

Abstract
Erythropoietin (Epo) has been used for many years in neonates for the treatment of anemia of prematurity. Epo has also been proposed for treatment of neonatal brain injury, as mounting evidence suggests neuroprotective properties for Epo. However, Epo’s neuroprotective mechanism of action is poorly understood. In this study we hypothesized that Epo may confer neuroprotection by enhancing cellular redox defense brought about by cellular glutathione (GSH). This was examined in cultures of differentiated cortical neural stem cells and using the B104 cell line as model systems. Our data shows that Epo causes a time- and dose-dependent increase in expression and activity of system Xc−, the transporter responsible for uptake of cystine for the production of glutathione. Cystine uptake increases 3–5 fold in differentiated neural stem cells and B104 cells treated with Epo. Exposure of cells to 100μM kainate suppressed cellular GSH and caused excitotoxicity, but GSH levels and cell viability was completely restored by Epo in the continued presence of kainate. This rescue effect of Epo vanished if system Xc− was inhibited pharmacologically using S4-CPG in the presence of Epo leading to marked cell death of B104 cells and cultured mouse cortical neural stem cells. This could also be achieved using xCT siRNA to decrease xCT expression. This data suggests that system Xc− activity and protein expression are positively regulated by Epo directly explaining its neuroprotective effect.
Keywords: Erythropoietin, Cystine Glutamate Exchanger, xCT, Neuroprotection
Introduction
Erythropoietin (Epo) is a glycoprotein hormone whose major function is to regulate erythropoiesis (Krantz, 1995). It is a member of the cytokine superfamily which also includes granulocyte-macrophage colony-stimulating factor (GM-CSF), ciliary neurotrophic factor (CNTF), interleukin 3 and 6, prolactin, cardiotrophin 1 (CT1), leukemia inhibitory factor (LIF) and thrombopoietin (Seko et al., 1998; Uppenkamp et al., 1998) in addition to its’ role in erythropoiesis, it appears to have an important role in the central nervous system.
Epo’s essential role in development became apparent in knock-out studies since animals with the targeted deletion of the Epo receptor died prematurely at embryonic day 13.5 (E13.5) from severe anemia (Yu et al., 2002). Although these and other studies demonstrate an absolute requirement for Epo signaling in hematopoiesis, it appears to exert a secondary neuroprotective role during brain development that is less well understood (Liu et al., 2005). Epo prevents neural apoptosis in the embryonic brain and mice lacking EpoR show signs of severe neuronal apoptosis. In EpoR knockout animals, neural progenitor cells were markedly reduced and culturing neurons with a functional receptor showed a marked increase in cell viability compared to knockouts.
Epo-induced neuroprotection has been associated with processes such as anti-inflammation, anti-apoptosis and neuroregeneration (Sola et al., 2005). These processes require multiple genes and signaling pathways. The pleiotropic effects of Epo are exerted through the EpoR by three distinct pathways. The common link in these three pathways is the phosphorylation of Janus Kinase (JAK) 2. Afterwards, there is activation of signal transducer and activator of transcription (STAT) 5, the phosphoinositide 3-kinase (PI3K)-serine-threonine kinase AKT and nuclear factor kappa B (NFκB) pathway (Sola et al., 2005). Currently, it is not known how the cell determines which pathway it utilizes to exert the effect of Epo. Speculatively, there may be interplay between the Epo receptor and other receptors that ultimately dictates the cumulative effect. Cell type may also play a major role in this process.
Epo is produced in the brain under conditions of nitrosative and oxidative stress and the Epo gene is upregulated under hypoxic conditions (Buemi et al., 2002). Oxygen and glucose deprivation (OGD) of neurons causes cell death in vitro. The neuronal death is attenuated by adding supernatant from astrocytes exposed to the same conditions. The effect was blocked by soluble EpoR demonstrating that Epo acts as a paracrine hormone (Ruscher et al., 2002). Excitotoxic conditions, for example exposure of cells to N-methyl-d-aspartate (NMDA) or nitric oxide (NO), which lead to the formation of free radicals which are attenuated in the presence of Epo (Digicaylioglu and Lipton, 2001) (Yamamoto et al., 2004). Calapai et al demonstrated that Epo decreases NO synthase activity in human vascular endothelial cells and protects neurons against ischemia-induced cell death (Calapai et al., 2000). Downstream effectors of Epo are poorly understood. However, one gene that is upregulated after Epo exposure is glutathione peroxidase, an enzyme involved in regenerating free glutathione (Genc et al., 2002).
In this study we set out to test the hypothesis that glutathione levels are increased in the presence of Epo due to an upregulation of the cystine glutamate exchanger, system Xc−, which is responsible for the cellular import of cystine for the synthesis of glutathione. System Xc− is a dimeric amino-acid transporter which couples to equimolar exchange of cystine with the release of glutamate. The transporter exists as a dimer composed of a regulatory subunit (4F2hc-CD98) and a catalytic subunit (xCT). The latter belongs to a family of amino acid transporters which has 12 transmembrane domain (Sato et al., 2000; Sato et al., 2002). The heavy chain 4F2hc is responsible for membrane translocation (Wells et al., 1992). Following uptake, cystine is reduced to cysteine, which is conjugated with glutamate and glycine, to form glutathione (L-γ-glutamyl-L-cysteinylglycine=GSH). GSH is a ubiquitous tripeptide involved in the regulation of the cells redox state, and protects cells from reactive oxygen (ROS) and nitrogen species. Potential reactive oxygen species include nitric oxide, hydrogen peroxide, superoxide anion, and hydroxyl radicals, many of which are generated in hypoxia-ischemia, excitotoxicity and infection. Thus, it is plausible that dysfunction or developmental expression of system Xc− could be responsible for decrease ability of precursor cells to defend against reactive oxygen species.
