

Abstract
Cardiomyopathy (CM) is a primary degenerative disease of myocardium and is traditionally categorized into hypertrophic and dilated CMs (HCM and DCM) according to its gross appearance. Cardiomyopathic hamster (CM hamster), a representative model of human hereditary CM, has HCM and DCM inbred sublines, both of which descend from the same ancestor. Herein we show that both HCM and DCM hamsters share a common defect in a gene for δ-sarcoglycan (δ-SG), the functional role of which is yet to be characterized. A breakpoint causing genomic deletion was found to be located at 6.1 kb 5′ upstream of the second exon of δ-SG gene, and its 5′ upstream region of more than 27.4 kb, including the authentic first exon of δ-SG gene, was deleted. This deletion included the major transcription initiation site, resulting in a deficiency of δ-SG transcripts with the consequent loss of δ-SG protein in all the CM hamsters, despite the fact that the protein coding region of δ-SG starting from the second exon was conserved in all the CM hamsters. We elucidated the molecular interaction of dystrophin-associated glycoproteins including δ-SG, by using an in vitro pull-down study and ligand overlay assay, which indicates the functional role of δ-SG in stabilizing sarcolemma. The present study not only identifies CM hamster as a valuable animal model for studying the function of δ-SG in vivo but also provides a genetic target for diagnosis and treatment of human CM.
Cardiomyopathy (CM) manifests dyspnea, cardiac failure, or sudden death, causing serious morbidity and mortality. Clinical features and molecular genetic studies of CM demonstrate a wide variety of possible genetic causes of this disease but the causative genes and pathogenesis are poorly understood (1–3). Medical treatment for this progressive disease are only palliative with poor prognosis. Syrian hamsters with CM are known to inherit both CM and muscular dystrophy as an autosomal recessive trait but the genetic cause still remains to be elucidated (4–6). Recent studies on muscular dystrophy revealed the genetic importance of sarcoglycans (SGs), a subcomplex of dystrophin-associated glycoprotein complex (DAGC), in this disease (7–10).
Distinct sublines of Syrian hamster manifesting hypertrophic CM (HCM; BIO 14.6 and its descendant UMX7.1) or dilated CM (DCM; TO-2) have been established from the original line BIO1.50 (5, 6). We have reported to the DDBJ (DNA Data Base of Japan) that no mutation exists in the coding regions of cDNAs of BIO14.6 for α-, β-, or γ-SGs, all of which are lost in cardiac and skeletal muscles of this animal, where dystrophin is normally expressed (11). Our latest study revealed that these SGs are also deficient in UMX7.1 and TO-2 (vide infra), suggesting a hypothesis that both HCM and DCM share the loss of SG subcomplex as a common causative feature in hamster. In addition, δ-SG, which was identified recently, seemed to constitute DAGC together with α-, β-, and γ-SGs (12). These facts prompted us to identify the causative gene common to HCM and DCM with a particular focus on δ-SG.
MATERIALS AND METHODS
Animals.
Normal Golden hamsters were purchased from SEASCO (Saitama, Japan). BIO14.6 and TO-2 lines from Bio Breeders (Fitchburg, MA). UMX7.1 subline was maintained in our laboratory (5). All the animals used in the present study were male, aged 4–6 months old, and were deeply anesthetized before experiments. Electrocardiograms were recorded by Cardio Auto FD-35 (Fukuda Denshi, Tokyo).
Antibodies and Western Blot Analysis.
Anti-α- and anti-δ-SGs, and anti-β-dystroglycan (β-DG) polyclonal antibodies were raised in rabbits against the synthetic peptides HIDKGFTLWAAEP, GPKAVEAYGKKFEVKT, and PKNMTPYRSPPPY, respectively, and affinity-purified. Anti α-DG mouse monoclonal antibody (clone VIA4–1) was purchased from Upstate Biotechnology. Two hundred fifty micrograms of protein from whole homogenates from left ventricles (LVs) was subjected to SDS/PAGE and transferred to nitrocellulose membrane for Western blot analysis. Target proteins were detected with ECL kit (Amersham).
Cloning and Analysis of cDNAs for α-, β-, γ-, and δ-SGs and Genomic DNA for δ-SG in Hamster.
A cDNA library from Golden hamster heart was constructed in λZAPII phage vector (Stratagene; ref. 13) and screened with cDNA fragments of rabbit heart α-, β-, or γ-SGs (7–10) or human heart δ-SG (12), which were amplified by reverse transcription-coupled PCR (RT-PCR) from Quick-Clone heart cDNA (CLONTECH) with desired primer sets (for detail, see below). Golden hamster λFIXII genomic library (Stratagene) was screened independently with the 5′ end ScaI fragment (70 bp) and the adjacent ScaI–EcoRI fragment (231 bp) of the cloned Golden hamster δ-SG cDNA (DDBJ accession no. AB001508). The authentic transcription initiation site of δ-SG in the LV of Golden hamster was determined by the Primer Extension system (Promega) using a PE primer, 5′-GTGGCGGATGAGAGTCCCCGTGGTC-3′ (as a primer-extension primer; see Fig. 5). Sequences of cDNA or genomic DNA fragments, subcloned into pBluescript (Stratagene), were determined by model 4000LS (LI-COR) and/or model 373S (Applied Biosystems) automated DNA sequencers. RNA blot and Southern blot analyses were performed (13), by using 20 μg of total RNA from LV or biceps femoris and 15 μg of completely digested genomic DNA prepared from liver, respectively.
