help to address this question. Answering this question is important for identifying the pathways menin controls to suppress tumorigenesis in endocrine organs, and for understanding how to potentially manipulate those pathways for therapeutic intervention.
In the current studies, mouse embryonoc fibroblasts (MEFs) in which Men1 could be conditionally deleted were generated. Unlike viral oncogene-immortalized Men1−/− MEFs (18), these MEFs could be synchronized to determine the effect of menin on cell cycle progression. In addition, we established a model system in which the floxed Men1 locus could be excised in a temporally controlled fashion that the acute effect of in vivo Men1 excision on cell proliferation could be examined. These technical advances allowed us to establish a crucial role for menin in repressing cell cycle entry into S phase in vitro as well as suppressing proliferation of normal pancreatic islet cells in vivo.
MATERIALS AND METHODS
Mouse breeding, genotyping, and excision of the floxed Men1 locus
All animal studies were approved by ULAR (University Laboratory Animal Resources), the University of Pennsylvania’s committee on animal care, and were carried out in accordance with the mandated standards. Men1 ΔN/ΔN mice (designated Men1l/l; mixed FVB;129Sv background) were kindly provided by Dr. Francis Collins at NIHGR (17). The pan-active human UBC9 promoter-driven Cre-ERT2 (19, 20) was introduced into murine fertilized eggs to generate Cre-ER transgenic mice (E. Brown, unpublished data), using the method of lentitransgenesis (21). Breeding was carried out by crossing Men1l/l and Cre-ER mice. Men1l/l;Cre-ER mice were genotyped by PCR using the 3 primers shown in Fig. 5A: P1, 5′-ccc aca tcc agt ccc tct tca gct −3′, P2, 5′-aag gta cag cag agg tca cag ag-3′, and P3, 5′-gac agg att ggg aat tct ctt tt-3′. The primers for genotyping Cre-ERT2 were 5′-tac acc aaa att tgc ctg cat tac cgg-3′ and 5′-ttt cca tga gtg aac gaa cct ggt-3′. Men1l/l ;Cre-ER or Men1+/+;Cre-ER mice at 12 weeks of age were first fed with tamoxifen (Sigma, St. Louis, MO) at a dose of 200 mg/kg body weight/day for 2 consecutive days, followed by a day off and then for a second 2 consecutive days at the same dose. Seven, 14, and 30 days later, the mice were sacrificed for analysis. In total, 22 mice, 11 male and 11 female, were analyzed, with the male and female mice randomly distributed between the two groups.
Fig. 5.


Excision of floxed Men1 results in increased islet cell proliferation. A, A schema for the floxed Men1 locus (Men1l/l, previously Men1ΔN/ΔN, (17)) and the excised product (Men1Δ/Δ). One of the 2 floxed Men1 alleles is depicted. In the absence of tamoxifen, primers 2 and 3 amplify fragment 1(F1); following tamoxifen treatment and excision of the floxed Men1 allele, primers 1 and 3 amplify fragment 2 (F2). B, Inducible and effective excision of the floxed Men1 locus. Mice of 12 weeks of age were fed with tamoxifen and genotyped as described in Materials and Methods. C–J, One month after tamoxifen treatment, Men1+/+;Cre-ER control mice and Men1l/l;Cre-ER mice were intraperitoneally injected with BrdU 2 h prior to harvesting pancreata for immunofluorescent staining. Pancreatic sections were stained with anti-menin and anti-BrdU antibodies to determine menin expression and the proliferative index of cells. DAPI staining was used to visualize nuclei. Images were acquired using the 20X objective lens. Merged image of F and J correlates expression of menin and the uptake of BrdU. The islet is circled by a dashed line. K, Quantification of BrdU-positive islet cells from 3 tamoxifen-treated Men1+/+; Cre-ER (control) mice and 4 Men1l/l;Cre-ER mice. L–O, BrdU-positive cells express insulin. Pancreatic sections from tamoxifen-fed (one month after feeding) Men1+/+; Cre-ER mice (L) and Men1l/l; Cre-ER mice (N) were co-stained with the anti-BrdU antibody (red) and the anti-insulin antibody (green). In the right panels (M and O), pancreatic sections were co-stained with the anti-BrdU antibody (red) and the anti-glucagon (green) antibody as indicated. Images were captured using the 20X objective lens.
