

Abstract
Alternative splicing plays an important role in generating proteome diversity. The polypyrimidine tract–binding protein (PTB) is a key alternative splicing factor involved in exon repression. It has been proposed that PTB acts by looping out exons flanked by pyrimidine tracts. We present fluorescence, NMR, and in vivo splicing data in support of a role of PTB in inducing RNA loops. We show that the RNA recognition motifs (RRMs) 3 and 4 of PTB can bind two distant pyrimidine tracts and bring their 5′ and 3′ ends in close proximity, thus looping the RNA. Efficient looping requires an intervening sequence of 15 nucleotides or longer between the pyrimidine tracts. RRM3 and RRM4 bind the 5′ and the 3′ pyrimidine tracts, respectively, in a specific directionality and work synergistically for efficient splicing repression in vivo.
Keywords: alternative splicing, polypyrimidine tract–binding protein, protein–RNA interactions
Alternative splicing is a highly regulated biological process that plays a crucial role in generating high proteomic diversity. It has been estimated that > 90% of human genes are alternatively spliced (1). Alternative splicing occurs frequently in cells, and most RNA-binding proteins that influence alternative splicing were found to be nonspliceosomal (2). The polypyrimidine tract (PPT)–binding protein (PTB) is one of the major trans-acting factors involved in splicing regulation. PTB is most often associated with its role as a splicing repressor (3–5), but it is also involved in other aspects of mRNA processing including 3′ end processing (6, 7), mRNA localization and stability (8), and internal ribosome entry site (IRES)-mediated translation (9).
PTB is a 58-kDa member of the hnRNP family consisting of four RNA recognition motifs (RRMs) joined by three linkers (10, 11). PTB recognizes PPTs in the RNA target containing CU-rich elements (12, 13). The mechanism by which PTB promotes exon exclusion is poorly understood. Our NMR structure of RNA-bound PTB has suggested a potential mechanism of PTB action in splicing whereby RRM3 and RRM4 bind the PPTs flanking an alternative exon and loop out the intervening RNA, thus repressing the exon (Fig. 1A) (14). The two RRM-bound PPTs appear in opposite direction as if forming a loop to exclude the intervening exon or the branched adenosine from the spliceosomal machinery. Other mechanistic models for PTB repression have proposed a direct (5) and an indirect (5, 15) competition between PTB and other splicing factors like U2AF65, corepression with Raver-1 (16) and PTB preventing exon (15) or intron definition (17). However, all proposed mechanisms are consistent with RNA looping between RRM3 and RRM4.
Fig. 1.

Structure of RNA-bound PTB34 and native gel. (A) Schematic representation of the RNA looping mechanism by PTB34. The figure shows two possible looped conformations: with the intervening RNA on the same side of the PTB34 interdomain linker (Left) or the opposite (Right). (B) FRET-based nondenaturing gel electrophoresis of the fluorophore-labeled PPTs in presence and absence of PTB34, as shown. Donor emission in the figure is represented as green and acceptor emission as red. RNA and RNA–protein complexes are assigned as shown.
Here, we have sought to test and characterize this suggested looping mechanism using FRET, NMR spectroscopy, and in vivo splicing assays.
Results
PTB34 Binds PPTs and Brings Their 5′ and 3′ Ends into Close Proximity.
First, we tested the binding of RRM3 and RRM4 of PTB (PTB34, Fig. 1A) to several model RNAs using a FRET-based gel shift assay (18). We prepared a series of single-stranded PPTs separated by a variable size loop [5′-CUCUCU(A)NCUCUCU-3′, N = 5–30, named PPT-N], and labeled their 5′ and 3′ ends with fluorescein and tetramethylrhodamine, respectively. FRET measures the distance between the fluorophores (18, 19), and it enables monitoring RNA conformational changes during the binding reaction: If PTB34 loops the RNA, it should bring the RNA 5′ and 3′ ends into close proximity resulting in a FRET increase. With this assay, we could detect both the gel shift of our labeled RNAs upon PTB34 binding and the RNA conformation by measuring the fluorescence intensity of the FRET pair. Distant fluorophores yield green bands, whereas close ones appear red (18). Fig. 1B shows the results for PPT-5, -10, -15, -20, and -30 in the absence and presence of PTB34. The free RNAs migrate as single bands, demonstrating the labeled RNA purity. As the RNA size increases, the corresponding bands migrate more slowly and change color from red (PPT-5) to yellow (PPT-30), as expected. Bound to PTB34, the bands become red and migrate slower indicating the formation of the protein–RNA complex. For PPT-5 and -10, a second supershifted band is also visible (80 and 30% intensity, respectively), but for PPT-15, -20, and -30, it does not exceed 10% intensity. Based on the gel mobilities, we assigned the first band to a monomeric complex and the second band to a dimeric complex with two RNAs and two PTB34s (Fig. 1B).