Figure 5.

Sequences of the authentic and alternative first exons, and the second exon of δ-SG gene. The primers used in primer-extension experiment (PE), PCR (exIF, alexIF, alexIR, exIIF, exIIR1, and exIIR2), and 5′ RACE (M1 and M2) are located by arrows. (Upper Left) The authentic first exon of δ-SG. The major transcription initiation site in the LV of normal hamsters is located at position +1 in the sequence. One of the several binding sequences for Sp1 is boxed. (Lower Left) The alternative first exon of δ-SG. (Right) The sequences for the second exon and the deduced amino acids for δ-SG. The single transmembrane domain (TMD) and one of the three potential N-glycosylation sites are indicated by a box and an asterisk, respectively.
Determination of the Genomic Breakpoint in Cardiomyopathic Hamsters (CM Hamsters).
Two hundred micrograms of the CM hamster genomic DNAs was sequentially digested with SpeI and PstI and subjected to agarose gel electrophoresis. The digested DNAs fractionating between 1.5 and 2.0 kb were recovered from the gel and were checked for hybridization with the 2.7-kb Int 1b probe (see Fig. 3). One microgram of the enriched DNAs was ligated to Marathon cDNA adaptor (CLONTECH) and amplified by PCR with Marathon adapter primer and a GS primer, 5′-CCTTGCAAAGTTTATAGCAGTGCC-3′ (as a gene-specific primer; see Fig. 3). The sequence of this amplified DNA fragment was directly determined in both strands by several inner primers and compared with that of the 2.7-kb Int 1b fragment of normal hamsters (see Fig. 3 Bottom).
Figure 3.
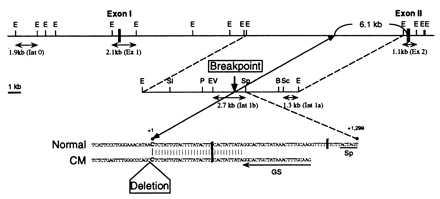
Identification of the deletion breakpoint of δ-SG gene in CM hamster genomes. (Top and Middle) Partial restriction maps of normal hamster (Normal) δ-SG gene. (Bottom) The sequence around the genomic breakpoint of the three CM hamsters (CM). The breakpoint (C in the sequences) locates 6.1 kb 5′ upstream from the 5′ end of the second exon of δ-SG gene. Genomic probes (Ex 1, Ex 2, Int 0, Int 1a, and Int 1b) and the GS primer used for cloning of the polymorphic SpeI–PstI genomic fragment of the three CM hamsters are indicated by double and single arrows, respectively. E, EcoRI; Sl, SalI; P, PstI; EV, EcoRV; Sp, SpeI; B, BstXI; Sc, SacI.
Multiplex RT-PCR.
Ten micrograms of total RNA from LV was treated with DNase, randomly reverse-transcribed with Superscript II (GIBCO/BRL), ethanol-precipitated, and dissolved in 100 μl of double-distilled water. A series of diluted samples ranging from 2/1, 2/4, 2/16, 2/64, to 2/256 μl were subjected to PCR (see Fig. 6). The cDNAs for both GAPDH (glyceraldehyde 3-phosphate dehydrogenase; as an internal standard) and the second exon of δ-SG were simultaneously amplified in the same tube with the primer sets 5′-CCACAGGAGCACCATGCCCAGCTC-3′/5′-ACTTTGAGAATCCAGATGGTCATG-3′ and 5′-ACCACAGTCCATGCCATCAC-3′ (exIIF)/5′-TCCACCACCCTGTTGCTGTA-3′ (exIIR2), respectively. PCR was carried out with the GeneAmp PCR system 9600 (PE Applied Biosystems) under the following conditions: preheating at 94°C for 2 min and then 40 cycles of 94°C for 20 sec, 60°C for 30 sec, and 72°C for 1 min. The yields of PCR products were quantified by nih image software after being stained by ethidium bromide and captured by a charge-coupled device camera.
Figure 6.
