

Abstract
Delta (™)-catenin is a brain specific member of the adherens junction complex that localizes to the post-synaptic and dendritic compartments. This protein is likely critical for normal cognitive function; its hemizygous loss is linked to the severe mental retardation syndrome, Cri-du-Chat, and it directly interacts with Presenilin-1 (PS1), the protein most frequently mutated in familial Alzheimer's disease. Mice lacking normal ™-catenin display severe impairments in learning and memory tasks and synaptic plasticity. Here we examine dendritic structure and cortical function in vivo in mice lacking ™-catenin. We find that in cerebral cortex of 5-week-old mice dendritic complexity, spine density, and cortical responsiveness are similar between mutant and littermate controls; thereafter, mutant mice experience progressive dendritic retraction, a reduction in spine density and stability, and concomitant reductions in cortical responsiveness. Our results indicate that ™-catenin regulates the maintenance of dendrites and dendritic spines in mature cortex but does not appear to be necessary for the initial establishment of these structures during development.
Introduction
Cell adhesion molecules regulate the establishment and maintenance of cell morphology. In the nervous system the highly polarized structures of neurons result, in part, from direct interactions between cadherins, a class of calcium-dependent adhesion molecules that span the plasma membrane, and catenins, intracellular proteins that bind to cadherin cytoplasmic domains (Benson and Tanaka, 1998; Fannon and Colman, 1996; Inoue and Sanes, 1997).
™-catenin is a member of the P120 subfamily of catenins that is expressed almost exclusively in neurons (Ho et al., 2000; Paffenholz and Franke, 1997; Zhou et al., 1997). Expression of ™-catenin begins at embryonic day 10 and peaks at postnatal day 7. In the mature neurons ™-catenin localizes to dendrites and dendritic spines (Kosik et al., 2005) with expression predominantly in the cerebral cortex, hippocampus, olfactory bulb, and to a lesser degree in the thalamus and cerebellum (Alan Brain Atlas).
In dendritic spines ™-catenin provides a link to the actin cytoskeleton via direct interactions with cortactin (Martinez et al., 2003) and the Rho-family GTPase, p190RhoGEF (Kim et al., 2008). Additionally, it interacts with synaptic scaffolding and receptor binding proteins including PSD-95 (Kim et al., 2002), S-SCAM (Ide et al., 1999), AMPA receptor binding protein (ABP), and GluR-interacting protein (GRIP) (Silverman et al., 2007). Because neural activity can regulate these interactions, ™-catenin is positioned to mediate activity-dependent alterations in synaptic adhesion and spine morphology thought to underlie adaptive changes in neural connectivity (Matsuzaki et al., 2004; Takeichi and Abe, 2005).
™-catenin's role in regulating neural structure is not fully established. In cultured hippocampal neurons dendritic branching is enriched or impoverished, respectively, by ™-catenin over- or under-expression (Arikkath et al., 2008; Elia et al., 2006; Kim et al., 2008). However, homozygous loss of ™-catenin in vivo does not appear to grossly impact neuronal development; cortical and cerebellar lamination are grossly normal as is hippocampal formation. Synaptic ultra-structure is also qualitatively normal in these mice (Israely et al., 2004).
Mutations in ™-catenin severely impair cognitive function and underlie some forms of mental retardation and Alzheimer's disease (Israely et al., 2004; Kim et al., 2006; Liauw et al., 2002; Medina et al., 2000). In humans ™-catenin is localized to chromosome 5p15.2, the critical region for the severe mental retardation syndrome Cri-du-Chat (CDCS), which accounts for 1% of all mentally retarded individuals. In patients with CDCS, the severity of mental retardation correlates with hemizygous loss of the ™-catenin gene (Medina et al., 2000). This link is further supported by studies demonstrating severe deficits in learning, memory, and synaptic plasticity in mice with a targeted disruption of ™-catenin (Israely et al., 2004). In addition, ™-catenin was first identified through its interaction with the loop domain of Presenilin-1 (Tanahashi and Tabira, 1999; Zhou et al., 1997). The gene encoding Presenilin-1 (PS1) is the most frequently mutated gene in familial Alzheimer's disease – a disease characterized by the progressive loss of synapses and dendrites that precedes cognitive decline.
To examine how the structure and function of cortical circuits is altered by the loss of ™-catenin in vivo, we used vital imaging techniques to track changes in the structure of dendrites, the stability of spines on these dendrites, cortical responsiveness, and the organization retinotopic maps in visual cortex of mice with a germ line deletion of ™-catenin. In 5-week-old ™-catenin mutant mice, dendritic complexity, spine density, and cortical responsiveness were all normal. Thereafter these mice experienced a progressive loss of dendrites and spines that was mirrored by decrements in cortical responsiveness and retinotopic organization. Our observations indicate that, in the neocortex, ™-catenin is critical for the maintenance, though not the establishment of neuronal structure and function in vivo.