We confirmed binding in solution with steady-state FRET (ssFRET). Fig. 2A shows the fluorescence spectrum of PPT-15 in the absence (in blue) and presence (in red) of PTB34. In the free RNA, the donor intensity (520 nm) is larger than the acceptor (580 nm), indicating that the fluorophores, and therefore the 5′ and 3′ ends, are distant. Bound to PTB34, the donor intensity decreases while the acceptor increases, indicating that the 5′ and 3′ ends are brought into close proximity in the complex. The calculated FRET ratio for PPT-15 increases from 0.38 to 0.66 (Fig. 2B), in agreement with the gel shift assay. Similar results were obtained for all PPTs except PPT-5, which did not show a significant FRET increase. The initial FRET ratio in absence of PTB34 decreases with increasing RNA length from 0.61 to 0.27. Bound to PTB34, all FRET ratios increase to approximately 0.65. Small differences in the final FRET ratios are likely caused by differences in the binding affinities of each RNA.
Fig. 2.

Evidence of RNA looping by PTB34. (A) Fluorescence emission spectra of labeled PPT-15 RNA in the absence (blue) and presence of 100 nM PTB34 (red). (B) FRET ratios for PPT-5 through PPT-30 in the absence (blue) and presence of 100 nM PTB34 (red). (C) FRET ratio of PPT-15 as a function of [PTB34]. The black line is a fit to a modified Hill equation (Methods). Error bars stem from three independent assays. (D) Apparent KD as a function of the intervening loop size. Error bars stem from the standard deviation to the fit as shown in C.
We tested for the specificity of the PPT–PTB34 interaction using a fluorophore-labeled RNA lacking a PPT, which did not show any FRET increase upon addition of PTB34. We also tested whether nonspecific RNA binding proteins could loop the PPTs using the small protein B (SmpB), a basic protein that binds tmRNA in bacteria (20). Although SmpB can bind PPT-15 nonspecifically, we did not observe any appreciable FRET change upon addition of 1 μM SmpB, confirming that the observed FRET increase is specific to PTB34 binding.
PTB34 Looping Efficiency Depends on RNA Loop Size.
We have used the observed FRET increases to quantify the binding affinity of PTB34 to PPTs (Fig. 2B). A fit of the PPT-15 titration to a modified Hill equation yields a dissociation constant KD = 11 ± 5 nM and a cooperativity coefficient n = 0.7 ± 0.2 (Fig. 2C). The KD is expected to decrease in vivo because of the presence of two additional RRMs (1 and 2) in the full-length PTB. The cooperativity coefficient near unity confirms that PTB34 binds stoichiometrically.
Similarly, we determined the KD for all six PPTs (Fig. 2D and Fig. S1). The resulting KD values decrease with increasing loop size and plateau for PPT-15 and higher, indicating that PTB34 requires larger than or equal to 15-nt-long intervening sequences to loop the RNA effectively (14). The KDs obtained for PPT-5 and -10 are global average constants for the monomer and dimer. The cooperativity coefficients of all PPTs ≥15 are near unity, confirming the stoichiometric nature of these complexes.
All RNA–Protein Complexes Have Similar Conformations.