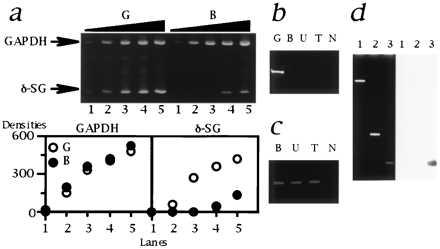
Analysis of δ-SG transcript in the CM hamsters. (a) Multiplex RT-PCR. (Upper) The sizes of PCR products for GAPDH and δ-SG (indicated by arrows) are 452 and 150 bp, respectively. Lanes: 1, 2/1 μl; 2, 2/4 μl; 3, 2/16 μl; 4, 2/64 μl; 5, 2/256 μl of template cDNA. The forward (exIIF) and reverse (exIIR2) primers were both located in the second exons. (Lower) The densities of each band quantified by nih image software are plotted. Note that GAPDH, an internal standard, was equivalently amplified. The amount of δ-SG transcript in B (•) was estimated to be 20–40 times lower than that of G (○). The similar data were also obtained for U and T (data not shown). (b) RT-PCR targeted for the authentic first and the second exons of δ-SG. The forward (exIF) and reverse (exIIR1) primers were located in the authentic first and the second exons, respectively. Note that a 217-bp band was detected only for G. N, no template. (c) 5′ RACE. The size of the bands was 0.5 kb. N, no template. (d) Localization of the alternative first exon of δ-SG. (Left) Ethidium bromide staning. (Right) Southern blot analysis with an alternative first exon probe (AlEx 1; 274-bp PCR products amplified with a primer set of alexIF and alexIR). Lanes: 1, 13.3-kb EcoRI–EcoRI fragment of the first intron of normal δ-SG gene containing the deletion breakpoint (Fig. 3 Middle); 2, 1.1-kb Ex 2 fragment (Fig. 3 Upper); 3, AlEx 1 probe as a positive control for hybridization. Note that AlEx 1 probe did not hybridize the genomic fragments of normal hamsters corresponding to lanes 1 and 2 that cover the genomic region between the deletion breakpoint and the second exon of δ-SG of the CM hamsters.
Analysis of an Alternative First Exon of δ-SG in CM Hamsters.
RT-PCR targeted for the authentic first and the second exons of δ-SG was carried out with the primer set 5′-GTGCAGGCAGGGCCTGCTCAC-3′ (exIF)/5′-CAGGCATCTTTTCCTCCAGC-3′ (exIIR1). An alternative first exon of δ-SG in the CM hamster LVs was identified by 5′ rapid amplification of cDNA ends (RACE) by using Marathon cDNA amplification kit (CLONTECH). Nested reverse primers 5′-GAGAGTACTGTTCCTGAGGC-3′ (M1) and 5′-TGAGCTGGGCATGGTGCTCC-3′ (M2) were used to ensure specificity and yield of amplification. A part of the alternative first exon (274 bp; AlEx 1 probe) was amplified by RT-PCR with the primer set 5′-GAATAAACCTACCACAAGAC-3′ (alexIF)/5′-TGGTTTCTTCTAGTGTCATC-3′ (alexIR) and hybridized against the 13.3-kb EcoRI–EcoRI fragment containing the genomic breakpoint of the CM hamsters (see Fig. 3 Middle).
In Vitro Pull-Down Binding Study and Ligand Overlay Assay.
For convenience, putative extracellular domains of α-, β-, γ-, and δ-SGs, and β-DG are designated herein as α-SG-N, β-SG-C, γ-SG-C, δ-SG-C, and β-DG-N (see Fig. 7). Inserts encoding the desired domains of hamster SGs or human DGs (14) were generated by PCR from cloned hamster SG cDNAs or human heart cDNA (Quick-Clone cDNA; CLONTECH), respectively, and were subcloned into pET33 or pCITE4 vector (Novagen). The primer sets used were as follow: α-SG-N (1–290), 5′-ATGGCAGCGACACTCACTTG-3′/5′-GGCATCTGCCAGGAAGTCTC-3′; β-SG-C (93–320), 5′-CGAATTGGGCCAAATGGCTG-3′/5′-CTAATGAGTGTTCCCACAGG-3′; γ-SG-C (61–291), 5′-ATGTGGTTTTCTCCAATAGG-3′/5′-TCAGAGACAGATGTGGCTGT-3′; δ-SG-C (57–289), 5′-AAAGTCATGAACTTCACAAT-3′/5′-TCAAAGGCAGACACTTGTGT-3′; α-DG (1–457), 5′-ATGAGGATGTCTGTGGGCCT-3′/5′-CCGGGGTGTCCGTGGTTTCT-3′; β-DG-N (458–750), 5′-CCAGTGCCCCGGGTCACCAC-3′/5′-GTAGACATCATCCTCACTGC-3′. The numbers in the parentheses denote the amino acid positions of the corresponding proteins.
Figure 7.

Specific interactions of SGs and DGs. The predicted hamster α-, β-, γ-, and δ-SG polypeptides comprised 387, 320, 291, and 289 amino acids, respectively, and exhibited the following essentially similar structures: (i) a large extracellular domain with a putative N-glycosylation site(s), (ii) a single transmembrane domain, and (iii) a relatively short intracellular domain. The N-terminal domain of α-SG is predicted to be extracellular, due to the presence of a signal sequence. The C-terminal domains of the β-, γ-, and δ-SGs are assumed to be extracellular. α-DG is an extracellular globular glycoprotein and β-DG is another membrane protein with a topography similar to α-SG (14). Accordingly, putative extracellular domains of α-, β-, γ-, and δ-SGs and β-DG are designated as α-SG N, β-SG C, γ-SG C, δ-SG C, and β-DG N. (a) In vitro pull-down study for SGs and DGs. Specific interaction of labeled (input indicated at the bottom) and immobilized (shown in the lanes) proteins is detected as a band. For SGs, reciprocal pull-down combinations were obtained as follows. α-SG-N bound β-SG-C and δ-SG-C. β-SG-C bound α-SG-N, γ-SG-C, and δ-SG-C. γ-SG-C bound β-SG-C and δ-SG-C. δ-SG-C bound α-SG-N, β-SG-C, and γ-SG-C. Strong signals indicate high affinities between the two proteins tested (such as β-SG-C vs. δ-SG-C). None of the intracellular domains of the four SGs and β-DG bound each other (data not shown). α-DG bound α-SG-N, β-SG-C, δ-SG-C, and β-DG-N. β-DG-N bound α-SG-N, β-SG-C, γ-SG-C, and δ-SG-C. In ×0.1 indicates 10% of the input. (b) Ligand overlay assay for DGs. Specific interaction of labeled (incubated; indicated at the bottom) and electrophoresed (shown in the lanes) proteins is detected as a band. CBB, Coomassie staining. The same binding profiles as in a were confirmed.