Immunofluorescent staining of pancreatic sections
Men1l/l;Cre-ER and control Men1+/+;Cre-ER mice (both tamoxifen-fed) were injected intraperitoneally with 50 mg BrdU (Sigma, St. Louis, MO)/kg body weight 2 h prior to sacrifice and dissection. Pancreata were isolated and processed for Hematoxylin and Eosin (H&E) staining, and 3 separate sections from each mouse were stained to quantify area of islets using Metamorph software (Molecular Devices Corporation, Sunnyvale, CA). For immunofluorescent staining, a rabbit anti-menin antibody (#80) (18) and a sheep anti-BrdU antibody (#2284, Abcam Inc,, Cambridge, UK) were used in combination with FITC-conjugated anti-rabbit IgG and TRITC-conjugated anti-sheep IgG secondary antibodies, together with DAPI (10ug/ml). Images were captured under a Nikon eclipse E800 fluorescent microscope equipped with a CCD digital camera and the BrdU positive cells among the total DAPI stained cells per islet were quantified. To co-stain BrdU with insulin or glucagon in islet cells, the following antibodies were used: monoclonal rat anti-BrdU (BU1/75-ICR1, Accurate Chemical & Scientfic Corp., Westbury, NY), Cy2-conjugated anti-rat IgG, guinea pig anti-insulin, rabbit anti-glucagon (Abcam Inc, Cambridge, UK), FITC-conjugated goat-anti-rabbit IgG (Molecular Probe Inc.) and FITC-conjugated goat anti-guinea pig IgG (Abcam Inc).
RT-PCR and Real-time TaqMan PCR
Total RNA was extracted from cell lines and pancreata using the RNeasy Mini Kit (Qiagen, Valencia, CA). One step RT-PCR was performed with RNA derived from pancreata, using the Titan One Tube RT-PCR System (Roche, Indianapolis, IN) following the manufacturer’s instructions. Real-Time TaqMan PCR quantification of gene expression was performed with RNA derived from cultured cell lines, using Taqman probes for p18Ink4c (Applied Biosystems, Foster City, CA; Mm00483243_m1), p27Kip1 (Mm00438167_g1), and GAPDH as an internal control (Mm99999915_g1). Analysis was performed using the relative quantification method according to instructions from the ABI.
Plasmid construction and production of recombinant viruses
Plasmids for generating recombinant retroviruses were constructed by inserting PCR-amplified human menin cDNA into the BamHI/NotI site of the retroviral vector pMX-puro to generate pMX-menin. The production of recombinant adenoviruses and retroviruses was as previously described (22). For complementation with wild-type menin, Men lΔ/Δ l cells were seeded on day 0, infected with various retroviruses (including GFP-expressing retroviruses as a control for infection efficiency) on day 1, and switched to fresh media on day 2 prior to selection with 2 μg/ml puromycin on day 4.
Generation of murine embryonic fibroblast (MEF) cell lines and fluorescence-activated Cell sorting (FACS) analysis
MEFs from Men1l/l embryos were isolated on embryonic day 14 (E14) and were immortalized using the 3T9 protocol (23). Briefly, 9 X105 MEFs were plated on a 60 mm plate and passaged every 3 days. After 30–35 passages, immortalized cells emerged. After immortalization, the cells were infected with adenoviruses expressing either GFP (Ad-GFP) or Cre recombinase (Ad-Cre), generating one control cell line (designated Men1l/l) and two menin-null cell lines (designated Men1Δ/Δ 1 and Men1Δ/Δ 2). After 2–3 passages of Men1 excision, the cells were seeded in MEF medium (22) at a density of 1.5 ×105 cells/100 mm dish on day 0 for cell cycle analysis. On day 1, cells were switched to medium containing only 0.1% FBS. On day 5, normal MEF medium containing nocodazole (Sigma, St. Louis, MO; 200 ng/ml) was added to cells, releasing them from arrest in G0/G1. At various time points after release, cells were pulsed with 10 μM BrdU for 2 hrs immediately before harvest and fixation. Cell pellets were processed for double staining with an anti-BrdU antibody (Pharmingen, San Jose, CA) and propidium iodide (10 μg/mL in PBS) (Sigma, St. Louis, MO), followed by analysis on a FACS Calibur (Becton-Dickinson, Franklin Lakes, NJ). Gating was performed to focus on the G1, S, and G2/M populations.
Antibodies and Western blotting
Whole cell lysates were prepared with ELB lysis buffer (0.1% NP-40, 160 mM NaCl, 50 mM Hepes pH 7.4, 5 mM EDTA, pH 8.0, 1mM DTT, 0.2mM PMSF) (24) supplemented with protease inhibitor cocktail Set (CalBiochem, San Diego, CA ), and subjected to Western blotting analysis as previously described (22). The primary antibodies used were rabbit anti-menin (BL-342, Bethyl Lab, Montgomery, TX), goat anti-actin (C-11, Santa Cruz, Santa Cruz, CA), mouse anti-p27Kip1 (BD Transduction, San Jose, CA), rabbit anti-p18 (N-20), rabbit anti- p21 (C-19), and rabbit anti-p16 (M-156; all Santa Cruz, Santa Cruz, CA).
CDK kinase assays
Cells were lysed in ELB lysis buffer and the lysates (250 μg) were immunoprecipitated by 5 μg of anti-mouse CDK2 antibody (Santa Cruz, Santa Cruz, CA) or control rabbit IgG. Immunoprecipitates were incubated with 2 μg histone H1 (Upstate Biotech, Norcross, GA), and 5 μCi γ32P-ATP for 30 minutes prior to SDS-PAGE separation, phosphoimaging analysis and quantification. Total histone substrate was visualized by Coomassie Blue staining.