Results
Acute imaging of layer 2/3 and 5 apical dendrites in vivo
Dendrites and dendritic spines were imaged in primary sensory cortex of transgenic mice expressing EGFP in a subset of pyramidal neurons in layers 2/3 and 5 (Feng et al., 2000). GFP expressing neurons in these mice receive a pattern of synaptic input indistinguishable from neighboring, unlabeled neurons (Holtmaat et al., 2005). In this line of mice, GFP expression in pyramidal neurons is suitably high for in vivo imaging around the third to fifth postnatal week in layer 5 and around the eighth to tenth postnatal week in layer 2/3. To determine how ™-catenin influences the development and maintenance of neural circuitry in primary sensory cortex we crossed the GFP-M line of mice into a ™-catenin mutant background (see Methods). All images of dendrites and dendritic spines were acquired in vivo.
In 5-week-old mice, measurements of layer 5 apical dendrite arbor size, segment number, tip number, and branching complexity were not significantly different between age-matched controls and mutants. When these measurements were made in 10-week-old mice, the differences were striking (Figure 1A-C). Relative to age-matched controls, total arbor length was reduced by approximately 30% in mutant mice. Dendritic geometry was similarly less complex in the mutants. The total number of dendritic segments and the number of dendritic tips were reduced by 30%.
Figure 1. Dendritic complexity of cortical pyramidal neurons in 5 and 10-week-old ™-catenin mutant and control mice.
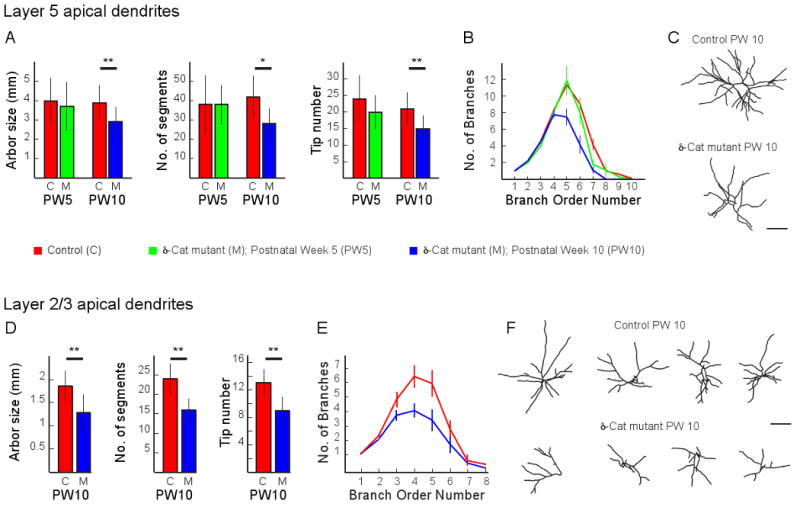
A) Measurements of total arbor size, segment number and tip number made from 3-dimensional image stacks of layer 5 apical dendrites acquired acutely in vivo from control (C, red) and d-catenin mutant (M) mice that were 5 or 10 weeks old (PW5, green or PW10, blue, respectively). Plots are mean and standard deviation. A single asterisk indicates significance at 0.05; two asterisks indicate significance at 0.01. CPW5, n = 9 neurons from 6 mice; MPW5, n = 8 neurons from 5 mice; CPW10, n = 30 neurons from 18 mice; MPW10, n = 13 neurons from 7 mice.
B) Plot of dendritic segment number as a function of branch order from the same dendrites analyzed in (A). Plots are mean and standard error. PW5 and PW10 controls are grouped for this plot and displayed as a single line. Asterisks indicate a significant decrease in branching complexity for neurons imaged in PW10 mutant mice.
C) Examples of reconstructions of the apical arbors of a layer 5 pyramidal neuron from a 10-week-old control (upper panel) and mutant mouse (lower panel). Scale bar is 100μm.
D) Measurements as in (A) from layer 2/3 apical dendrites PW10 control and mutant mice. CPW10, n = 8 neurons from 6 mice; MPW10, n = 6 neurons from 5 mice.
E) Plot of dendritic segment number as a function of branch order from the same dendrites analyzed in (D).
F) Example reconstructions of the apical dendrites of some of the layer 2/3 pyramidal neurons analyzed in (D) and (E). Scale bar is 100 μm.
Similar impairments in dendritic complexity were also observed in layer 2/3 pyramidal neurons in 10-week-old mutants (Figure 1D-F), indicating that these impairments are not laminar specific. Total dendrite length was reduced by more than half a millimeter in the mutants, representing a 30% loss of arbor length. Because layer 2/3 pyramidal neurons do not express GFP at 5 weeks of age in these mice, we were unable to examine these neurons at this younger age.
In both cortical layers the lengths of lower order branches were not measurably different between mutant and control mice (data not shown), suggesting that the simpler dendritic arbors observed in 10-week-old ™-catenin mutant mice resulted from a reduced number of higher order dendritic segments, not a global reduction in lengths of all segments.
In accordance with these observations, laser capture quantitative rtPCR showed that in wild-type mice ™-catenin RNA is expressed in both cortical layers and that its levels, normalized to actin RNA levels and expressed in arbitrary units, do not change significantly between 5 and 10 weeks of age (Layer 2/3: 5 weeks postnatal – 1.8±0.7, 10 weeks postnatal – 2.3±0.9, P = 0.44; Layer 5: 5 weeks postnatal – 1.4±0.5, 10 weeks postnatal – 1.3±0.23, P = 0.77; n=4 mice at each age, laser capture from 10 sections per mouse).