The primer sets for human α- and β-DGs were designed herein according to the general belief that a single polypeptide precursor of 895 amino acids is cleaved at Arg-457 to generate α- and β-DGs (14). All the PCR products were sequenced on both strands and care was taken to fuse cDNAs in-frame (15). Proteins with the 6×His-tag were expressed from pET constructs in Escherichia coli BL21 (DE3), purified, and immobilized onto TALON beads (CLONTECH). 35S-labeled proteins, fused with an S-tag comprising 15 amino acids, were generated from pCITE constructs by in vitro transcription/translation, using a TNT-coupled reticulocyte lysate system (Promega). Equivalent amounts of the labeled proteins were incubated with the 6×His-tagged proteins immobilized onto TALON beads, centrifuged, washed, and boiled in SDS sample buffer. The eluted proteins were resolved by SDS/PAGE and subjected to fluorography. For ligand overlay assay, 6×His-tagged fusion proteins were fractionated by SDS/PAGE, transferred to nitrocellulose membrane, renatured with guanidine hydrochloride, incubated with the 35S-labeled fusion proteins, washed, and then analyzed by fluorography.
RESULTS AND DISCUSSION
Clinical Features of Hearts from HCM and DCM Hamsters.
First, we compared the morphological and pathological aspects of the hearts from Golden, BIO14.6, UMX7.1, and TO-2 hamsters (Fig. 1). Degenerative changes such as necrosis, fibrosis, and calcification were detected in all the CM hamsters (Fig. 1a). The mass and wall thickness of the LV from BIO14.6 or UMX7.1 were dramatically or slightly increased, respectively. On the other hand, the LV wall of TO-2 was very thin (Fig. 1 b and c). The hypertrophic alteration was also reflected to the amplitude of QRS complex in electrocardiogram recordings (Fig. 1d). From these clinicopathological findings, it is reasonable to categorize BIO14.6 and UMX7.1 as HCM and TO-2 as DCM.
Figure 1.
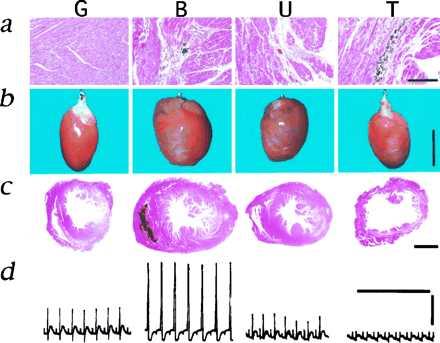
Microscopic and macroscopic features of CM hamster hearts. G, B, U, and T denote Golden, BIO14.6, UMX7.1, and TO-2 hamsters, respectively. More than 10 animals of each subline were analyzed and representative photographs are presented. (a) Hematoxylin and eosin (H&E) staining of the LVs. Note necrosis, fibrosis, and calcification in B, U, and T. (Bar = 200 μm.) (b) Macroscopic appearances of the whole hearts. The remaining blood within a heart was cleared by a Langendorff perfusion apparatus (16). The white streaks visible in B, U, and T indicate calcification. (Bar = 1 cm.) (c) Cross-section of the LVs and right ventricles stained with H&E. (Bar = 2 mm.) (d) The electrocardiogram recording from the first lead. The amplitudes (volts) of QRS complex for G, B, U, and T were 0.55 ± 0.06 (mean ± SEM, n = 11), 0.98 ± 0.15 (n = 7), 0.45 ± 0.05 (n = 17), and 0.22 ± 0.08 (n = 3), respectively. The vertical and horizontal bars indicate 0.5 V and 1 sec, respectively.
δ-SG as a Candidate Causative Gene Common to Both HCM and DCM Hamsters.
In myocardium of all the CM hamsters, α- and δ-SGs, as well as β- and γ-SGs (data not shown), were lost, whereas α- and β-DGs were dramatically reduced but detected (Fig. 2a). All these SGs were also missing in skeletal muscle of all the CM hamsters (data not shown). However, levels of mRNAs for α-, β-, and γ-SGs were equally expressed in all the hamsters including normal controls (Fig. 2b). In contrast, the transcript for δ-SG was not detected by RNA blot analysis in both cardiac and skeletal muscles of the CM hamsters (Fig. 2b). These findings suggested that the defect of δ-SG gene might be the common genetic cause for all the CM hamsters.