Results
To examine the potential role of Epo in protecting neural cells, we used two model systems from mouse and human that have been previously demonstrated to express Epo receptors (EpoR) were the mouse neural stem cells (NSCs) (Yu et al., 2002) and B104 human neuroblastoma cells (Ribatti et al., 2007) (Sartelet et al., 2007). We validated that in our hands, both express the EpoR at the mRNA and protein level (Fig. 1A & B) along with expression of xCT, the catalytic subunit of system Xc (Fig. 1A and 1B). Importantly, xCT and EpoR proteins colocalize as demonstrated by immunohistochemistry in Figs. 1C–H for both cell systems.
Figure 1.
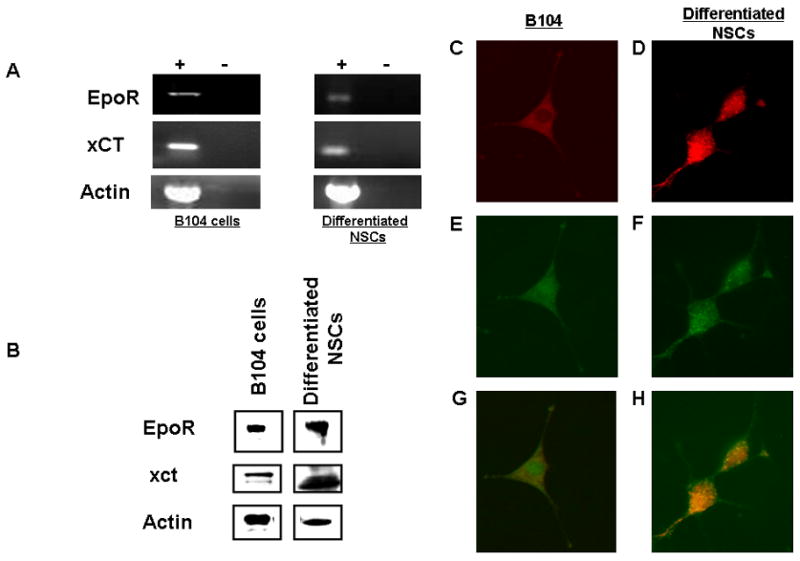
EpoR and xCT expression in B104 cells. (a.) RT-PCR of 2μg of total RNA collected from B104 cells (b.) Immunoblot of EpoR, xCT and Actin. B104 cells (c.) xCT (TRITC), (e.) EpoR (FITC), (g.) combined. Differentiated neural stem cells d.) xCT (TRITC), (f.) EpoR (FITC) and (h.) combined.
Glutamate toxicity data had not been established in our cell line so it was established in Figure 2A (NSCs) and Figure 2B (B104s). Both cell lines had an increase in apoptosis with increasing concentrations of glutamate. To demonstrate that Epo indeed confers neuroprotection, we challenged differentiated NSCs with 3 mM glutamate yielding a significant (p≤ 0.01), >75% loss of viable cells (Fig. 3A). Cultures that received cotreatment with 10 ng/ml Epo and glutamate challenge were essentially unaffected by 3 mM Glu showing a small, 10% reduction in cell viability. However, if Epo treated NSCs were also treated with 250 μM of the system Xc− inhibitor S-4CPG, 72% of cells died (p ≤ 0.01). These experiments suggest that Epo is protecting NSCs from Glu toxicity in a manner that engages system Xc activity. Since the latter is responsible for the cellular import of cystine to generate glutathione (GSH) we sought to examine the effect of these treatments on cystine uptake directly. Indeed, as illustrated in Fig. 3B, cystine uptake into differentiated NSCs was elevated 5-fold in the presence of Epo indicating an upregulation of system Xc expression or activity. Similar data was obtained in B104 cells (Figure 3C) with a 75% reduction in cell viability (p≤ 0.01) and > 70 % (p≤ 0.01) decrease in cell viability when exposed to S4-CPG. In Figure 3D, B104 cells had a 3-fold (p≤ 0.01) increase in cystine uptake in the presence of Epo.
Figure 2.