Statistical analysis and quantification
Microsoft Excel and GraphPad Prism software were used to prepare graphs and to perform statistical analyses. When appropriate, the student’s t test was utilized to determine significance of results.
RESULTS
Ablation of Men1 in vitro increases cell proliferation and transition from G0/G1 to S phase
We immortalized MEFs from mouse embryos with the floxed Men1 (17) using the 3T9 protocol (23), and then infected the cells with recombinant adenoviruses expressing either GFP (Ad-GFP) or Cre (Ad-Cre) that could excise the floxed Men1 from the genome. The cell lysates from the infected cells were subjected to Western blotting analysis. Ad-Cre (lanes 2–3), but not Ad-GFP (lane 1), abrogated expression of menin (Fig. 1A). Men1 excision was also confirmed by genotyping (Fig. 1B and Fig. 5A), since Men1 excision yielded a PCR fragment of the increased size. The Men1Δ/Δ 1 cells and the Men1Δ/Δ 2 cells, two independent pools of the MEFs infected by Ad-Cre, proliferated more quickly than the menin-expressing Men1l/l cells (2.7 and 2.9 × 106 versus 1.3 × 106 cells; for Men1l/l versus Men1Δ/Δ 1, p<0.03; for Men1l/l versus Men1Δ/Δ 2, p<0.02) (Fig. 1C). To further confirm this difference in cell proliferation in vitro, we excised Men11/l from one additional independent clone and similar results were obtained (R. Schnepp, data not shown).
Fig. 1.

Ablation of Men1 in MEFs results in increased cell proliferation in vitro. A, Cre-mediated excision of the Men1 flanked by the two lox P sites abrogates menin protein expression. The details of the floxed Men1 is described in Fig. 5A. Men1l/l cells were either infected with adenoviruses Ad-GFP (Men1l/l cell line) or Ad-Cre (Men1Δ/Δ 1 and Men1Δ/Δ 2, two independent pools of the infected cells), prior to detection of menin and control actin 5 days after infection. B, Excision of Men1 in the Men1Δ/Δ 1 and Men1Δ/Δ 2 cells was confirmed by genotyping, as shown in Fig. 5B. C, Deletion of Men1 in MEFs increases cell proliferation. Men1l/l, Men1Δ/Δ 1, and Men1Δ/Δ 2 cells were seeded in triplicate on day 0 and counted using a hemacytometer on day 4. Data were derived from the mean of triplicate cultures. This is representative of 3 independent experiments.
We next determined whether menin inhibits cell cycle progression, if it does, at what phase it inhibits cell cycle progression. Serum-starved menin-null or menin-expressing cells were stimulated by addition of serum and allowed to progress for various periods of time up to 24 hrs. Cells were harvested at various time points after release and processed for staining with anti-BrdU antibody and propidium iodide, followed by flow cytometry analysis. Following serum removal, Men1l/l cells were distributed at G0/G1 (48%) and G2/M (48.1%), with only 3.9% of the cells in S phase (Fig. 2A, top and left panel), as compared to that of asynchronous cells (55%, data not shown). Similarly, Men1Δ/Δ 1 cells were primarily distributed in G0/G1 and G2/M phases, with only 7.5% cells in S phase (Fig. 2A, bottom and left panel). Twelve hours after release from serum starvation, only 12.2% of the Men1l/l cells progressed from G0/G1 to S phase (Fig. 2A, top and center panel). In contrast, 32% of Men1Δ/Δ 1 cells entered S phase (Fig. 2A, bottom and center panel). At 24 h of release, 41.5% of Men1l/l cells reached S phase (top and right panel), while only 28% of the Men1Δ/Δ 1 cells were in S phase, as they had already passed their peak at S phase (18 h) and progressed to G2/M (Fig. 2A, bottom and right panel). The detailed kinetics of cell cycle progression for both Men1 l/l cells and Men1Δ/Δ 1 cells are shown in Fig. 2B. These results demonstrate that loss of menin expression accelerates progression from G0/G1 to S phase.
Fig. 2.

Ablation of Men1 in MEFs accelerates cell cycle progression from G0/G1 to S phase. A, Serum-starved Men1l/l and Men1Δ/Δ 1 cells were stimulated with the addition of serum and harvested 0, 6, 12, 18, and 24 h after release. The cells were pulsed with BrdU, harvested, and processed for analysis by flow cytometry. B, Detailed kinetics of cell cycle change in G0/G1, S, and G2/M phases in Men1l/l and Men1Δ/Δ 1 cell lines. Duplicate cultures were examined for each time point. This is representative of 2 independent experiments.