These observations indicate that germ-line deletion of ™-catenin does not impair the normal growth of cortical dendrites or dendritic spines during development, but is detrimental to the maintenance of these structures in mature cortex.
Longitudinal imaging of layer 5 apical dendrites and spines in vivo
To obtain a more complete and nuanced understanding of what is happening to dendrites between 5 and 10 weeks of age in mutant mice we repeatedly imaged the same cortical neurons in vivo each week.
Figure 2 summarizes the results of these chronic imaging experiments from 5 neurons imaged in control mice and 6 neurons imaged in ™-catenin mutant mice. Neurons in control mice lost between 1% and 4% of their total arbor length between 5 and 10 weeks of age. The fractional loss observed over the same time course in ™-catenin mutant mice ranged from 6% to 23% of total arbor length, and reached significance relative to controls at 7 weeks of age. Notably, all imaged neurons in mutant mice experienced a significant reduction of their apical arbor. However, for any given neuron, about 40% of its dendritic terminal branches experienced a retraction greater than 10μm, with the remaining 60% of terminal branches displaying only very minor losses of less than 10μm over a 5-week interval. Thus, the majority of dendritic length lost in ™-catenin mutant mice came from the retraction displayed on 40% of its dendritic branches. For those dendrites that did undergo a retraction, the retractions were of two types – a slow, progressive retraction from the tip, or a rapid retraction with single branches losing between 65μm and 175μm in the course of a single week and then remaining stable at their new length for many weeks (as an example, see Fig. 2C).
Figure 2. Timeline of dendritic retraction.
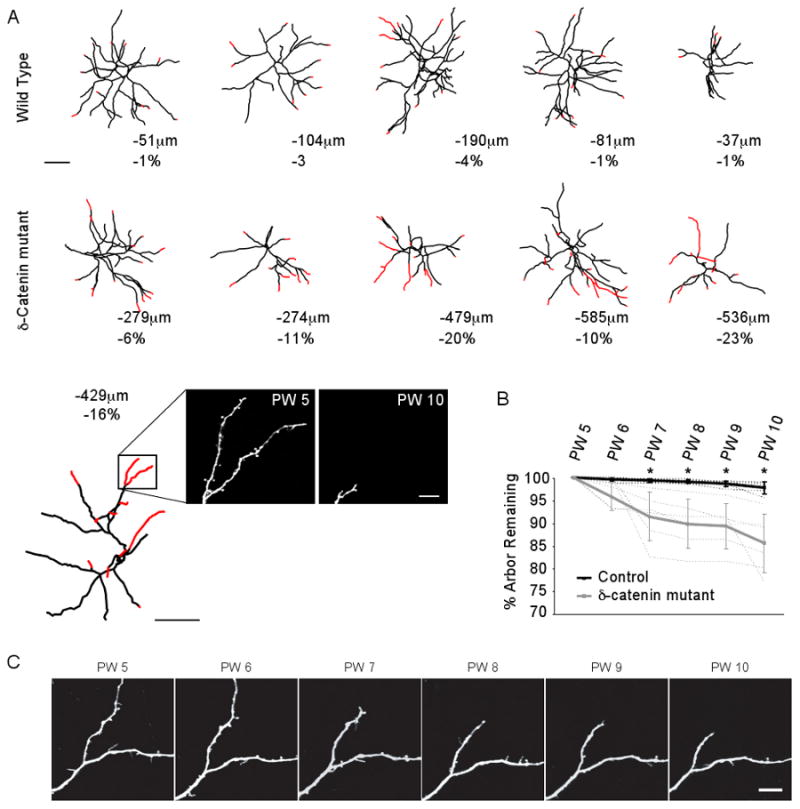
A) Neurolucida reconstructions of the apical arbors of layer 5 pyramidal neurons repeatedly imaged in vivo each week between postnatal weeks 5 and 10 in control (upper row) and mutant (lower row) mice. Red identifies dendritic regions that retracted between PW5 and PW10. Below each reconstruction the total length lost and fractional change in the size of the dendritic arbor is given. Scale bar is 100μm. In the last trace of the ™-catenin mutant series, actual images of one domain are presented showing the loss observed between postnatal week 5 (PW5) and PW10. Scale bar for the two images is 10μm.
B) Fractional change in dendrite length as a function of time in control (black line) and mutant (gray line) mice. Dashed lines indicate individual animals. Error bars are standard deviation. Note that the fractional loss observed in mutant mice becomes significant relative to controls at 7 weeks of age (asterisk).
C) Weekly times series of a dendritic region imaged in a mutant mouse. Scale bar is 10μm.
Dendritic spines also underwent a progressive change in their kinetics and density that mirrored the time course of dendrite loss (Figure 3). At 5 weeks of age spine stability was similar between mutants and controls. By 7 weeks of age, fractional spine loss significantly increased in ™-catenin mutant mice relative to controls (Figure 3D), though we observed no measurable difference in fractional spine gain between the two groups (Figure 3E). This elevated loss of spines resulted in a significant decrease in spine density by the 7th postnatal week (KO = 2.32 ± 0.37 10μm−1, WT = 3.64 ± 0.45 10μm−1, P = 0.03), which continued to drop through the 10th postnatal week (spine density: KO = 1.90 ± 0.45 10μm−1, WT = 3.13 ± 0.64 10μm−1, P < 0.001; Figure 3F). The density of long, thin, filipodia-like protrusions was not different between mutants and controls, though densities trended lower in mutant mice.