To characterize the global structure of all the PTB-RNA complexes, we used time-resolved FRET (trFRET) to accurately measure the distances between the two fluorophores (21, 22). trFRET consists of measuring the fluorescence lifetime of the donor fluorophore in the presence and absence of the acceptor. Fig. 3A shows the donor fluorescence decays for PPT-20. In the absence of PTB34 (compare black and blue curves), the donor lifetime decreases more rapidly in the presence of the acceptor, which is indicative of FRET. This difference is used to fit a distribution of distances between the dye pair (23). The resulting distribution reveals a bimodal distribution (Fig. 3A Inset, blue). We assign the major distribution (85%, 71 Å) to unfolded molecules where the 5′ and 3′ ends are distant. The minor distribution (15%, 31 Å) is attributed to a minor population of molecules transiently folding into hairpins or duplexes where the pyrimidines and adenosines from the linker form base pairs resulting in closer 5′ and 3′ ends of the RNA.
Fig. 3.

Time-resolved FRET analysis. (A) Donor fluorescence decay PPT-20 in the absence (black) and presence of the FRET acceptor (blue) and in the presence of PTB34 (red). Inset: Donor–acceptor distance distribution in the absence (blue) and presence (red) of PTB34. (B) Average donor–acceptor distance as a function of the intervening RNA length.
In the presence of PTB34, the donor lifetime decreases dramatically, indicating a large increase in FRET (Fig. 3A, compare red and blue curves). The calculated distribution collapses to a single distribution centered at 35 Å (Fig. 3A. inset, red), confirming the shorter distance between the 5′ and 3′ ends in the protein-RNA complexes observed in previous experiments.
We measured the donor–acceptor distance for all PPTs in the presence and absence of PTB34 and found similar results. Fig. 3B shows the mean distance of the major distribution in the absence of PTB34 as a function of loop size (blue). The distance between the two dyes increases linearly between 51 and 85 Å for PPT-5 and -30, respectively, with a slope of 1.4 ± 0.1 Å/nt. Upon PTB34 binding, all PPTs have a mean distance ranging 32 to 38 Å. For PPT-5 and -10, the observed mean distance corresponds to a weighted average between the monomer and the dimer (Fig. 1B).
RRM3 and RRM4 Bind the 5′ and 3′ PPTs, Respectively.
We next used NMR spectroscopy to investigate the complex between PTB34 and two pyrimidine tracts separated by 15 adenosines. Although the FRET data showed unambiguously that PTB34 forms RNA loops, the FRET data did not provide information on the directionality of binding: Two binding modes are possible with either RRM3 and RRM4 binding the 5′- and 3′-tracts, respectively, or the opposite (Fig. 1A).
Complex formation with CUCUCU(A)15CUCUCU, GGUCUCU(A)15CUCU and GGCUCU(A)15UCUCU resulted in almost identical spectra, indicating that all three RNAs bind PTB34 in the same orientation even if the pyrimidine tract is shorter at the 5′ end or the 3′ end (5′-GG was used for in vitro transcription). By comparing the spectra of PTB34 in complex with GGUCUCU(A)15CUCU (1∶1 stochiometry) and in complex with CUCUCU (1∶2 stochiometry), we could observe clear differences indicating contacts between the 15 adenine linker and the protein. Major chemical shift changes and line broadening are observed within the interdomain linker (Q421, Q424, and N432, Fig. 4) and in the loop between α-helix 2 and β-strand 4 in RRM4 (H514 and D515, Fig. 4). This indicates that the adenine linker interacts with this region of PTB34. Hence, the RNA takes the shortest path of the two possibilities (Fig 1A), where RRM3 and RRM4 are binding the 5′ and 3′ tracts, respectively (Fig. 4B). The distance measured in the structure between the 5′ nucleotide (nt) bound to RRM3 and the 3′ nt bound to RRM4 is 41 Å, which is in good agreement with the estimated FRET distance of 32 to 38 Å between the two dyes. The spectra of PTB34 bound with PPT-10 to -30 are very similar, indicating that the RNA is bound with this same orientation independently of the linker length up to 30 nt.
Fig. 4.

Chemical shift mapping and structure of RNA-bound PTB34. (A) Plot of the chemical shift differences (in parts per million, plotted against the residue numbers) between PTB34 bound to two 5′CUCUCU3′ and PTB34 bound to GGUCUCU(A)15CUCU. Chemical shift differences are calculated as Δδ = [ΔδHN + (ΔδN/6.51)2]1/2. (B) Structure of PTB34 bound to short pyrimidine tract (14) (RNA in orange and protein ribbon in gray). The nitrogen shown in blue indicates the residues for which chemical shift difference is greater than 0.1 ppm, and the nitrogen shown in cyan indicates resonances that are broadened or disappear upon binding of GGUCUCU(A)15CUCU.