Ablation of Men1 increases CDK2 activity, but decreases p18Ink4c and p27Kip1RNA and protein levels
The cell cycle is positively regulated by various CDKs and CDK2 plays a crucial role in controlling G0/G1 to S transition (26). Thus we determined whether menin inhibits CDK2 activity. Lysates from Men1l/l cells and Men1Δ/Δ 1 cells, at various time points of cell cycle progression, were immunoprecipitated with an anti-CDK2 antibody, and the precipitated kinase activity was detected using histone H1 as a substrate. Figure 3A (top panel) shows that CDK2 activity, as indicated by the amount of phosphorylation of histone H1, increased after Men1 was excised (lanes 5–7). Quantification of phosphorylation shows that the CDK2 activity was approximately two fold higher in Men1Δ/Δ 1 cells than that in Men1l/l cells at each corresponding time point (Fig. 3A, bottom). Menin expression did not alter the phosphorylation of the inhibitory Y15 residue nor the activating T160 residue of CDK2 (T. Cash, data not shown). This result suggests that loss of menin expression increases the CDK2 activity and promotes G0/G1 to S transition.
Fig. 3.

Ablation of Men1 results in increased CDK2 activity and decreased levels of p27Kip1 and p18Ink4c protein and RNA. A, Ablation of Men1 in MEFs increases CDK2 activity. As in Fig. 2A, serum-starved Men1l/l and Men1Δ/Δ 1 cells were released, and harvested for immunoprecipitation with an anti-CDK2 antibody to measure CDK2 activity. B, Excision of Men1 in MEFs decreases p18Ink4c and p27Kip1 protein levels. The indicated cells were released from serum starvation and harvested at the indated time points, and then subjected for Western blotting with the indicated antibodies. C, Ablation of Men1 decreases p18Ink4c and p27Kip1 RNA levels. Real-Time TaqMan PCR analysis was carried out using TaqMan probes for p18Ink4c, p27Kip1, and GAPDH. D, The p27Kip1and p18Ink4c mRNA levels decrease in Men1l/l;Cre-ER mice one month after tamoxifen treatment, as demonstrated by RT-PCR. Shown are representative samples of 4 mice for each of the genotypes.
Since the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 are implicated in menin-mediated repression of cell proliferation in transformed MEFs (10), and p27Kip1 and p18Ink4c proteins inhibit CDK2 activity (27), we determined whether menin regulates expression of various CDK inhibitors in our untransformed cells. Men1l/l and Men1Δ/Δ 1 cells were both released from serum starvation, and then monitored for 24 hrs for expression of various CDK inhibitors, using Western blotting analysis. Men1l/l cells expressed menin while Men1Δ/Δ 1 cells lost menin expression as expected (Fig. 3B). Expression of both p18Ink4c and p27Kip1 was higher in Men1l/l cells than that in Men1Δ/Δ 1 cells (Fig. 3B). In contrast, the expression levels of p21Cip1 and p16Ink4a were comparable between Men1l/l cells and Men1Δ/Δ 1 cells (Fig. 3B). In addition, the mRNA levels of p27Kip1 and p18Ink4c in Men1l/l cells are 2.5 (p< 0.02) and 3.5 fold (p<0.001) higher, respectively, than in Men1Δ/Δ 1 cells (Fig. 3C). Although menin-dependent transcription of p27Kip1 and p18Ink4c was recently reported (10), this is the first time that menin was shown to suppress cell cycle progression, repress CDK2 activity, and upregulate p27Kip1 and p18Ink4c in a well-controlled system. These data suggest that menin regulates CDK2 at least in part by regulating p27Kip1 and p18Ink4c.
Complementation of Men1Δ/Δ 1 cells with wild-type menin inhibits cell proliferation and G0/G1 to S phase progression and restores p18Ink4c and p27Kip1 protein and RNA levels
If Men1 excision leads to enhanced cell proliferation and G0/G1 to S phase transition, complementation of menin-null cells with menin should suppress cell proliferation and G0/G1 to S phase progression. Thus, we infected Men1Δ/Δ 1 cells with control vector retroviruses or retroviruses encoding wild type menin, and the resulting cells were monitored for cell growth, expression of p27Kip1 and p18Ink4c, and G0/G1 to S progression. By day 4, there were 2.5 ×106 vector-complemented cells versus 1.3 × 106 menin-complemented cells (Fig. 4A); these differences were significant (p< 0.0006). In addition, expression of p27Kip1 and p18Ink4c was higher in menin-complemented cells than in vector-complemented cells, at both the protein and mRNA (Figs. 4B, C) levels, consistent with a previous report (10) that menin is crucial for optimal expression of p27Kip1 (p<0.003) and p18Ink4c (p<0.0002).
Fig. 4.


Complementation of Men1Δ/Δ 1 cells with wild-type menin inhibits cell proliferation and restores p18Ink4c and p27Kip1 protein and RNA levels. A, Men1Δ/Δ1 cells were complemented with vector control retroviruses or retroviruses expressing menin. The resulting cell lines were seeded in triplicate on day 0 and counted on day 4 as described in Figure 1C. This is representative of 2 independent experiments. B, Western blotting analysis of cell lines indicates that complementation with menin results in increased p27Kip1and p18Ink4c protein levels. C, Complementation with menin results in increased p27Kip1 and p18Ink4c RNA levels. Real-Time TaqMan PCR analysis was carried out using TaqMan probes for p18Ink4c, p27Kip1, and GAPDH. D, Complementation with retroviruses expressing menin represses transition from G0/G1 to S phase. As in Fig. 2A, serum-starved cells were released for various periods of time to monitor cell cycle progression. E, Cell cycle profiles were determined at multiple time points as indicated, as described in Figure 2.