Figure 3. Spine kinetics, lengths, and volumes measured along layer 5 apical dendrites.

A) High magnification images of dendritic spines repeatedly imaged each week in vivo between postnatal weeks 5 and 10 in a control mouse. The age at image acquisition in postnatal weeks (PW) is stamped above each image. These same time stamps apply as well to the image panels in (B). In both, yellow arrowheads identify examples of spines that are stable throughout the entire imagining period. Red arrowheads identify examples of spines that are lost. Green arrowheads identify examples of spines that are gained. Scale bar is 5μm.
B) Images as in (A) acquired in a mutant mouse.
C) Survival fraction for spines imaged in control and mutant mice. The survival fraction plots the fraction of spines imaged at the first time point (here 5 weeks of age) that remain at each subsequent imaging interval. In this and panels D-G, measurements from control mice are plotted using black lines; those from mutant mice are plotted using gray lines. Dashed lines are measurements from individual neurons in each group. Error bars are standard deviation. Age, in weeks postnatal is plotted along the abscissa. Note that there is a significantly lower survival fraction of the mutants observed at 7 weeks of age.
D) Plot of the fraction of spines imaged at postnatal week 5 that have been lost at each subsequent imaging interval. Note the significantly higher loss in the mutants at 7 weeks of age.
E) Plot of the fraction of new spines present at each imaging interval that were not present at earlier intervals. Note there is no difference between the groups.
F) Plot of spine density as a function of age in control and mutant mice. Note the significant drop in spine density at 7 weeks of age in mutant mice.
G) Plot of filopodia density as a function of age in control and mutant mice.
H) Cumulative distribution of relative brightness of protrusions from dendritic domains in control mice (blue), and from dendritic domains in mutant mice that were stable over the 5-week imaging period (red) or that had retracted during this period (green).
I) Cumulative distribution of protrusion length, as in (H).
In all panels, a single asterisk indicates significance a P<0.05 and a double asterisk indicates significance at P<0.01. Number of spines analyzed: control, n = 4386; mutant, n = 1651; Number of filopodia analyzed: control, n = 714; mutant, n = 260.
We examined the morphology of spines along stable dendrites in control and mutant mice and along dendrites that ultimately retracted in mutants (Figure 3, panels H and I). Relative brightness is a measure of spine volume (Holtmaat et al., 2005). An examination of the relative brightness distribution in Figure 3H shows that very small volume protrusions (< 0.3 A.U.) are maintained at normal ratios, but mid-range brightness is lost (asterisks), resulting in a rightward shift towards brighter protrusions. Very thin protrusions are functionally immature, weak, often lack presynatpic partners, and are typically formed within 1-3 days of the image (Knott et al., 2006; Matsuzaki et al., 2001; Trachtenberg et al., 2002; Zito et al., 2009). This observation that young spines are represented at normal levels along dendrites that ultimately retract further supports the conclusion from Figure 3E that new spine growth is unaffected in mutant mice. Consistent with a role for ™-catenin in the maintenance of dendritic spines, medium sized spines are preferentially lost in mutant mice prior to the retraction of the dendritic domain. No differences in spine lengths were found between any groups (Figure 3I; KS test, significance set to 0.05).
Cortical responses to visual stimulation at 5 and 10 weeks of age
Do these dendritic changes alter cortical function? To examine this question we acquired movies of intrinsic optical signals as the mice viewed a bar of light moving at a fixed frequency (Figure 4A). Fourier analysis of each pixel location's change in reflectance generated two maps: a magnitude map of response amplitude at stimulus frequency (Figure 4B) and a phase map (Figure 4C) representing the time of the cortical response in the stimulus cycle (Grinvald et al., 1986; Kalatsky and Stryker, 2003). The phase value of each pixel was converted to degrees of visual angle and displayed according to a color scale corresponding to the functional retinotopic map. To evaluate the functional organization of the phase maps, we measured ‘map scatter’, the average difference in phase between each pixel in the map and the 24 surrounding pixels. In this metric, measured in degrees, less scatter is indicative of a smoother map and greater precision in receptive field organization (Cang et al., 2005).
Figure 4. Cortical responsiveness driven by vision or direct stimulation in vivo.

A) Schematic showing the size of the visual stimulus presented to each mouse in degrees of visual space. The stimulus was a horizontal bar drifting at 0.125Hz.
B) Example maps of cortical responses to visual stimuli, measured as changes in reflectance, in control and mutant mice at 5 and 10 weeks of age. Scale bar is 1mm.
C) Example phase maps from control and mutant mice as in (B). Below each map is shown a magnified view of the 25×25 pixel arrays highlighted by the black boxes in the phase maps. Note the greater difference in local phase value in the mutant mice at both ages.