Figure 2.
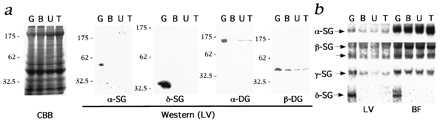
δ-SG as a candidate causative gene common to both HCM and DCM hamsters. (a) Western blot analyses of α- and δ-SGs and α- and β-DGs for LVs. CBB; Coomassie staining. Note that reduction of α-DG was more prominent than that of β-DG in all the CM hamsters. (b) RNA blot analyses of α-, β-, γ-, and δ-SGs. The transcript sizes are 1.5 kb for α-, 4.5 and 3.0 kb for β-, 1.7 kb for γ-, and 9.5 kb for δ-SGs. Besides the 9.5-kb transcript, there were also 4.3-, 2.3-, and 1.4-kb δ-SG transcripts, none of which was detectable in any CM hamsters. Full-length cDNAs for Golden hamster α-, β-, γ-, and δ-SGs were used as probes. BF, biceps femoris.
Deletion of δ-SG Gene in Both HCM and DCM Hamsters.
Genomic Southern blot analyses by several cDNA probes of δ-SG detected the same fragments among normal and all the CM hamsters (data not shown), which strongly indicated that a mutation of δ-SG gene of the CM hamsters reside around its promoter region. We cloned genomic DNAs from normal hamsters covering the first and the second exons of δ-SG gene (Fig. 3). A series of Southern blot analyses revealed a genomic deletion disrupting normal δ-SG gene in all the CM hamsters (Figs. 3 and 4). A 1.1-kb EcoRI restriction fragment that includes the second exon (Ex 2 probe; Fig. 3) detected the same bands in normal and all CM hamsters (Fig. 4c). On the other hand, a 2.1-kb EcoRI fragment containing the first exon (Ex 1 probe; Fig. 3) failed to detect the corresponding genomic fragments in any CM hamsters (Fig. 4b). These observations suggest that the 3′ end of a genomic deletion resides between the first and the second exons (i.e., the first intron) of δ-SG gene in all the CM hamsters.
Figure 4.
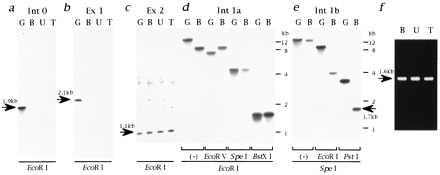
Cloning of the genomic DNA containing the deletion breakpoint of the CM hamsters. Genomic DNAs digested with EcoRI were hybridized by the Int 0 (a), Ex 1 (b), or Ex 2 (c) probes. The sizes of hybridized bands are shown by arrows. (d) Genomic DNAs digested with EcoRI followed by EcoRV, SpeI, or BstXI were hybridized by the Int 1a probe. Note that restriction fragment length polymorphisms were detected for EcoRI/EcoRV but not for EcoRI/SpeI digestion. (e) Genomic DNAs digested with SpeI followed by EcoRI or PstI were hybridized by the Int 1b probe. Note that the 1.7-kb polymorphic band (shown by an arrow) was detected for SpeI/PstI digestion. The same results as d and e were obtained for both U and T (data not shown). (f) Amplification of polymorphic PstI–SpeI fragments of the CM hamster genomes. The size of the amplified fragment (1.6 kb; shown by an arrow) is smaller than that of the SpeI–PstI restriction fragment length polymorphism band (1.7 kb), because the GS primer used for this cloning is located 80 bp 5′ upstream from the SpeI cleavage site (Fig. 3 Bottom).
A 1.3-kb SacI–EcoRI fragment (Int 1a probe; Fig. 3) hybridized with the same fragments digested with EcoRI and SpeI in both Golden and BIO14.6 hamsters, whereas the same Int 1a probe exhibited restriction fragment length polymorphism for genomic DNAs digested with EcoRI and EcoRV for BIO14.6 hamster (Fig. 4d). The same results were obtained for UMX7.1 and TO-2 hamsters (data not shown). These results indicate that the genomic breakpoint is located between EcoRV and SpeI restriction sites of the first intron of δ-SG in all the CM hamsters.
A 2.7-kb EcoRV–SpeI fragment (Int 1b probe; Fig. 3) detected 1.7-kb polymorphic bands for genomic DNAs digested with SpeI and PstI for UMX7.1 and TO-2 hamsters (data not shown) as well as for BIO14.6 hamster (Fig. 4e). We further amplified these 1.7-kb SpeI–PstI polymorphic genomic fragments from all the CM hamsters (Fig. 4f). Comparison of the sequences of the polymorphic fragments with that of Int 1b probe located the deletion breakpoint at 1,299 bp 5′ upstream from the underlined thymidine in the SpeI recognition sequence (ACTAGT) for all the CM hamsters (Fig. 3 Bottom). Finally, a 1.9-kb EcoRI fragment located at 27.4 kb 5′ upstream from the breakpoint (Int 0 probe; Fig. 3) detected no genomic band in any CM hamsters (Fig. 4a). Thus, with the fact that the SpeI restriction site was located at 4.8 kb 5′ upstream the 5′ end of the second exon of δ-SG gene (Fig. 3 Middle), we concluded that, in all the CM hamsters, more than 27.4 kb of the genomic region including the first exon of δ-SG is deleted, the 3′ end of which is located at 6.1 kb 5′ upstream of the 5′ end of the second exon of δ-SG gene (Fig. 3 Top).