A Mutant Reveals a Synergistic Role Between RRM3 and RRM4 for Efficient RNA Looping.
The NMR structure of PTB34 free (11) and bound (14) has revealed interactions between RRM3 and RRM4 involving 27 amino acids. We sought to test the function of these interactions in the looping of RNA by PTB34. To this aim, we used the FRET-based gel shift assay with a 6-fold mutant of PTB34 that was shown to prevent the interaction between RRM3 and RRM4 (I356K, F446E, I449K, E502K, V505E, and I509K) (11). It is noteworthy that none of these mutations affect the protein surfaces interacting with RNA. Fig. 5A shows that PTB34 mutant does not form the RNA–protein complex with PPT-15 as readily as the wild-type (WT) PTB34 (Fig. 1). This result suggests an active role for the interdomain interactions between RRMs 3 and 4 in PTB for looping and implicates a synergistic effect between the two domains for efficient looping in vivo. The resulting dissociation constant (KD = 105 ± 4 nM) is approximately 10-fold higher than the WT, and the large cooperativity coefficient (n = 7 ± 1) indicates the presence of higher-order oligomers, which result in the observed high FRET value, similarly to PPT-5 and -10 (Fig. 2). The formation of stoichiometric complex is expected to occur at much lower concentrations than the dimer, but almost no FRET increase is observed below 100 nM (compare Figs. 2 and 5), indicating that the FRET ratio of the stoichiometric mutant complex is similar to that of the free RNA, also supporting the idea that the mutant does not loop the RNA. The observed KD is, therefore, an average value of multiple species in solution, and likely to be an upper estimate for the stoichiometric complex. When the linker length increases (PPT-30), the affinity for the mutant protein increases (KD = 38 ± 6 nM, Fig. S2) due to an increase of negative charges, whereas it remains almost the same for the WT PTB (KD = 10 ± 3 nM, Fig. S1). The cooperativity coefficient decreases, too (n = 1.6 ± 0.4), indicating the presence of fewer higher-order complexes. However, a FRET gel shift assay reveals no specific looped complex as the WT (Fig. S2). These results indicate that the interdomain interactions between RRM3 and RRM4 provide RNA looping and only slightly higher affinity.
Fig. 5.

Mutant PTB34 exhibits a different conformation than the WT. (A) Nondenaturing gel electrophoresis of PPT-15 in the presence and absence of PTB34-m. A clear band shift was not observed as for the WT (Fig. 1B). ssFRET titration of PTB34-m and PPT-15. (B) Schematic diagram of the single-molecule experiment. (C) Single-molecule FRET histograms for RNA only (Top), the WT protein (Middle), and the 6-fold mutant (Bottom). Histograms are built from the observed FRET ratios of hundreds of single molecules. The 0-FRET peak corresponds to molecules without an acceptor. The fits to a Gaussian distribution (red lines) are only intended to guide the eye.
Single-molecule FRET is a powerful tool to study the conformational dynamics of protein-RNA complexes (24). We used this technique to study the differences in binding of PTB34 and the 6-fold mutant with PPT-15. A biotinylated RNA and fluorophore-labeled PPT-15 was surface-immobilized on a PEG-passivated quartz slide via a biotin-streptavidin bridge. FRET histograms are built from the observed FRET ratio of hundreds of individual molecules. In the absence of PTB34 (Fig. 5C, Top panel), the majority of molecules exhibit low FRET (0.2–0.4) and only few molecules transiently reach the high FRET range. Such a broad FRET distribution is expected for a single-stranded RNA without a well-defined secondary structure, consistent with our trFRET data (Fig. 3A). Addition of 10 nM PTB34 results in the appearance of a major peak at 0.55 FRET (Fig. 5C, Center panel). We assign this major peak to the PTB34–RNA complex, consistent with the bulk FRET experiments. In the presence of 10 nM 6-fold PTB34 mutant, the observed FRET distribution resembles the RNA-only distribution (Fig. 5C, Bottom panel). A minor broad distribution ranging from 0.4–0.8 FRET and centered at higher FRET than the WT is also present. We assign this distribution to the formation of the mutant PTB34–RNA complex. This result is consistent with both the weaker binding of the mutant PTB34 and the lack of interactions between RRM3 and RRM4. The observed FRET differences between the WT and the mutant distributions suggest that in the absence of interactions at the interface between RRM3 and RRM4, the mutant binds in a different conformation.