To further confirm the crucial role of menin in regulating cell cycle progression, we tested if complementing cells with menin can rescue the role of menin in suppressing transition from G0/G1 to S phase. Following serum starvation, 12.5% of vector-complemented cells and 10% of menin-complemented cells were in S phase (Fig. 4D). Notably, 12 h after release, 33% of vector-complemented cells were in S phase, as compared to 16% in menin-complemented cells (Figure 4D). Twenty-four hours after release, vector-complemented cells progressed out of the peak of S phase (31%), while a greater percentage of menin-complemented cells remained in S phase (36%; Fig. 4D). A more detailed cell cycle profile at multiple time points after release further supports the role of menin in slowing down G0 to S phase transition (Fig. 4E).
Men1 excision in pancreatic islets acutely results in increased islet cell proliferation and size
The above studies in cultured cells demonstrated a crucial role for menin in controlling S phase entry. However, it is still unclear whether this role of menin also applies to in vivo endocrine cells such as pancreatic islet cells, in which a germline mutation in only one Men1 allele predisposes the patient to the development of insulinomas (28). In addition, since it takes approximately 6 months for mice carrying a Men1 mutation to develop insulinomas, which have a high proliferation index (17), an important unresolved question is whether time-controlled Men1 excision can quickly lead to enhanced proliferation of pancreatic islet cells. To address these questions, we bred mice with the Men1 locus flanked by loxp sites (Men1l/l, previously Men1ΔN/ΔN) (17) with mice (Men1+/+) expressing Cre-ER (estrogen receptor) driven by a pan-active UBC9 promoter (E. Brown, unpublished data), to generate mice with the Men1l/l ;Cre-ER genotype. Cre-ER expressed from a transgene can be activated by tamoxifen, resulting in excision of genes flanked by lox P sites (29). Both control mice (Men1+/+) expressing Cre-ER and the Men1l/l;Cre-ER mice were fed with tamoxifen, and then pancreata were harvested to determine excision of the conditional Men1 locus. Tamoxifen effectively induced Men1 excision in the pancreata of the Men1l/l;Cre-ER mice (Fig. 5A and B, lane 2), but not in Men+/+; Cre-ER mice (Fig. 5B, lanes 1). Conversely, in the absence of tamoxifen, the floxed Men1 remained intact in the pancreata of Men1l/l;Cre-ER mice, indicating no leakiness in excision of the Men1 locus in the absence of tamoxifen (Fig. 5B, lane 3). Given the effective control of Men1 excision, further experiments were performed using Men1+/+;Cre-ER and Men1l/l;Cre-ER mice to control for any nonspecific effects of tamoxifen treatment.
To detect islet cell proliferation after Men1 excision, BrdU was injected into Men1+/+;Cre-ER mice and Men1l/l;Cre-ER mice one month after the tamoxifen treatment. Pancreata from the mice were processed for staining with anti-menin and anti-BrdU antibodies to determine the relationship between Men1 excision and BrdU uptake by pancreatic islet cells. Islet cells from the Men1+/+;Cre-ER mice expressed menin (Fig. 5C), but contained only one BrdU-positive cell (Fig. 5D and F). In addition, menin appeared to be expressed preferentially in islet cells as compared to the adjacent exocrine cells (Fig. 5C). In contrast, islet cells from Men1l/l;Cre-ER mice largely lost menin expression, but contained multiple BrdU-positive cells (Fig. 5G, H, J). Quantification of the BrdU-positive cells from islets of multiple mice indicates that approximately 0.4% of islet cells were BrdU-positive in Men1+/+;Cre-ER mice, but notably 2.0% of cells were BrdU-positive in Men1l/l;Cre-ER mice (Fig. 5H and K, p < 0.008). To determine whether the BrdU-positive cells are either insulin-secreting β-cells or glucagon-secreting α-cells, pancreatic sections were co-stained with the anti-insulin antibody or the anti-glucagon antibody. In the islet from tamoxifen-fed Men1l/l; Cre-ER mice, there were two BrdU-positive cells, both co-stained with the anti-insulin antibody (red in the nucleus, Fig. 5N). Conversely, BrdU-positive cells were not co-stained with the anti-glucagon antibody (Fig. 5O). Together, these results indicate that Men1 excision leads to increased proliferation of islet cells well before the development of insulinomas. These results further support the in vitro results that menin represses G0/G1 progression or S phase entry of cultured cells (Fig. 2).