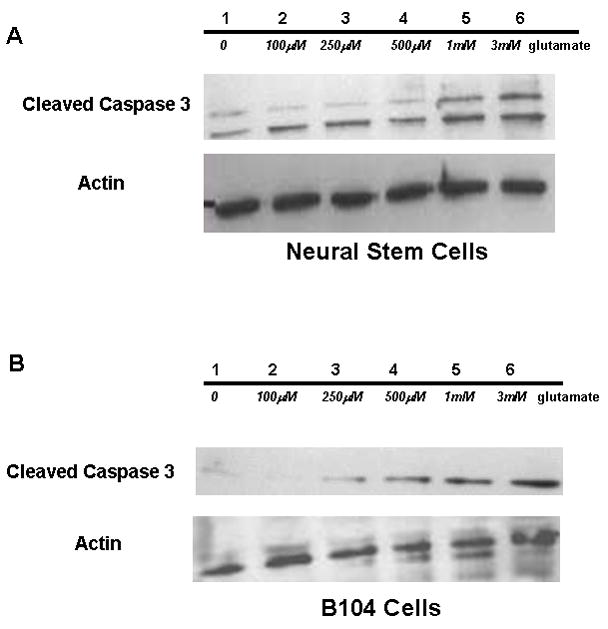
Glutamate Toxicity in NSCs and B104. (a.) Immunoblot of NSC protein extract probed for cleaved caspase 3 and actin at increasing concentrations of glutamate (1-0 glu, 2–100 μM glu, 3–250 μM glu, 4–500 μM glu, 5-1mM glu, 6-3mM glu). (b.) Immunoblot of B104 cell extract probed for cleaved caspase 3 and actin at increasing concentrations of glutamate (1-0 glu, 2–100 μM glu, 3–250 μM glu, 4–500 μM glu, 5-1mM glu, 6-3mM glu).
Figure 3.
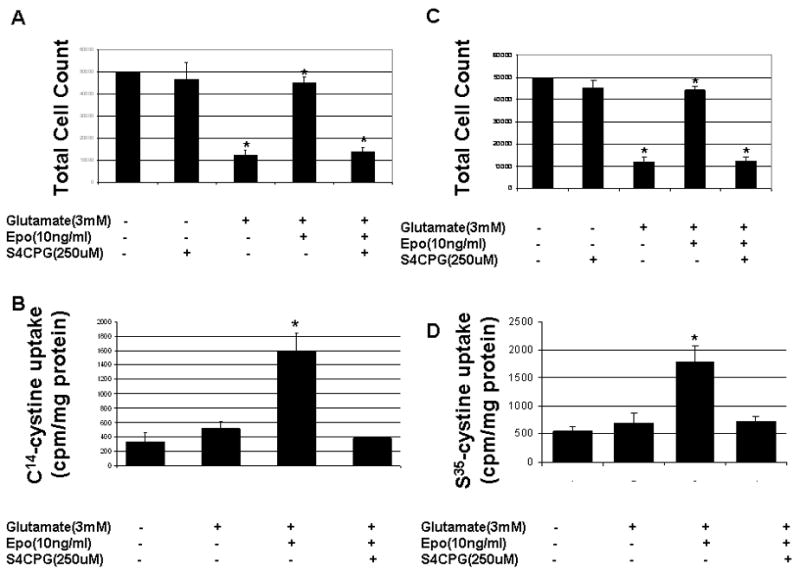
Cell viability and cystine uptake in differentiated neural stem cells. (a.) Total cell count in control, 3mM glutamate, 3mM glutamate + Epo (10ng/ml), 3mM glutamate + Epo (10ng/ml), 3mM glutamate + Epo (10ng/ml) + S4-carboxyphenylglycine (250μM). (b.) Cystine uptake in differentiated NSCs –control, 3mM glutamate and 3mM glutamate + Epo (10ng/ml). Cell viability and cystine uptake in B104 cells (c.) Total cell count in control, 3mM glutamate, 3mM glutamate + Epo (10ng/ml), 3mM glutamate + Epo (10ng/ml), 3mM glutamate + Epo (10ng/ml) + S4-carboxyphenylglycine (250μM). (d.) Cystine uptake in differentiated B104 cells –control, 3mM glutamate and 3mM glutamate + Epo (10ng/ml). * p ≤ 0.05 using the paired student t-test.
To examine whether the increase cystine uptake resulted from an increase in system Xc− expression, we next studied the time-dependence of xCT protein levels following exposure of cells to 10ng/ml Epo for 4–24h along with actin used as a loading control. Similar blots were obtained from 3 different experiments in both cell lines and in each case the protein concentration was normalized to actin levels allowing evaluation of changes in protein concentration. NSCs treated with 10ng/ml of Epo (Figure 4A) showed a statistical increase in protein level at 12 and 24 hours, >1.2 fold (p≤ 0.05) increase and >1.6 fold (p≤0.01) increase. The resulting data, shown in Fig. 4B show a significant increase in xCT expression as early as 4 hours after addition of Epo. The xCT/actin ratios in Figure 4B, expressed as fold increase were significant at 4, 8, 12 and 24 hour, respectively. The concentration of Epo chosen (10ng/ml) was because it was closest to the half-maximal effect of B104 cells. B104 cells would be used for all experiments including the siRNA experiments.
Figure 4.