D) Mean and standard deviation of the response magnitude to visual stimuli at all thresholds for control (values at PW5 and PW10, shown at solid gray lines, were statistically indistinguishable, and pooled and plotted as a single black line) and mutant mice at PW 5 (dashed line; middle trace) and PW10 (dotted line; lower trace).
E) Plot of mean and standard deviation of the scatter, in degrees, for phase maps in control and mutant mice. Line styles are the same as in (D). Higher values indicate greater scatter and worse retinotopic organization. Control PW5, n = 6; mutant PW5, n = 13; control PW10, n = 8; mutant PW10, n = 9.
F&G) Examples of intrinsic optical responses, measured as a change in reflectance, to direct cortical stimulation at 150μA in PW5 (F) and PW10 (G) control and mutant mice. Scale bar in F and G is 1mm.
H) Plots of strength of cortical response to stimulus intensities of 50, 100, and 150μA in 5-week-old mice. The strength is measured as the average change in reflectance for all of the pixels included in each threshold. The strength at threshold 1 is the peak pixel value in the region of interest; the strength at threshold 0.5, for example, is the average strength of each pixel whose magnitude is greater than 50% of the peak pixel value. Each line is the average across all mice in that group. In these panels it is difficult to see the gray bars representing measurements from mutant mice because they sit almost perfectly below the lines from the control mice. Control PW5, n = 5; mutant PW5, n = 6.
I) Plot as in H, but in 10-week-old control and mutant mice. Control postnatal week10, n = 9; mutant postnatal week 10, n = 6. Error bars are standard deviation.
In control mice, we did not observe significant changes between 5 weeks of age and 10 weeks of age in measures of cortical responsiveness or functional organization (Figure 4B-E). In ™-catenin mice, visually driven cortical responses were degraded by 5 weeks of age relative to age-matched controls, and became progressively more impaired by 10 weeks of age (Figure 4B-E).
Cortical responses to direct stimulation at 5 and 10 weeks of age
Because the mutant mice have a germ-line deletion of ™-catenin, we wondered whether the reduced cortical responses to visual stimuli were cortical in origin or reflected decrements in the responsiveness of the retina or the lateral geniculate nucleus of the thalamus, the earlier stages of visual processing. We examined this by measuring the strength of the intrinsic signal optical response to direct cortical stimulation (Figure 4F-I). Here we inserted a bipolar stimulating electrode into the cortex and passed 50, 100 and 150 micro amps of current while measuring intrinsic optical responses. In 5-week-old mutants, we found no change in cortical responsiveness relative to age-matched controls (Figure 4F,I). By 10 weeks of age, however, cortical responsiveness was significantly reduced in the ™-catenin mutant mice at all stimulus intensities (Figure 4G,I). These latter measurements support a view in which the loss of cortical responsiveness parallels the loss of dendrites rather than precedes it.
Discussion
The anatomical and functional data we present suggest that ™-catenin is required for the maintenance of dendrites and dendritic spines in mature neocortex, but does not appear to be required for the initial establishment of normal dendrite complexity and length and spine density in vivo. This conclusion is somewhat at odds with recent reports documenting the necessity of δ-catenin for normal dendritic growth and spine formation of cultured hippocampal neurons (Arikkath et al., 2008; Elia et al., 2006; Kim et al., 2008). Because all of the data we present here documents changes in mature cortex in vivo, we can make no statement on whether our conclusions can be applied to developing hippocampus. Our results also support a view in which proper cortical function, and by extension, cognitive function, depends on the maintenance of the full complexity of each neuron's dendritic tree and the maintenance of the synaptic input to these dendrites.
Changes in neuronal dendritic morphology are induced by changes in cytoskeletal dynamics. The Rho family of small GTPases are highly conserved regulators of the cytoskeleton (Hall, 1998) and in neurons are important intermediates of extracellular stimuli and dynamic changes in neuronal morphology and connectivity (Elia et al., 2006; Leemhuis et. al., 2004; Martinez et. al., 2003; Nakayama et. al., 2000; Parrish et. al., 2007). δ-catenin in vitro can promote actin dependent dendritic branching by regulating Rho activity (Abu-Elneel et al., 2008; Martinez et al., 2003; Kim et al., 2008; Kim et al., 2008b). Activation of Rho results in a rapid loss of distal but not primary processes in hippocampal neurons in vitro (Nakayama et al., 2000; Kim et al., 2002). δ-catenin mediates Rho activity through an inhibitory interaction with the Rho activator, p190RhoGEF (Abu-Elneel et al., 2008; Kim et al., 2008; Kim et al., 2008b). The loss of distal, but not primary dendritic branches we report here is similar to the loss observed with activation of Rho.