RNA Looping by PTB34 is Essential for Efficient Splicing Regulation In Vivo.
To test how functionally important is the interface between RRM3 and RRM4, we used a splicing reporter assay (25) in HeLa cells with a reporter gene containing two separate PPTs separated by a long linker (DS9-175) (26). In a control experiment without protein over expression, the alternative exon is 80% included, whereas only 34% is included when PTB is transfected and overexpressed. This assay illustrates the repressive role of PTB in the regulation of this alternative exon (Fig 6).
Fig. 6.

In vivo splicing assay showing the importance of the interaction of RRM3 and RRM4 of PTB on splicing repression. (A) In vivo splicing of reporter gene DS9-175 (26) containing two PPTs separated by a long linker. nPTB, PTB WT, and PTB 3Mut were overexpressed in HeLa cells and after RT-PCR developed on a Typhoon PhosphorImager. Similar expression levels of proteins were verified by Western blot (C). Whereas overexpression of PTB WT shows repression of exon inclusion (34%), the mutant has lost the ability to repress this inclusion (74%). (B) Histogram of the splicing-assay. Each RT-PCR product was quantified with the ImageQuant software and the exon-inclusion ratio calculated (exon included product/(exon excluded product + exon included product)). Standard deviation was computed from three independent experiments. (C) Western blot corresponds to the same splicing-assay shown on the Left and verifies similar expression levels of proteins. Consistent with published data (28), the expression level of nPTB in HeLa cells was very low and near detection limit. (D) Graph displays relative protein levels of PTB WT and PTB 3Mut to GAPDH as quantified from Western blot using ImageQuant software. Although PTB 3Mut has a 10% higher expression than PTB WT, it represses splicing much weaker.
We then transfected the cells with a PTB mutant (PTB 3Mut) with three mutations in helix 2 of RRM4 (E502K, V505E, and I509K) that we know are sufficient to prevent the interaction between RRM3 and RRM4 (11). Here, 74% of the exon is included, indicating that PTB 3Mut is much less repressive than PTB WT (Fig. 6). To confirm that the loss of repression is not due to a lower expression of PTB 3Mut, we performed a Western blot for the same splicing assay (Fig. 6C). PTB 3Mut shows an even higher expression (10%) than PTB WT, confirming that the loss of repression is not due to a lower expression level. As a control, overexpression of neuronal PTB (nPTB) under the same condition confirms earlier reports (27) that nPTB appears less repressive than PTB (56% inclusion), but still more repressive than PTB 3Mut. However, in the case of nPTB, loss of repression is due to a much lower expression level, as reported (28). Because the interaction between RRM3 and RRM4 is essential for RNA looping, these in vivo data support the idea that splicing repression by PTB depends on the interface between RRM3 and RRM4 and consequently on the ability of PTB to mediate RNA looping.