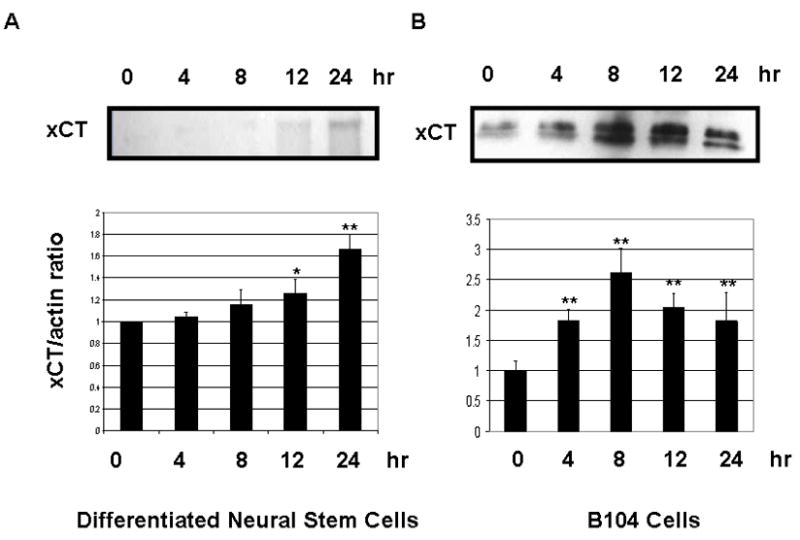
Erythropoietin Time Course. (a.) Representative Western Blot of system Xc− and actin in B104 cells treated with 10ng/ml of Erythropoietin. (b.) Relative xCT concentration (normalized to actin) at 0, 4, 8, 12 and 24hr time points. 25μg of protein was added to each well. *p≤0.05 and **p≤0.01 using the paired student t-test.
To examine whether Epo showed a dose-dependence in its effect on system Xc− expression we administered Epo to NSCs and B104 cells at increasing concentrations, using 0, 5, 10, 25 and 50 ng/ml Epo respectively. In Figure 5A, a representative Western blot shows the corresponding increases in xCT expression over 24 hours. Normalizing xCT expression to actin expression (xCT/actin) shows a statistically significant increase in xCT expression at all concentrations tested. In NSCs, fold increase over control was statistically significant at 5, 10, 25 and 50 ng/ml but only statistically significant at 25 and 50ng/ml in B104 cells.
Figure 5.
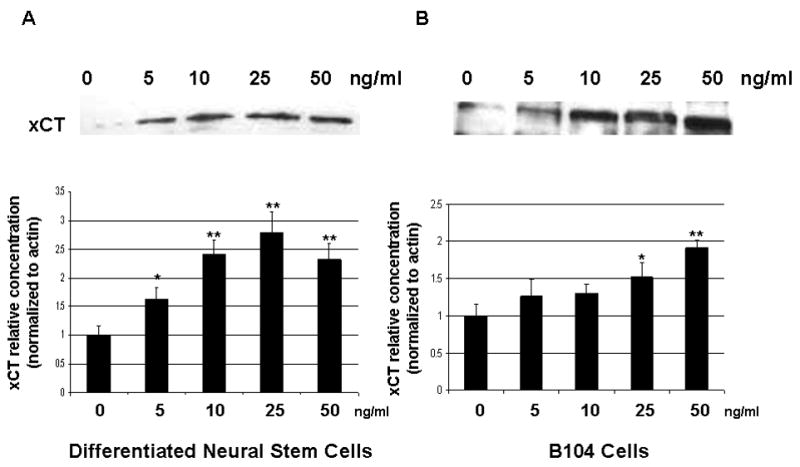
Epo Dose Response. (a.) Representative Western blot of system Xc− in B104 cells with 0, 5, 10, 25, 50 ng/ml Epo. (b.) Relative xCT concentration (normalized to actin) at same concentrations listed in (a.). * p ≤ 0.05 using the paired student t-test.
Since system Xc− provides the cystine as substrate for glutathione production synthesis, these data suggest that glutathione levels may actually increase following Epo exposure. To examine this directly, we measured total GSH levels in cells treated with 100μM kainate acid in the presence or absence of Epo (Fig. 6). Kainate was used in these experiments to negate the role of glutamate in the formation of glutathione. Total glutathione levels were normalized to protein concentrations and expressed as fluorescence/mg protein. Kainate alone reduced GSH levels by 74% but GSH levels were restored to control values with 10ng/ml Epo and to 67% of control with 5ng/ml Epo supplementation.
Figure 6.
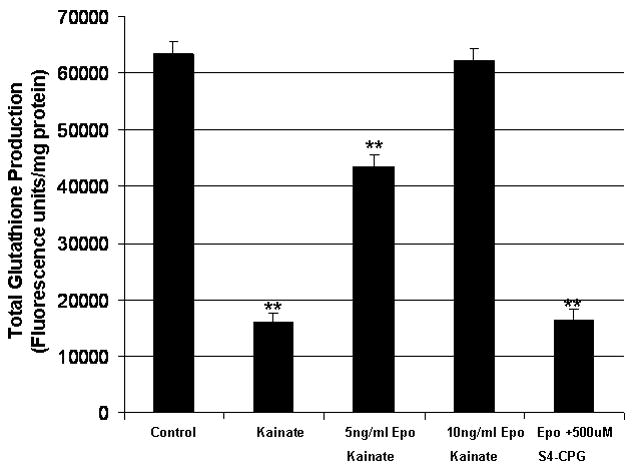
Epo effect on total glutathione. B104 cells treated with kainate in the presence (5ng/ml and 10ng/ml) or absence of Epo. Glutathione is expressed as fluorescence/mg protein. *p ≤ 0.05 using the paired student t-test.