δ-catenin likely regulates spine maintenance and stability through its interactions with other proteins in the synaptic adherens junction. Loss of δ-catenin results in decreased protein levels of α-catenin, β-catenin, and N-cadherin (Benson et al., 1998; Fannon & Coleman 1996; Israely et al., 2004). P120 catenin family members, including δ-catenin, stabilize adherens junction components in vitro (Davis et al., 2003). Although in vivo data from δ-catenin mutant mice is consistent with a functional role for δ-catenin in stabilizing synaptic adherens junction components, it remains unclear whether the observed reduction of these components in vivo is caused by decreased stability or simply a decrease in the number of dendritic spines. Despite these uncertainties, the altered spine dynamics observed in the δ-catenin mutant mice are consistent with both increased activation of Rho in mature neurons and down regulation of adherens junction components (Nakayama et al., 2000; Tashiro et al., 2000; Govek et al., 2004; Takeichi et al., 2007; Bozdagi et al., 2004; Bozdagi et al., 2000; Abe et al., 2004; Okamura et al., 2004; Tang et al., 1998; Inoue and Sanes, 1997; Iwai et al., 2002; Tai CY et al., 2007).
Notably, our data do not support a role for δ-catenin in the formation of new dendritic spines. The rate of spine growth was unaffected in mutant mice, and the cumulative distribution of spine volumes along retracting dendrites showed no change in the fraction of small volume spines, which have very short lifetimes and have typically grow in the 1-3 day period preceding the image (Holtmaat et al., 2005; Knott et al., 2006; Trachtenberg et al., 2002).
The observed progressive decline in dendritic length, complexity, and spine density in the δ-catenin mutant mice decreases the connectivity of neural circuits. This likely underlies the loss of cortical responsiveness to sensory and direct stimulation, and impairs the integration and processing of information in these circuits (Hausser et al., 2000). Loss or abnormal connectivity in the cortex has long been suggested to underlie mental retardation (Kaufmann et al., 2000). In humans, δ-catenin is localized on chromosome 5p15.2, the critical region for the severe mental retardation syndrome, Cri-du-Chat (CDCS). Breakpoint analysis in patients with 5p terminal deletions indicates that the severity of mental retardation correlated with hemizygous loss of the δ-catenin gene (Medina et al., 2000). It is not clear how dendrites are compromised in CDCS, and attributing any dendritic anomalies to δ-catenin loss is problematic – semaphorin F, a regulator of axon pathfinding, is also localized to the same chromosomal region. However, our studies suggest that CDCS, and other forms of mental retardation could result from a progressive degeneration of neural connectivity rather than, or in addition to a deficit of circuit establishment.
Supporting this view, δ-catenin mutant mice display severe impairments in hippocampal dependent learning paradigms, including the Morris water maze and contextual fear conditioning (Israely et al., 2004). This impaired cognitive function likely has a cortical component as well. We observed progressive functional impairments in cortical visual information processing that precede the timing of the behavioral studies in Israely (2004). It is likely that the degraded sensory processing resulting from dendritic retraction contributes to the observed spatial learning deficit in the delta catenin mutant mice. In particular, the Morris water maze relies on the mouse using distal extramaze visuo-spatial cues to orient itself and locate the hidden platform. To control for visual ability, the visible platform task is used. One problem with this setup is that the intramaze visual cues that signal the platform location are closer than the extramaze visual cues used in the hidden version of the task. A reduction in visual acuity would impair navigation based on the more distant visual cues used in the hidden version of the task. Thus, the visual impairment we observe would accentuate the magnitude of the spatial the learning deficit.
These results have additional implications in the pathogenesis of Alzheimer's disease. δ-catenin was first identified through its interaction with the loop domain of Presenilin-1 (PS1), a region containing a concentration of Familial Alzheimer's Disease (FAD)-linked point mutations (Zhou et al., 1997). FAD is a disorder characterized in its earliest stages by cognitive decline, followed by the accumulation of extracellular amyloid plaques, neurofibrillary tangles, degeneration of neuronal architecture, and eventual neuronal loss (Selkoe, 2001). Notably, over-expression of wild type PS1 inhibits δ-catenin induced cellular branching and promotes δ-catenin processing and turnover (Kim et al., 2006), and FAD mutations in the PS1 loop domain, M146V and L286V, accelerate the degradation of δ-catenin (Kim et al., 2006). The progressive retraction of dendrites and loss of presumptive synapses in the δ-catenin mutant mice bears strong similarity to the pathogenesis of FAD. It is possible that δ-catenin degradation is enhanced in FAD, resulting in a progressive decline in protein levels. This, in turn, may contribute to the observed morphological and pathological changes that occur in the pathogenesis of FAD.
Taken together, this work indicates a novel role for δ-catenin in the maintenance of neural function by regulating the stability of dendritic and synaptic structures in vivo. Our data lends support to the importance of the maintenance of proper connectivity in cognitive function and provides insight into novel mechanisms that may underlie severe mental retardation and diseases of the nervous system.
Methods
Generation of ™-catenin mutant mice
™-catenin mutant mice, expressing a fusion of the N terminus of the protein and a GFP-stop cassette, were generated as described in Israely et. al., 2004. This fusion protein lacks the armadillo repeats and PDZ domains essential for localization and function. These mice were bred with Thy1-GFP mice (Feng et al., 2000). Heterozygous offspring were then crossed to produce ™-catenin mutant;Thy1-GFP +/- mice. ™-catenin +/+;Thy1-GFP littermates were used as controls.