Discussion
Despite the diverse and crucial roles PTB plays in many processes, its molecular mechanism of action remains elusive. We have used FRET, NMR spectroscopy, and in vivo splicing assays to study the ability of PTB to remodel RNA structure. The FRET data unambiguously show the ability of PTB34 to bring together (< 40 Å) the RNA 5′ and 3′ ends, and to form RNA loops, as suggested (14). However, the spacer between the two PPTs must be ≥15 nt. The FRET data also provide the affinity of these RNAs for PTB34, which is < 50 nM when the linker is ≥15 adenosines. Compared to the affinity obtained for each domain separately (KD ≈ 1 μM) (29), this indicates that the two pyrimidine tracts bind synergistically even with a 30 nt long linker. These results prompted us to look further into how PTB34 could favor RNA looping. Our NMR investigation of PTB34 bound to pyrimidine tracts linked by 15 adenines showed that the RNA is bound to PTB34 with a specific directionality, with the 5′ and the 3′ pyrimidine tracts bound to RRM3 and RRM4, respectively. These data support earlier footprinting experiments of the intron upstream of the alternative-exon 9 of GABA-g2 that showed the binding of RRM4 to the 3′ pyrimidine tracts and of the rest of the protein to the 5′ pyrimidine tract (30). This RNA binding directionality might be due to the presence of several positively charged residues (R366, R418, R437, K439, K440, and K444) in a region of the protein that interact with the RNA linker nonsequence specifically. Finally, we showed that the interaction between RRM3 and RRM4 is essential for efficient RNA binding, looping and for splicing repression in vivo. However, the question remains whether the differences in splicing repression between the mutant and the WT arise from defective RNA looping or from its lower binding affinity. The mutant binds PPT-15 with 10-fold lower affinity than the WT, whereas the decrease for PPT-30 was only 3- to 4-fold. These KD values are average values of different species in solution and likely to be an upper estimate for the stoichiometric complex. Additionally, the single-molecule and trFRET measurements (Fig. 5 and Fig. S2) confirm that the structure of the mutant PTB34–RNA complex is different from the WT. Therefore, the observed FRET increases in Fig. 5 and Fig. S2 are not due to the formation of looped RNA, even if the apparent average FRET ratios are similar to the WT. This is further supported by the much higher cooperativity coefficients observed in the mutant titrations, which indicate the presence of higher-order complexes. The stoichiometric RNA–protein complexes, which should be formed at lower concentrations than the higher-order complexes, yield no apparent FRET increase, indicating the lack of RNA looping. Based on these results, we expect that the mutant will bind pyrimidine tracts with longer spacers, such as DS9-175 used in the splicing assay, with an affinity similar to the WT. Therefore, the defective RNA looping, and not its weaker binding affinity, is the most likely reason for the lower splicing repression of the mutant compared to the WT.
Altogether, our study confirms the unique ability of PTB34 to bring distant pyrimidine tracts into close proximity. These findings strongly support the model that PTB could repress alternative exons by looping out the exon or the associated branch point (31, 32). It also explains the RNA-remodeling role proposed for PTB in IRES-mediated translation regulation, because PTB34 could bring distant pyrimidine tracts into close proximity and, therefore, influence the IRES structure (31).
Methods
RNA Purification and Labeling.
RNA samples with 5′ fluorescein and 2′ OH protection groups were purchased from Keck Foundation Biotechnology Resource Laboratory at Yale University School of Medicine, deprotected, purified, and labeled, as described (18, 33).
Protein Expression and Purification.
PTB34 and mutant proteins were overexpressed in Escherichia Coli and purified, as described (11, 14).
FRET Gel Shift Assays.
15% nondenaturing polyacrylamide (29∶1 acrylamide∶bisacrylamide ratio) gel electrophoresis was performed in 20 mM NaOAc and 10 mM Sodium Phosphate (pH 6.5) using low-fluorescence glass plates, as described (18). 10 pmol doubly labeled RNA samples were annealed and allowed to equilibrate with 40 pmol PTB34 for 5 min before loading. After electrophoresis at 4 °C, the gel was scanned in a Typhoon Imager by exciting the donor with a 532-nm laser. The fluorescence emissions of the donor (526 nm) and acceptor (580 nm) were analyzed with Fluorsep (Amershan Bioscience).
ssFRET.
ssFRET measurements of doubly labeled PPTs were carried out in a spectrofluorometer. A 25-nM RNA sample was annealed in standard buffer, and fluorescence was measured in the absence and presence of PRB34. Fluorescein was excited at 490 nm (10-nm bandwidth), and fluorescein and rhodamine emissions were measured at 520 and 580 nm (5-nm bandwidth), respectively. Relative FRET efficiencies were calculated as I580/(I580 + I520), where I520 and I580 are the fluorescein and rhodamine intensities, respectively. The global dissociation constant (KD) and cooperativity coefficients were obtained by plotting FRET as a function of [PTB34], and fitting to the modified Hill equation:
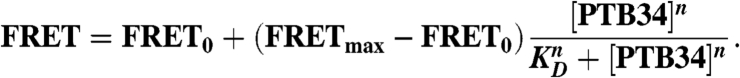 |
trFRET for Distance Measurements.