To extend our in vitro findings regarding the role of menin in upregulating p27Kip1 and p18Ink4c to the in vivo organ such as pancreatic islets, we also determined whether loss of Men1 affects p27Kip1 and p18Ink4c expression in the murine pancreata. Pancreata were harvested from Men1+/+;Cre-ER mice and Men1l/l;Cre-ER mice that were fed with tamoxifen. Quantification of various mRNAs from the pancreata by RT-PCR shows that Men1 expression was detectable in Men1+/+;Cre-ER mice (Fig. 3D, lane 1, top panel) but greatly reduced in Men1 l/l;Cre-ER mice (lanes 2–3). Similarly, expression of p27Kip1 and p18Ink4c was also markedly decreased in Men1 l/l;Cre-ER mice, as compared to the control mice, while expression of control GAPDH was comparable between Men1+/+;Cre-ER and Men1 l/l;Cre-ER mice (Fig. 3D). These data show that menin may also regulate p27Kip1 and p18Ink4c levels in vivo.
Enhanced islet cell proliferation after Men1 excision may affect the size of the islets after certain period of enhanced proliferation. Notably, the size of islets from Men1l/l;Cre-ER mice was, on average, larger than that of the control mice one month after tamoxifen treatment (Fig. 6A). The mean of the area of the islets from the Menl/l;Cre-ER mice was approximately 3.5-fold larger than that from the control mice (Fig. 6B, 0.50 versus 1.73, p < 0.0001). Collectively, these results indicate that deletion of Men1, within a month, leads to enhanced cell proliferation and enlargement of pancreatic islets, a tissue commonly affected in MEN1 syndrome.
Fig. 6.

Excision of the floxed Men1 results in enlargement of pancreatic islets. A, Enlargement of pancreatic islets after excision of Men1. H & E staining of pancreatic sections prepared from tamoxifen-fed Men1+/+; Cre-ER and Men1l/l;Cre-ER mice one month after tamoxifen treatment. Images were acquired using the 20X objective lens. I, denotes islets. B, Quantification of the size of islets derived from 3 Men1+/+; Cre-ER and 4 Men1l/l; Cre-ER mice, as described in Materials and Methods. Each circle represents a value of the area for a single islet in arbitrary units. The line indicates the mean of areas of all the measured islets.
To further determine how soon after Men1 deletion BrdU uptake increases in islet cells, we further examined pancreata at 7 and 14 days following tamoxifen treatment. At 7 days, approximately 0.2% of islet cells in Men1+/+;Cre-ER mice were BrdU-positive, in comparison to 0.6% of islet cells in Men1l/l;Cre-ER mice (Fig. 7A, p< 0.005), indicating a significant increase in islet cell proliferation 7 days after Men1 excision. At 14 days, 0.2% of islet cells from control mice were BrdU-positive, as compared to 1.4% of islet cells in Men1l/l;Cre-ER mice (Fig. 7A, p < 0.4×10−5). The mean islet area was not significantly different between Men1+/+;Cre-ER and Men1l/l;Cre-ER mice on day 7. However, on day 14, the mean of the area of the islets from the Menl/l;Cre-ER mice was approximately 1.5-fold larger than that from the control mice ( Fig. 7B, 0.84 versus 1.22, p < 0.005). These results strongly suggest that deletion of Men1 acutely results in increased cell proliferation, which may accelerate the accumulation of islet cells, resulting in islet enlargement and hyperplasia 14 days after Men1 excision.
Fig. 7.
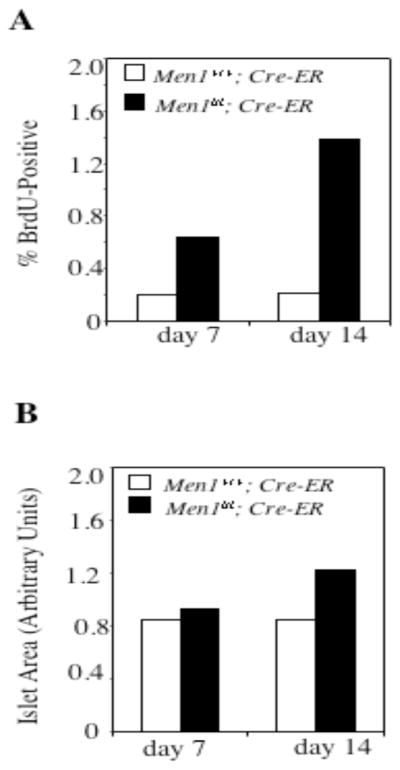
Excision of Men1 acutely accelerates islet cell proliferation. A, Seven and fourteen days after tamoxifen treatment, pancreata from mice (4 Men1+/+; Cre-ER and 3 Men1l/l; Cre-ER mice for day 7; 4 Men1+/+; Cre-ER and 4 Men1l/l; Cre-ER mice for day 14) were processed for BrdU staining as described in Fig. 6C–J. B, Quantification of pancreatic islets from mice (4 Men1+/+; Cre-ER and 3 Men1l/l; Cre-ER mice for day 7; 4 Men1+/+; Cre-ER and 4 Men1l/l; Cre-ER mice for day 14) was performed as described in Fig. 6B.