These data suggest that in neural stem cells Epo protects through the regulation of system Xc−. As all the above data point to system Xc− being an important downstream target of Epo and the likely candidate that confers cellular protection in the presence of Epo, we employed the use of siRNA to suppress system Xc− expression in B104 cells. B104 cells were transfected with xCT siRNA at 5 ng, 10 ng and 20ng/ml and samples compared to a control and scrambled siRNA. Cells were plated at 500,000 per 6 well plates. In cells transfected with scrambled siRNA there was a 18% decrease in cell number. xCT siRNA transfected cells showed a 38%, 66% and 74% decrease in cell number after 24 hours of transfection for 10 ng, 25 ng and 50 ng/ml xCT siRNA, p≤ 0.001 (Figure 7A). The relative decrease in cell number for each sample was 1.2, 1.67, 2.88 and 3.84 fold for scrambled, 5 ng/ml, 10ng/ml and 20ng/ml xCT siRNA. All cells were treated with 10ng/ml of Epo. A representative Western blot is shown to demonstrate the inhibition of xCT expression (Figure 7B).
Figure 7.
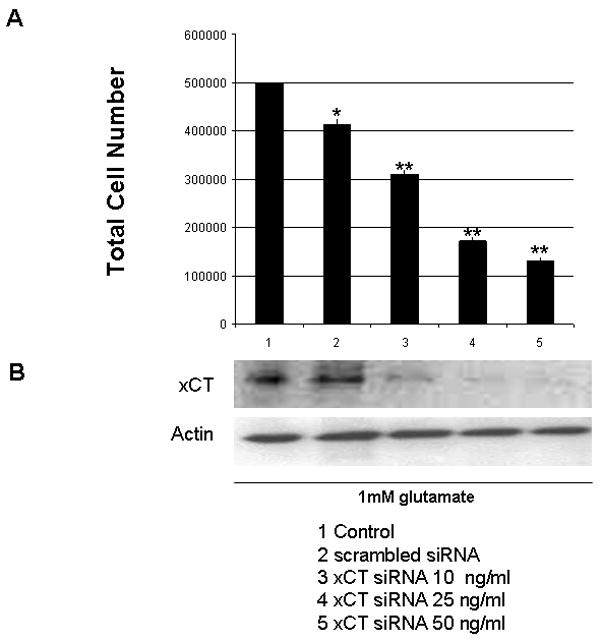
xCT siRNA effect on cell viability. Lane 1–5 include (1) Control, (2) scrambled siRNA 50ng/ml, (3) xCT siRNA 10ng/ml, (4) xCT siRNA 25ng/ml and (5) xCT siRNA 50ng/ml. (a) Total Cell Count. *p<0.05 and **p<0.01. (b) xCT and actin immunoblot, 25μg protein loaded to each well.
Discussion
Epo has been shown to be protective in many systems including liver (Joyeux-Faure, 2007), pancreatic islet cells (Fenjves et al., 2004), intestine (Guneli et al., 2007), pheochromocyctoma cells (Wu et al., 2007), vascular injury (Chong et al., 2003) and heart failure (Latini et al., 2008). In these systems and others Epo is known to be cytoprotective and its mechanism leads to decreased oxidative stress and decrease lipid peroxidation (Szczepanski et al., 2006). Mechanisms of Epo-induced cytoprotection involve multiple pathways including endothelial nitric oxide synthase (d’Uscio et al., 2007) and the induction of heme oxygenase 1 (Katavetin et al., 2007).
In brain, multiple pathways are involved in Epo-induced neuroprotection including the modulation of antiapoptotic factors (Chong et al., 2007) such as caspase 1, 3, and caspase 8 (Ghezzi and Brines, 2004). Other mechanisms involve reduced lipid peroxidation and increased glutathione peroxidase (Katavetin et al., 2007) (Genc et al., 2002). These studies link glutathione modulation as a potential pathway of protection but no studies have examined the rate limiting step of glutathione synthesis as potential regulatory target of Epo.
This study was motivated by prior suggestions that Epo, in addition to its pleiotropic effects on a variety of cell types in the body may also confer some neuroprotection (Hasselblatt et al., 2006). While previous studies could not delineate the sole mechanism by which Epo confers neuroprotective effects, our findings propose that it may be the result of a positive regulation of the cystine glutamate exchanger, system Xc−. Its upregulation causes an increase in cystine uptake and much enhanced production of the cellular antioxidant GSH. Addition of Epo in vitro showed a dramatic increase in xCT protein expression in a time dependent manner and we found an increase in xCT after addition of Epo in vitro at all time points studied. Importantly, Epo effects occurred relatively fast. Of course one may argue that in order for Epo to have a protective effect it must be quick to upregulate expression of genes that could be protective. We did find a 2-fold increase in system Xc− expression as early as 4 hours after the insult (Figure 4B) in B104 cells and this increase was maintained for at least 24 hours. The time course is clinically relevant. Epo, in these experiments, was administered concomitantly with glutamate. Clinically, this is still a useful strategy. There are a fair number of neonates who suffer episodes such as severe hypoxemia which could benefit from an agent like Epo with neuroprotection as early as 4 hours.