Gene Structure, Tissue Distribution, and Predicted Polypeptide of δ-SG.
Comparison of the sequences of genomic DNA and cDNA revealed that the authentic first exon of δ-SG gene consists of the 5′ untranslated region and the second exon begins just at the translation initiation codon (Fig. 5). The hamster δ-SG gene lacks a TATA box but contains several consensus binding sequences for the transcriptional activator Sp1 (17) around the major transcription initiation site, one of which is presented in Fig. 5.
RNA blot analysis of various tissues of normal hamsters, by using the full-length δ-SG cDNA as a probe, detected a predominant transcript of 9.5 kb in cardiac and skeletal muscles (Fig. 2b) and fainter bands of the same size in stomach and uterus (data not shown), indicating that δ-SG is expressed in not only striated but also smooth muscles, where no pathological change was observed by light microscopy in any CM hamsters (data not shown). The reason why the tissues other than cardiac and skeletal muscles are not affected in the CM hamsters was not extensively analyzed in the present study.
Sequencing of δ-SG cDNA predicted that hamster δ-SG is a polypeptide of 289 amino acid residues with a single transmembrane domain, exhibiting 93.8% amino acid identity with human δ-SG (12). The C-terminal 233 amino acids were predicted to be extracellular because no N-terminal signal sequence (18) was found. Three potential N-glycosylation sites (19) were identified at this putative extracellular domain. The structures of the other SG polypeptides predicted from the cloned cDNAs are described in Fig. 7.
δ-SG Transcript in CM Hamsters.
Conservation of the protein coding region of δ-SG gene starting just from the second exon (Fig. 5 Right) in the CM hamsters raised a question if δ-SG transcript might be present. Indeed, semi-quantitative multiplex RT-PCR targeted to the second exon did detect δ-SG transcript in LVs of all the CM hamsters but estimated the amount as significantly (20–40 times) lower than that of normal hamsters (Fig. 6a), which is consistent with our RNA blot analysis data (Fig. 2b). Failure to amplify the δ-SG cDNAs with another primer set, the forward primer of which resides in the first exon (exIF in Fig. 5; Fig. 6b), indicates that the first exon of δ-SG in the CM hamsters is not identical to the authentic one used by normal hamsters. We cloned and elucidated this “alternative” first exon comprising 481 bp by 5′ RACE method (Figs. 5 and 6c). The “alternative” first exon seems to be located 5′ upstream of the deleted region of δ-SG in the CM hamsters, because it did not hybridize with the normal genomic DNA fragments encompassing the deletion breakpoint and the second exon of δ-SG gene of the CM hamsters (Fig. 6d).
These results suggest that, in all the CM hamsters, transcription of δ-SG gene does operate from an alternative or “cryptic” promoter. However, such alternative δ-SG transcripts are probably inadequate or unable to compensate for the authentic δ-SG transcripts, which are all missing in all the CM hamsters, and the consequent loss of δ-SG polypeptide (Fig. 2a) might be the common pathogenetic cause of CM and muscular dystrophy in all the CM hamsters. Another example of such genomic deletion specific to a gene promoter region is one form of human X chromosome-linked DCM. In this setting, the muscle-specific promoter of dystrophin gene is deleted and the preserved brain-specific promoter of the same gene cannot compensate for this deleted promoter in cardiac muscle with consequent loss of dystrophin (20).
A Scheme for CM Development Secondary to δ-SG Deficiency.
To understand the pathological role of δ-SG in CM, we elucidated the precise molecular architecture of DAGC (14, 21). α- and β-DGs (14), the other constituents of DAGC, link extracellular matrix laminin with an intracellular membrane protein dystrophin, the loss of which causes Duchenne muscular dystrophy (21). First, we analyzed the structure of the SG subcomplex by in vitro pull-down binding study (Fig. 7a). The four SGs bound one another at the putative extracellular (EC) domains (Fig. 8 Left), consistent with the reported importance of the EC domains of the SGs, where almost all the identified mutations of every SG gene reside (7–10, 24). Next, we investigated possible interactions of the SGs with α- and β-DGs by in vitro pull-down binding study (Fig. 7a) and ligand overlay assay (Fig. 7b). α-DG interacted with the EC domain of every SG except γ-SG, along with the EC domain of β-DG. On the other hand, β-DG bound all the SGs at the EC domains. Thus, with the fact that the SGs form a distinct integral component of DAGC on a stoichiometrically equal basis (22), we propose a molecular model for the structure of DAGC (Fig. 8 Left).
Figure 8.