2-photon in vivo imaging and analysis
Surgery, imaging, and analysis were performed as detailed in (Chow et al., 2009). Briefly, for acute imaging experiments the skull was thinned to a thickness of 20-50μm under isoflurane anesthesia. For chronic imaging experiments, the skull was removed and replaced with a 3mm diameter glass cover slip. Images of GFP-expressing pyramidal neurons in cortex were obtained using a custom-built 2-photon laser-scanning microscope. A Ti:Sapphire pulsed laser (SpectraPhysics Mai Tai BB) tuned to 910nm was used to excite GFP. Emitted photons were collected using an Olympus 40× 0.8NA IR water-dipping objective lens and were detected using a Hamamatsu R3896 photomultiplier tube. Control of the microscope and image collection was carried out using ScanImage software (Pologruto et al., 2003). Dendrites were traced in three dimensions from image stacks and analyzed using Neurolucida's Image Stack module.
Intrinsic signal optical imaging
Delta-catenin mutant mice and wild-type littermates were anesthetized with halothane (5% for induction, 1–2% for maintenance) and mounted in a stereotaxic frame. The eyes were covered with silicon oil. The scalp covering the right occipital cortex was resected and the skull covered with agar and a glass coverslip. The region of the skull over V1 was illuminated with 546 nm light to acquire an image of the surface vasculature. For ISI the preparation was illuminated with 700nm light and imaged with a tandem lens macroscope defocused 600μm into the brain. Images were acquired with a 12-bit CCD camera (Dalsa 1M30), frame grabber (Matrox Meteor II/Dig) and custom software. The visual stimulus was a white horizontal bar, 2 degrees in height, which drifted up or down at 0.125 Hz on a black background. An 8-min-long movie was taken for each direction (up/down), for a total of two movies. Acquisition was at 30 frames per second, and the 12-bit frames were binned in software four times temporally and 2×2 spatially, resulting in 16-bit image files. From movies of these 16-bit files, Fourier analysis of each pixel column generated maps of magnitude and phase at 0.125 Hz (Kalatsky et al., 2003). Scatter was computed on phase maps that were corrected for hemodynamic delay using time reversal (Kalatsky et al., 2003; Cang et al., 2005). The region of interest was defined using the magnitude map after it had been smoothed using a 5×5 Gaussian filter. The phase value for each pixel in the masked phase map was compared to the average of its neighbors in a 5×5 box, and the average difference for each map was calculated. In practice, this method is capable of detecting retinotopic maps even when response magnitude is weak (Smith et al., 2007). Maximum and mean magnitude values were taken from unfiltered magnitude maps.
Direct cortical stimulation and imaging
Mice were anesthetized with Halothane (1%-2%) in oxygen. Body temperature was maintained using a regulated heating pad (AD Instruments; ML295M) A 2mm diameter region of the skull overlying the primary visual cortex was thinned using a high-speed micro drill (Fine Science Tools, 18000-17). Drilling was stopped every 10 seconds to prevent thermal damage to the underlying cortex. A scalpel (Surgistar; 38-6900) was used to finish thinning the skull to a thickness of approximately 50μm. A small burr hole less than 200μm was made in the center of the thinned region through which a bipolar stimulating electrode (FHC; CBBEP50) was inserted at an angle of 45 degrees until the tip reached a depth of 250μm below the pial surface. Current was passed using an AMPI stimulus isolation unit triggered once every 8 seconds for 8 minutes by a Master-8. The intrinsic signal optical response to direct cortical stimulation was monitored using 630nm illumination. An 8-minute movie was generated at 7.5 frames per second. Each frame of the movie was 256×256 pixels at 16 bits. A Fourier-transform was performed on each pixel column in the movie to generate a map of cortical responsiveness, as detailed in (Smith and Trachtenberg, 2007). For each animal, 3 runs each at 50μA, 100μA, and 150μA were acquired and averaged to produce one average map for each mouse at each of the three stimulus intensities. Maps were analyzed using custom MatLab routines as described in (Faguet et al., 2009).