In earlier work cystine deprivation was shown to be toxic to oligodendroglial precursor cells (Back et al., 1998) which suggests system Xc− activity may be vital for neural precursor cell survival and cellular protection. Other studies also confirm that inhibition of cystine uptake is neurotoxic by a receptor independent pathway (Rosin et al., 2004) which includes system Xc−. Glia which overexpressed Nrf2, a regulator of system Xc−, protected neurons from oxidative stress and showed that even at a density of < 1% of these glia in mixed culture could protect neurons from oxidative stress (Shih et al., 2003). Conversely, in microglia, increased glutamate release was seen by system Xc− and its inhibition prevented neurotoxicity caused by microglia. Taken together, this may suggest that system Xc− under certain circumstances may be neuroprotective and may have a cell specific role in glutamate metabolism.
We used B104 cells as a convenient model system for study since EpoR is highly expressed in these cells and being a cell line, biochemical studies are readily feasible. However, the major findings of our study were confirmed in differentiated neural stem cells, the cell type most likely at risk of injury in premature infants who routinely receive Epo. An important question is the developmental expression of system Xc− and how it may alter Epo’s ability to confer neuroprotection. The use of the mouse cortical neural stem cells clearly suggests that the immature brain will benefit from Epo, yet it remains to be seen whether mature neurons, harmed for example after a stroke, would also be positively influenced by Epo. Such studies are underway.
Inhibition of xCT can be accomplished pharmacologically using for example S4-CPG at a 250μM concentration. When xCT was inhibited we found a mild decrease in cell viability in the absence of an excitotoxic insult. xCT has been reported to be neuroprotective when overexpressed (Shih et al., 2006) and its inhibition causes apoptosis in tumors (Chung et al., 2005). The fact that Epo looses any neuroprotective effects when xCT function is inhibited pharmacologically or using siRNA suggest very strongly that the observed upregulation of system Xc− expression by Epo is a necessary step in the observed neuroprotection. The scrambled siRNA used in the experiment showed a 17% decrease in viability but was used at the highest concentration of 50 ng/ml. This concentration of siRNA could have a toxic effect on cells. The decrease in total glutathione when S4-CPG is administered is supporting evidence that there is a substantial contribution to glutathione biosynthesis by system Xc−. Indeed, the knockdown experiments provide direct evidence for the molecular regulation of Epo-induced neuroprotection by xCT.
Taken together, data presented suggest that Epo is a positive regulator of system Xc−, which in turn confers neuroprotection. A logical next step is to examine this activity across multiple neural cells and if possible use in vivo model systems that examine excitotoxic injury in neonatal brain. If successful, these may provide strong rationale for the clinical use of Epo in prenatal cerebral insults. Moreover, other synergistic drugs may be identified that could further enhance the positive transcriptional effects on system Xc−.
Materials and Methods
All equipment used was from BioRad and chemicals from Sigma Chemical Company (St. Louis, MO) unless stated otherwise. RT-PCR primers were ordered from IDTDNA.
B104 Neuroblastoma Cells and Cortical Neural Stem Cells
B104 cells were ordered from ATCC. Total cell count was determined using a standard hemocytometer and Trypan Blue in all experiments. B104 media consists of 0.75g sodium bicarbonate, 10% newborn calf serum placed in Dulbecco’s Modified Essential Media (DMEM). B104 cells were grown to 80% confluence and then split 1:6 every 3 days and plated in T25cm flasks. Epo treatment was given at the time of kainate/glutamate administration with equal cell counts. Treatment for all experiments was 24 hours.
Neural stem cells were ordered from Chemicon as Cryopreserved Mouse Cortical Neural Stem Cells (Catalog Number SCR029). Polyornithine-Laminin coated plates were made using concentrations of polyornithine (10mg/ml) and laminin (5μg/ml). Plates were incubated with polyornithine overnight and then washed and laminin solution added and allowed to incubate overnight then used directly or stored at −80 C. Protocol for culturing neural stem cells was followed per protocol. In brief, cells were grown in Neural Stem Cell Expansion Medium (Chemicon Neural Stem Cell Medium, 20ng/ml Fibroblast Growth Factor 2 (FGF-2), 20ng/ml Epidermal Growth Factor (EGF) and heparin 2μg/ml) and would be passaged every 3–5 days. Cells were allowed to grow to 80% confluence then split 1:3 in T25cm flasks. Differentiation was done after passage 3. Differentiation was induced by withdrawing EGF, FGF-2 and heparin.