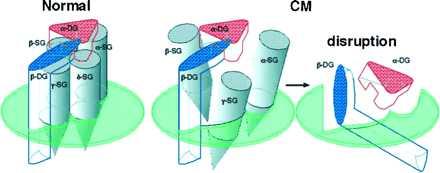
Hypothetical model of the DAGC architecture in normal cardiac sarcolemma and its disruption caused by deficiency of δ-SG in CM hamsters. The horizontal planes indicate sarcolemma, above which is the extracellular space. α-DG directly binds extracellular matrix laminin (14) and β-DG directly binds dystrophin with the C-terminal intracellular domain (25). (Left) In normal hamsters, the four SGs bind one another at the extracellular domains and constitute the SG subcomplex, which serves as a molecular stabilizer for the DG subcomplex. (Center and Right) Deficiency of δ-SG in the CM hamsters could disrupt the SG subcomplex (Center) because, for example, α-SG does not bind to γ-SG, and eventually the whole DAGC (Right), rendering cardiomyocytes more susceptible to mechanical stress generated by contraction of cardiac muscle. Molecular weights of α-, β-, γ-, and δ-SGs and α- and β-DGs are 50, 43, 35, 35, 156, and 43 kDa, respectively (7–10, 12, 14).
The SG subcomplex may function as a molecular stabilizer for the DG subcomplex. Marked reduction in α- and β-DGs in the LVs of all the three CM hamsters, along with the loss of α-SG (Fig. 2a), is in good agreement with the previous report (11), and our DAGC model predicts it from the loss of δ-SG (Fig. 8 Center and Right). The secondary deficiency of the DG subcomlex (Fig. 8 Right) may explain the disruption of functional linkage between laminin and dystrophin (11, 23), leading to cellular degeneration or necrosis. Compared with α-DG, β-DG was relatively well preserved in the CM hamsters (Fig. 2a), probably because β-DG is directly anchored by dystrophin (25). In humans, the defect of any one of the four SG genes causes limb-girdle muscular dystrophy (LGMD) with secondary loss of all the other SGs (22). Overall, LGMDs rarely accompany CM (26), but there are several unequivocal case reports for LGMD with CM where SGs are missing instead of dystrophin (27). Further study concerning a possible mutation of one of the four SG genes in human CM should support the general importance of SGs in CM.
Another Genetic Defect in DCM Hamster?
The striking finding in the present study is that both HCM and DCM could be caused by mutation of the same gene in hamster. Hypertrophy in HCM is considered as a natural compensatory reaction, because the surviving cardiomyocytes surrounding a necrotic cell in CM suffer from hemodynamic overload that induces cardiac hypertrophy to maintain the cardiac output (28). In other words, DCM hamsters might have another genetic defect in this compensatory hypertrophic mechanism. The present study not only suggests the usefulness of CM hamsters for understanding the functions of DAGC in vivo but also provides a target for genetic diagnosis and significant insight into pathogenesis of human CM.
Acknowledgments
We thank Dr. M. Imamura for helpful discussion; Ms. R. Kaku and Mr. M. Yoneyama for technical assistance; and Drs. L. Proschek, M. Yanagisawa, Y. Nonomura, M. Kato, and M. Nagano for continuous encouragement. A.S. is the Special Postdoctoral Researcher of The Institute of Physical and Chemical Research. This work was supported in part by the Special Grant for Promotion of Research from The Institute of Physical and Chemical Research (A.S.), a grant from the Mitsui Life Social Welfare Foundation (A.S.), and a Grant-in-Aid for Encouragement of Young Scientists from the Ministry of Education, Science, Sports and Culture, Japan (A.S.).
ABBREVIATIONS
- CM
cardiomyopathy
- HCM
hypertrophic CM
- DCM
dilated CM
- DAGC
dystrophin-associated glycoprotein complex
- SG
sarcoglycan
- DG
dystroglycan
- EC
extracellular
- LV
left ventricle
- CM hamster
cardiomyopathic hamster
- RT-PCR
reverse transcription-coupled PCR
- RACE
rapid amplification of cDNA ends
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the DDBJ, EMBL, and GenBank databases [accession nos. D83651 (hamster α-SG cDNA), D83652 (hamster β-SG cDNA), D83653 (hamster γ-SG cDNA), AB001508 (hamster δ-SG cDNA), AB007020 (alternative first exon of hamster δ-SG), AB001509 (hamster δ-SG genomic DNA around the first exon), AB007021 (hamster δ-SG genomic DNA around the second exon), AB007022 (hamster δ-SG genomic DNA within the first intron) and AB007023 (CM hamster genomic DNA around the deletion breakpoint)].