DISCUSSION
To understand how Men1 mutations result in MEN1 syndrome, it is important to elucidate how menin regulates cell cycle progression, proliferation of pancreatic islet cells, and the acute effect of Men1 deletion on islet cell proliferation. In this study we initially examined the role of menin in regulating cell cycle progression in vitro and then extended the study to the proliferation of pancreatic islet cells in vivo. Using MEFs with homozygous conditional Men1 alleles, we showed that Men1 excision accelerated S-phase entry for 4–5 hrs, the first direct evidence linking menin to inhibition of cell cycle progression. Accelerated S-phase entry in Men1-excised cells was accompanied by decrease of p18Ink4c and p27Kip1 in protein levels (Fig. 3B). This is consistent with a recent report that expression of p18Ink4c and p27Kip1 is decreased in mouse MEN1 insulinomas as well as in an insulinoma cell line expressing low level of menin (11).
CDK2 plays an important role in G1 to S transition (26), and p18Ink4c and p27Kip1 can either directly or indirectly inhibit CDK2 (27). Consistent with this, we found that Men1 excision led to elevated CDK2 activity, concomitant with downregulation p18Ink4c and p27Kip1 and earlier entry to S phase in MEFs. To our knowledge this is the first time that loss of menin expression is simultaneously linked to downregulation of p18Ink4c and p27Kip1, increased CDK2 activity, and accelerated S phase entry. Although it is attractive to hypothesize that increased CDK2 activity in menin-null cells contributes to the accelerated S phase entry, the increased CDK2 activity may not directly cause the accelerated S phase entry. Our findings that menin-mediated expression of p18Ink4c and p27Kip1 correlates with the delayed entry to S phase are also in agreement with the report that double mutation of p18Ink4c and p27Kip1 enhances transition from G0 to S phase in muscle cells (30). However, our findings do no necessarily mean that p18Ink4c and p27Kip1 are the major effectors of menin-mediated inhibition of G0 to S phase transition. It is possible that the potential effect of Men1 excision on cell cycle withdrawal and maintenance of quiescence also in part contributes to the quicker transition to S phase. In addition, we previously also showed that menin interacted with and functionally inhibited ASK, an essential component of protein kinase complex cdc7/ASK that is required for S-phase entry (7). Thus, the menin and ASK interaction may also contribute to repression of entry to S-phase.
To extend our findings on the role of menin in repressing cell proliferation and S phase entry to pancreatic islets in vivo, we determined whether deletion of Men1 quickly results in increased islet cell proliferation. Within 7 days of Men1 excision, pancreatic islet cells displayed increased BrdU uptake, a reliable indicator of entry into S phase. Consistent with the crucial role of menin in keeping proliferation of islet cells in check, the mean islet size gradually increased from day 14 to day 30 after Men1 excision. These results establish for the first time that loss of menin expression acutely results in enhanced cell proliferation in islet cells, a tissue commonly affected in the MEN1 patient. Thus, these results indicate that menin normally represses proliferation of islet cells, and an acute and early effect of the Men1 mutation is enhanced proliferation of islet cells including β-cells. However, these results do not exclude the possibility that secondary effects, such as defects in genome instability and apoptosis, following Men1 mutation, also contribute to MEN1 tumorigenesis.
An important related observation is that excision of Men1 accelerated proliferation of islet cells, but not the adjacent exocrine cells, indicating a tissue-specific role for menin in regulating proliferation of the endocrine pancreata. The floxed Men1 was effectively excised in the pancreas including both the exocrine and endocrine cells, based on analysis of the genomic DNA for the floxed Men1 (Fig. 5B). The floxed Men1 locus was also effectively excised from other tissues such as bone marrow and the mouse tail (data not shown). This is consistent with the expected broad expression of the Cre-ER transgene. These results are also in accordance with a recent study showing that inactivation of Men1 in the liver, a tissue not affected in MEN1 syndrome, does not result in tumorigenesis (31). Thus, menin may play an especially critical role in suppressing cell proliferation in endocrine organs. It is not clear how menin specifically represses the proliferation of islet cells but not the adjacent exocrine cells.
The current studies lead to a model in which menin normally regulates the levels of p18Ink4c and p27Kip1 to repress CDK2 activity and limit islet cell proliferation. Accordingly, mutation of Men1 results in increased islet cell proliferation. Men1 excision quickly results in increased pancreatic islet proliferation, which may help to initiate development of islet hyperplasia. The enhanced proliferation of pancreatic islet cells, perhaps in combination with decreased apoptosis and genome stability, may further accelerate the rate of secondary genetic and/or epigenetic alterations, leading to the development of insulinomas. However, the current studies do not rule out that additional menin-related regulators, such as Jun D (3), cyclin D1 (32), and ASK (5) are also involved in the regulation of islet cell proliferation. In conclusion, these studies demonstrate that menin plays an essential role in the tissue-specific suppression of pancreatic islet cell proliferation and in inhibition of the G0/G1-S transition. Furthermore, these findings suggest the possibility that targeting the CDK2 axis may prove useful in treating MEN1 islet tumors, and set the stage to further investigate precisely how menin specifically upregulates the transcription of the p18Ink4c and p27Kip1 genes.