Cystine Uptake
Sodium-independent cystine uptake was done using conditions described in Chung et al., 2005. In brief, collect cells and plate 1×106 in a six well plate, allowing the cells to adhere for two hours. Cells were treated with experimental conditions and incubated overnight. After centrifugation, cells were placed in cystine uptake buffer (CUB) (122mM choline-chloride, 1.8mM KCl, 1.3mM CaCl2, 1.2mM potassium phosphate, 25mM Triethyammonium bicarbonate, 10mM glucose, 0.4mM magnesium sulfate adjusted to pH 7.4) and warmed to 37°C. 500μl of CUB is added to the pellet. 100μl of sample is used for protein determination. S35/C14-Cystine is added to the remaining 400μl volume of CUB and incubated for 10 minutes. Equal volume of acid (0.3N HCl) and base (0.3N NaOH) are added for 10 minute with a wash between steps. The samples are allowed to equilibrate for 10 minutes. Radioactivity was detected using a S35/C14 detection program in a scintillation counter.
Protein Concentration Assay
The Bio-Rad DC Protein Assay was used in order to determine the protein concentrations of the samples. The absorbencies were read on a spectrophotometer at 750nm.
Western Blot
Cells were triturated and pelleted then lysed with RIPA buffer. 50 μg of protein was collected and ran on a Bio-Rad Ready Gel Tris 10% HCL. Samples were run at 120 V for 45 minutes on ice. Gel was transferred to nitrocellulose using the Trans Blot Semi-Dry apparatus for 15 minutes at 15 V. Blots were washed three times with TBS-0.1% Tween (TBST). Membranes were blocked for 30 minutes with 3% milk solution. Primary antibodies were used at 1:1000 dilutions in 3% milk. Primary antibodies were allowed to incubate overnight on a rotator. Primary antibodies used were Cleaved Caspase 3 (Asp175) (Cell Signaling), Actin (MAB1501-Chemicon), System Xc (Novus Biological). Secondary antibody was used at a 1:1000 dilution for 1 hour on a rotator. After washing, blots were developed using the Pierce Super Signal detection kit and developed on film with the Kodak developer.
siRNA Production
Primers designed from the Block-It siRNA program (Invitrogen) for xCT siRNA (S1–5′-GGATCCTAATACGACTCACTATAGAT-3′, S2–5′-AAGTTATTCTGTCTGATAATCTATAGT-3′, S3–5′-GGATCCTAATACGACTCACTATAGTT-3′, S4–5′-AAGATTATCAGACAGAATAACTATAG-3′) were made using the Ribomax T7 RNA Polymerase System. Cells were then transfected with siRNA using the CodeBreaker siRNA Transfection Reagent protocol. Cells were grown for 24–48 hours then cells lysed for Western Blot analysis after total cell count. RNA was normalized using the Quant-it Ribogreen RNA quantification kit.
Reverse Transcriptase Polymerase Chain Reaction
Total RNA was prepared using the Trizol reagent and protocol. 1μg of total RNA was used for each experiment. The Qiagen One Step RT-PCR kit was used. Actin, EpoR and xCT primers ordered from IDTDNA with the following sequence: Actin; forward 5′-ATCATGTTTGAGACCTTCAACAC-3′, reverse 5′-TCTGCGCAAGTTAGGTTTTGTC-3′; EpoR, forward 5′-GTCTGACTTGGACTCAAAGC-3′, reverse 5′-CTCAGTCTGGGACCTTCTGC-3′ and xCT, forward 5′ GCAGTCGCAGGACTGATTTA-3′, reverse 5′-GAGAGGCACCTTGAAAGGAC-3′. The conditions were optimized and ran on a Thermacycler. 100bp EZ load molecular ruler was used for size determination.
Acknowledgments
Funding for this project was provided by the Robert Wood Johnson Harold Amos Medical Faculty Development Award, the CHRC K12 NICHD Training Grant, and by NIH R01 NS052634-05 (HS).
Abbreviations
- Epo
Erythropoietin
- CUB
cystine uptake buffer
- GM-CSF
Granulocyte-monocyte colony stimulating factor
- CNTF
ciliary neurotrophic factor
- LIF
leukemia inhibitory factor
- NO
nitric oxide
- NMDA
N-methyl D-aspartate
- System Xc
cystine glutamate exchanger
- xCT
catalytic subunit of System Xc−
- GSH
glutathione
- PFA
paraformaldehyde
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Back SA, et al. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–53. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buemi M, et al. Erythropoietin and the brain: from neurodevelopment to neuroprotection. Clin Sci (Lond) 2002;103:275–82. doi: 10.1042/cs1030275. [DOI] [PubMed] [Google Scholar]
- Calapai G, et al. Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur J Pharmacol. 2000;401:349–56. doi: 10.1016/s0014-2999(00)00466-0. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, et al. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res. 2003;71:659–69. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, et al. Vascular injury during elevated glucose can be mitigated by erythropoietin and Wnt signaling. Curr Neurovasc Res. 2007;4:194–204. doi: 10.2174/156720207781387150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–10. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Uscio LV, et al. Essential role of endothelial nitric oxide synthase in vascular effects of erythropoietin. Hypertension. 2007;49:1142–8. doi: 10.1161/HYPERTENSIONAHA.106.085704. [DOI] [PubMed] [Google Scholar]
- Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–7. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- Fenjves ES, et al. Adenoviral gene transfer of erythropoietin confers cytoprotection to isolated pancreatic islets. Transplantation. 2004;77:13–8. doi: 10.1097/01.TP.0000110422.27977.26. [DOI] [PubMed] [Google Scholar]
- Genc S, et al. Erythropoietin restores glutathione peroxidase activity in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in C57BL mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci Lett. 2002;321:73–6. doi: 10.1016/s0304-3940(02)00041-1. [DOI] [PubMed] [Google Scholar]
- Ghezzi P, Brines M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ. 2004;11(Suppl 1):S37–44. doi: 10.1038/sj.cdd.4401450. [DOI] [PubMed] [Google Scholar]
- Guneli E, et al. Erythropoietin protects the intestine against ischemia/reperfusion injury in rats. Mol Med. 2007;13:509–17. doi: 10.2119/2007-00032.Guneli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselblatt M, et al. The brain erythropoietin system and its potential for therapeutic exploitation in brain disease. J Neurosurg Anesthesiol. 2006;18:132–8. doi: 10.1097/00008506-200604000-00007. [DOI] [PubMed] [Google Scholar]
- Joyeux-Faure M. Cellular protection by erythropoietin: new therapeutic implications? J Pharmacol Exp Ther. 2007;323:759–62. doi: 10.1124/jpet.107.127357. [DOI] [PubMed] [Google Scholar]
- Katavetin P, et al. Antioxidative effects of erythropoietin. Kidney Int Suppl. 2007:S10–5. doi: 10.1038/sj.ki.5002482. [DOI] [PubMed] [Google Scholar]
- Krantz SB. Erythropoietin and the anaemia of chronic disease. Nephrol Dial Transplant. 1995;10(Suppl 2):10–7. doi: 10.1093/ndt/10.supp2.10. [DOI] [PubMed] [Google Scholar]
- Latini R, et al. Do non-hemopoietic effects of erythropoietin play a beneficial role in heart failure? Heart Fail Rev. 2008;13:415–23. doi: 10.1007/s10741-008-9084-z. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke. 2005;36:1264–9. doi: 10.1161/01.STR.0000166180.91042.02. [DOI] [PubMed] [Google Scholar]
- Ribatti D, et al. Erythropoietin/erythropoietin receptor system is involved in angiogenesis in human neuroblastoma. Histopathology. 2007;50:636–41. doi: 10.1111/j.1365-2559.2007.02653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin C, et al. Excitatory amino acid induced oligodendrocyte cell death in vitro: receptor-dependent and -independent mechanisms. J Neurochem. 2004;90:1173–85. doi: 10.1111/j.1471-4159.2004.02584.x. [DOI] [PubMed] [Google Scholar]
- Ruscher K, et al. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci. 2002;22:10291–301. doi: 10.1523/JNEUROSCI.22-23-10291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartelet H, et al. Expression of erythropoietin and its receptor in neuroblastomas. Cancer. 2007;110:1096–106. doi: 10.1002/cncr.22879. [DOI] [PubMed] [Google Scholar]
- Sato H, et al. Molecular cloning and expression of human xCT, the light chain of amino acid transport system xc. Antioxid Redox Signal. 2000;2:665–71. doi: 10.1089/ars.2000.2.4-665. [DOI] [PubMed] [Google Scholar]
- Sato H, et al. Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J Neurosci. 2002;22:8028–33. doi: 10.1523/JNEUROSCI.22-18-08028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seko Y, et al. Vascular endothelial growth factor (VEGF) activates Raf-1, mitogen-activated protein (MAP) kinases, and S6 kinase (p90rsk) in cultured rat cardiac myocytes. J Cell Physiol. 1998;175:239–46. doi: 10.1002/(SICI)1097-4652(199806)175:3<239::AID-JCP1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Shih AY, et al. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–23. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola A, et al. Erythropoietin after focal cerebral ischemia activates the Janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatr Res. 2005;57:481–7. doi: 10.1203/01.PDR.0000155760.88664.06. [DOI] [PubMed] [Google Scholar]
- Szczepanski M, et al. Erythropoietin prevents oxidative stress and lipid peroxidation process in human umbilical vein endothelial cells induced by tumor necrosis factor alpha. Pol Merkur Lekarski. 2006;21:534–9. [PubMed] [Google Scholar]
- Uppenkamp M, et al. Thrombopoietin serum concentration in patients with reactive and myeloproliferative thrombocytosis. Ann Hematol. 1998;77:217–23. doi: 10.1007/s002770050446. [DOI] [PubMed] [Google Scholar]
- Wells RG, et al. The 4F2 antigen heavy chain induces uptake of neutral and dibasic amino acids in Xenopus oocytes. J Biol Chem. 1992;267:15285–8. [PubMed] [Google Scholar]
- Wu Y, et al. Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis via the Akt/GSK-3beta/caspase-3 mediated signaling pathway. Apoptosis. 2007;12:1365–75. doi: 10.1007/s10495-007-0065-9. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, et al. Effect of erythropoietin on nitric oxide production in the rat hippocampus using in vivo brain microdialysis. Neuroscience. 2004;128:163–8. doi: 10.1016/j.neuroscience.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Yu X, et al. Erythropoietin receptor signalling is required for normal brain development. Development. 2002;129:505–16. doi: 10.1242/dev.129.2.505. [DOI] [PubMed] [Google Scholar]