Statistics
Significance was computed using the Wilkoxon rank sum test. For comparisons of cumulative distributions, a Komogorov-Smirnov test was used. P values were corrected for multiple comparisons using the Bonferroni method.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arikkath J, Israely I, Tao Y, Mei L, Liu X, Reichardt LF. Erbin controls dendritic morphogenesis by regulating localization of delta-catenin. J Neurosci. 2008;28:7047–7056. doi: 10.1523/JNEUROSCI.0451-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DL, Tanaka H. N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci. 1998;18:6892–6904. doi: 10.1523/JNEUROSCI.18-17-06892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow DK, Groszer M, Pribadi M, Machniki M, Carmichael ST, Liu X, Trachtenberg JT. Laminar and compartmental regulation of dendritic growth in mature cortex. Nat Neurosci. 2009;12:116–118. doi: 10.1038/nn.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia LP, Yamamoto M, Zang K, Reichardt LF. p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron. 2006;51:43–56. doi: 10.1016/j.neuron.2006.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faguet J, Maranhao B, Smith SL, Trachtenberg JT. Ipsilateral eye cortical maps are uniquely sensitive to binocular plasticity. J Neurophysiol. 2009;101:855–861. doi: 10.1152/jn.90893.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannon AM, Colman DR. A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron. 1996;17:423–434. doi: 10.1016/s0896-6273(00)80175-0. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Grinvald A, Lieke E, Frostig RD, Gilbert CD, Wiesel TN. Functional architecture of cortex revealed by optical imaging of intrinsic signals. Nature. 1986;324:361–364. doi: 10.1038/324361a0. [DOI] [PubMed] [Google Scholar]
- Ho C, Zhou J, Medina M, Goto T, Jacobson M, Bhide PG, Kosik KS. delta-catenin is a nervous system-specific adherens junction protein which undergoes dynamic relocalization during development. J Comp Neurol. 2000;420:261–276. [PubMed] [Google Scholar]
- Holtmaat A, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Ide N, Hata Y, Deguchi M, Hirao K, Yao I, Takai Y. Interaction of S-SCAM with neural plakophilin-related Armadillo-repeat protein/delta-catenin. Biochem Biophys Res Commun. 1999;256:456–461. doi: 10.1006/bbrc.1999.0364. [DOI] [PubMed] [Google Scholar]
- Inoue A, Sanes JR. Lamina-specific connectivity in the brain: regulation by N-cadherin, neurotrophins, and glycoconjugates. Science. 1997;276:1428–1431. doi: 10.1126/science.276.5317.1428. [DOI] [PubMed] [Google Scholar]
- Israely I, Costa RM, Xie CW, Silva AJ, Kosik KS, Liu X. Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr Biol. 2004;14:1657–1663. doi: 10.1016/j.cub.2004.08.065. [DOI] [PubMed] [Google Scholar]
- Kalatsky VA, Stryker MP. New paradigm for optical imaging: temporally encoded maps of intrinsic signal. Neuron. 2003;38:529–545. doi: 10.1016/s0896-6273(03)00286-1. [DOI] [PubMed] [Google Scholar]
- Kim H, Han JR, Park J, Oh M, James SE, Chang S, Lu Q, Lee KY, Ki H, Song WJ, Kim K. Delta-catenin-induced dendritic morphogenesis. An essential role of p190RhoGEF interaction through Akt1-mediated phosphorylation. J Biol Chem. 2008;283:977–987. doi: 10.1074/jbc.M707158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Bareiss S, Kim KK, Tatum R, Han JR, Jin YH, Kim H, Lu Q, Kim K. Presenilin-1 inhibits delta-catenin-induced cellular branching and promotes delta-catenin processing and turnover. Biochem Biophys Res Commun. 2006;351:903–908. doi: 10.1016/j.bbrc.2006.10.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Sirota A, Chen Yh YH, Jones SB, Dudek R, Lanford GW, Thakore C, Lu Q. Dendrite-like process formation and cytoskeletal remodeling regulated by delta-catenin expression. Exp Cell Res. 2002;275:171–184. doi: 10.1006/excr.2002.5503. [DOI] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Donahue CP, Israely I, Liu X, Ochiishi T. Delta-catenin at the synaptic-adherens junction. Trends Cell Biol. 2005;15:172–178. doi: 10.1016/j.tcb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Liauw J, Nguyen V, Huang J, St George-Hyslop P, Rozmahel R. Differential display analysis of presenilin 1-deficient mouse brains. Brain Res Mol Brain Res. 2002;109:56–62. doi: 10.1016/s0169-328x(02)00491-6. [DOI] [PubMed] [Google Scholar]
- Martinez MC, Ochiishi T, Majewski M, Kosik KS. Dual regulation of neuronal morphogenesis by a delta-catenin-cortactin complex and Rho. J Cell Biol. 2003;162:99–111. doi: 10.1083/jcb.200211025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina M, Marinescu RC, Overhauser J, Kosik KS. Hemizygosity of delta-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome. Genomics. 2000;63:157–164. doi: 10.1006/geno.1999.6090. [DOI] [PubMed] [Google Scholar]
- Paffenholz R, Franke WW. Identification and localization of a neurally expressed member of the plakoglobin/armadillo multigene family. Differentiation. 1997;61:293–304. doi: 10.1046/j.1432-0436.1997.6150293.x. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JB, Restituito S, Lu W, Lee-Edwards L, Khatri L, Ziff EB. Synaptic anchorage of AMPA receptors by cadherins through neural plakophilin-related arm protein AMPA receptor-binding protein complexes. J Neurosci. 2007;27:8505–8516. doi: 10.1523/JNEUROSCI.1395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SL, Trachtenberg JT. Experience-dependent binocular competition in the visual cortex begins at eye opening. Nat Neurosci. 2007;10:370–375. doi: 10.1038/nn1844. [DOI] [PubMed] [Google Scholar]
- Takeichi M, Abe K. Synaptic contact dynamics controlled by cadherin and catenins. Trends Cell Biol. 2005;15:216–221. doi: 10.1016/j.tcb.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Tanahashi H, Tabira T. Isolation of human delta-catenin and its binding specificity with presenilin 1. Neuroreport. 1999;10:563–568. doi: 10.1097/00001756-199902250-00022. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS. Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport. 1997;8:1489–1494. doi: 10.1097/00001756-199704140-00033. [DOI] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K. Rapid functional maturation of nascent dendritic spines. Neuron. 2009;61:247–258. doi: 10.1016/j.neuron.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]