Acknowledgments
This work was in part supported by NIH grants (R01 CA113962 and CA100912 to XH) and a scholar award from the Rita Allen Foundation. We thank Dr. Francis Collins at NIHGR for generously providing the mice with the floxed Men1 locus, Drs. David Tuvson and Dr. Jake Kushner at the University of Pennsylvania for Ad-Cre and the anti-BrdU antibody (BU1/75-ICR1 for Fig. 5L and N), respectively.
References
- 1.Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276(5311):404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 2.Lemmens I, Van de Ven WJ, Kas K, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6(7):1177–83. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal SK, Novotny EA, Crabtree JS, et al. Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc Natl Acad Sci U S A. 2003;100(19):10770–5. doi: 10.1073/pnas.1834524100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci U S A. 2001;98(7):3837–42. doi: 10.1073/pnas.061358098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schnepp RW, Hou Z, Wang H, et al. Functional interaction between tumor suppressor menin and activator of S-phase kinase. Cancer Res. 2004;64(18):6791–6. doi: 10.1158/0008-5472.CAN-04-0724. [DOI] [PubMed] [Google Scholar]
- 6.Kumagai H, Sato N, Yamada M, et al. A novel growth- and cell cycle-regulated protein, ASK, activates human Cdc7-related kinase and is essential for G1/S transition in mammalian cells. Mol Cell Biol. 1999;19(7):5083–95. doi: 10.1128/mcb.19.7.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masai H, Matsui E, You Z, Ishimi Y, Tamai K, Arai K. Human Cdc7-related kinase complex. In vitro phosphorylation of MCM by concerted actions of Cdks and Cdc7 and that of a criticial threonine residue of Cdc7 bY Cdks. J Biol Chem. 2000;275(37):29042–52. doi: 10.1074/jbc.M002713200. [DOI] [PubMed] [Google Scholar]
- 8.Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13(4):587–97. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 9.Yokoyama A, Wang Z, Wysocka J, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24(13):5639–49. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milne TA, Hughes CM, Lloyd R, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102(3):749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102(41):14659–64. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol. 2003;17(9):1880–92. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- 13.Crabtree JS, Scacheri PC, Ward JM, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98(3):1118–23. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Libutti SK, Crabtree JS, Lorang D, et al. Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Res. 2003;63(22):8022–8. [PubMed] [Google Scholar]
- 15.Biondi CA, Gartside MG, Waring P, et al. Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Mol Cell Biol. 2004;24(8):3125–31. doi: 10.1128/MCB.24.8.3125-3131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res. 2003;63(16):4836–41. [PubMed] [Google Scholar]
- 17.Crabtree JS, Scacheri PC, Ward JM, et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol Cell Biol. 2003;23(17):6075–85. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin S, Mao H, Schnepp RW, et al. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003;63(14):4204–10. [PubMed] [Google Scholar]
- 19.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci U S A. 1996;93(20):10887–90. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237(3):752–7. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 21.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295(5556):868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 22.Schnepp RW, Mao H, Sykes SM, et al. Menin induces apoptosis in murine embryonic fibroblasts. J Biol Chem. 2004;279(11):10685–91. doi: 10.1074/jbc.M308073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todaro G, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozar K, Ciemerych MA, Rebel VI, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118(4):477–91. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 25.Sheaff RJ. Regulation of mammalian cyclin-dependent kinase 2. Methods Enzymol. 1997;283:173–93. doi: 10.1016/s0076-6879(97)83015-7. [DOI] [PubMed] [Google Scholar]
- 26.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60(14):3689–95. [PubMed] [Google Scholar]
- 27.Franklin DS, Godfrey VL, O’Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. 2000;20(16):6147–58. doi: 10.1128/mcb.20.16.6147-6158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marx SJ. Multiple Endocrine Neoplasia Type I. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. New York: McGraw-Hill; 1998. pp. 489–506. [Google Scholar]
- 29.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–57. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 30.Myers TK, Andreuzza SE, Franklin DS. p18INK4c and p27KIP1 are required for cell cycle arrest of differentiated myotubes. Exp Cell Res. 2004;300(2):365–78. doi: 10.1016/j.yexcr.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Scacheri PC, Crabtree JS, Kennedy AL, et al. Homozygous loss of menin is well tolerated in liver, a tissue not affected in MEN1. Mamm Genome. 2004;15(11):872–7. doi: 10.1007/s00335-004-2395-z. [DOI] [PubMed] [Google Scholar]
- 32.Ratineau C, Bernard C, Poncet G, et al. Reduction of menin expression enhances cell proliferation and is tumorigenic in intestinal epithelial cells. J Biol Chem. 2004;279(23):24477–84. doi: 10.1074/jbc.M401835200. [DOI] [PubMed] [Google Scholar]