References
- 1.Toyo-oka T, Nagayama K, Suzuki J, Sugimoto T. Circulation. 1992;86:295–301. doi: 10.1161/01.cir.86.1.295. [DOI] [PubMed] [Google Scholar]
- 2.Tanigawa G, Jarcho J A, Kass S, Solomon S D, Vosberg H-P, Seidman J G, Seidman C E. Cell. 1990;62:991–998. doi: 10.1016/0092-8674(90)90273-h. [DOI] [PubMed] [Google Scholar]
- 3.Kelly D P, Strauss A W. N Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 4.Homburger F, Baker J R, Nixon C W, Whitney R. Med Exp. 1962;6:339–345. [Google Scholar]
- 5.Jasmin G, Eu H Y. Ann NY Acad Sci. 1979;317:46–58. doi: 10.1111/j.1749-6632.1979.tb56509.x. [DOI] [PubMed] [Google Scholar]
- 6.Sole M J. Hamster Information Service. 1986;8:3–6. [Google Scholar]
- 7.Roberds S L, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson R D, Lim L E, Lee J C, Tomé F M S, Romero N B, Fardeau M, Beckmann J S, Kaplan J -C, Campbell K P. Cell. 1994;78:625–633. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- 8.Lim L E, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, Tomé F M S, Fardeau M, Jackson C E, Beckmann J S, Campbell K P. Nat Genet. 1995;11:257–265. doi: 10.1038/ng1195-257. [DOI] [PubMed] [Google Scholar]
- 9.Bönnemann C G, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally E M, Duggan D J, Angelini C, Hoffman E P, Ozawa E, Kunkel L M. Nat Genet. 1995;11:266–272. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- 10.Noguchi S, McNally E M, Othmane K B, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bönnemann C G, Gussoni E, Denton P H, Kyriakides T, Middleton L, Hentati F, Hamida M B, Nonaka I, Vance J M, Kunkel L M, Ozawa E. Science. 1995;270:819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- 11.Roberds S L, Ervasti J M, Anderson R D, Ohlendieck K, Kahl S D, Zoloto D, Campbell K P. J Biol Chem. 1993;268:11496–11499. [PubMed] [Google Scholar]
- 12.Nigro V, Piluso G, Belsito A, Politano L, Puca A A, Papparella S, Rossi E, Viglietto G, Esposito M G, Abbondanza C, Medici N, Molinari A M, Nigro G, Puca G A. Hum Mol Genet. 1996;5:1179–1186. doi: 10.1093/hmg/5.8.1179. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto A, Yanagisawa M, Sakurai T, Takuwa Y, Yanagisawa H, Masaki T. Biochem Biophys Res Commun. 1991;178:656–663. doi: 10.1016/0006-291x(91)90158-4. [DOI] [PubMed] [Google Scholar]
- 14.Ibraghimov-Beskrovnaya O, Milatovich A, Ozcelik T, Yang B, Koepnick K, Francke U, Campbell K P. Hum Mol Genet. 1993;2:1651–1657. doi: 10.1093/hmg/2.10.1651. [DOI] [PubMed] [Google Scholar]
- 15.Sakamoto A, Yanagisawa M, Sawamura T, Enoki T, Ohtani T, Sakurai T, Nakao K, Toyo-oka T, Masaki T. J Biol Chem. 1993;268:8547–8553. [PubMed] [Google Scholar]
- 16.Ono K, Delay M, Nakajima T, Irisawa H, Giles W. Nature (London) 1989;340:721–724. doi: 10.1038/340721a0. [DOI] [PubMed] [Google Scholar]
- 17.Kadonaga J T, Jones K A, Tjian R. Trends Biochem Sci. 1986;11:20–23. [Google Scholar]
- 18.von Heijne G. Nucleic Acids Res. 1986;14:4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard S C, Ivatt R J. Annu Rev Biochem. 1981;50:555–583. doi: 10.1146/annurev.bi.50.070181.003011. [DOI] [PubMed] [Google Scholar]
- 20.Muntoni F, Cau M, Congiu R, Ganau A, Arvedi G, Mateddu A, Marrosu M G, Cianchetti C, Realdi A, Cao A, Melis M A. N Engl J Med. 1993;329:921–925. doi: 10.1056/NEJM199309233291304. [DOI] [PubMed] [Google Scholar]
- 21.Ahn A H, Kunkel L M. Nat Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- 22.Jung D, Duclos F, Apostol B, Straub V, Lee J C, Allamand V, Venzke D P, Sunada Y, Moomaw C R, Leveille C J, Slaughter C A, Crawford T O, McPherson J D, Campbell K P. J Biol Chem. 1996;271:32321–32329. doi: 10.1074/jbc.271.50.32321. [DOI] [PubMed] [Google Scholar]
- 23.Iwata Y, Nakamura H, Mizuno Y, Yoshida M, Ozawa E, Shigekawa M. FEBS Lett. 1993;329:227–231. doi: 10.1016/0014-5793(93)80227-l. [DOI] [PubMed] [Google Scholar]
- 24.Nigro V, Moreira E S, Piluso G, Vainzof M, Belsito A, Politano L, Puca A A, Passos-Bueno M R, Zatz M. Nat Genet. 1996;14:195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- 25.Jung D, Yang B, Meyer J, Chamberlain J S, Campbell K P. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- 26.Perloff J K, De Leon A C, Jr, O’Doherty D. Circulation. 1966;33:625–648. doi: 10.1161/01.cir.33.4.625. [DOI] [PubMed] [Google Scholar]
- 27.Fadic R, Sunada Y, Waclawik A J, Buck S, Lewandoski P J, Campbell K P, Lotz B P. N Engl J Med. 1996;334:362–366. doi: 10.1056/NEJM199602083340604. [DOI] [PubMed] [Google Scholar]
- 28.Zak, R. (1974) Circ. Res. 35, Suppl. 2, 17–26. [PubMed]