The distance between the two fluorophores was measured using trFRET, as described (18). A 250-nM solution of donor-only labeled RNA in standard buffer was annealed, and fluorescein was excited at 470 nm (30-nm band-pass dichroic filter with a Yb-doped fiber laser (5 ps pulses, 40 MHz). Donor isotropic emission was collected at 520 nm (20-nm band-pass dichroic filter) using a microchannel photomultiplier tube feeding a time-correlated single photon counting card under magic angle polarization (54.7°) to > 40,000 peak counts. Fluorescence decays were collected in 4816 channels with 12.2 ps/channel time resolution. Nondairy coffee creamer solution was used to measure the instrument response function. The donor-only fluorescein emission decay was deconvoluted from the instrument response function and fit to a sum of three exponential decays characterized by their lifetime τi with fractional contributions αi. Then, the time-resolved fluorescence decay of a 250-nM solution of the doubly labeled RNA (IDA) in standard buffer was measured similarly. The donor–acceptor distance distributions (18, 19) were obtained by fitting
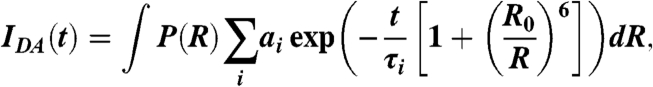 |
where P(R) is the distance distribution, αi and τi are the donor-only lifetime parameters, and R0 is Forster distance (55 Å). The distance distribution was modeled as a three-dimensional Gaussian P(R) = 4πR2N exp(-(R - μ)2/σ2), where N is normalization constant, and μ and σ describe the Gaussian’s shape.
Single-Molecule FRET Experiments.
PPT-15 [5′-CUCUCU(A)15CUC UCU dTAA-3′] was labeled with 5′ Cy3 and 3′ biotin and purified as discussed. Cy5 was attached to an amino linker on dT following the manufacturer′s protocol. The biotinylated single-stranded RNA was annealed in standard buffer, and surface-immobilized on a PEG-passivated quartz slide, as described (34, 35). The donor and acceptor intensities (ID,IA) from single molecules were used to calculate apparent FRET efficiencies as IA/(IA + ID) in the presence and absence of 10 nM PTB34 or 6-fold mutant.
NMR Spectroscopy.
Complex formation for NMR spectroscopic measurement was carried out as described (14). Resonance assignments for the 15N-13C labeled PTB34 in complex with GGUCUCU(A)15CUCU were obtained with the TROSY-HNCA experiment at 313 K.
In Vivo Splicing Assay.
DNA Constructs.
PTB1, nPTB, and reporter plasmid DS9-175 were prepared as described (26). The three mutations in PTB1 (PTB 3Mut) were made with two consecutive PCR reactions using the primers 2Mut-F, 2Mut-R, 1Mut-F, and 1Mut-R (Table S1). The PCR protocol included 20 cycles: 95 °C for 30 s, 55 °C for 1 min, and 72 °C for 7.5 min. The PCR followed a digestion with Dpn1 for 1 h at 37 °C.
Transfections.
HeLa cells were cultured in Eagle’s minimum essential medium supplemented with 10% FBS on six well plates and grown to an approximately 90% confluence. DNA-Lipofectamine 2000 complexes were assembled according to the manufacturer’s protocol. The complexes were added to the cells and incubated for 5 h. After, medium was changed and cells were grown for 19 h.
RNA-Isolation and RT-PCR.
Both were carried out following the instructions of the Cells-to-cDNA™ II Kit. After RT-PCR, 5 μL of the resulting cDNA was used in a 50-μL PCR-reaction with 10 μM of the: γ-32P-labeled Forward-Primer Dup1a and Reverse-Primer Dup8 (Table S1). The PCR protocol included 35 cycles: 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 30 s. Samples of the PCR reaction were then loaded on a 6% denaturing acrylamide gel. The gel was exposed to a PhosphorImager, and the bands were quantified using ImageQuant software.
Western Blot.
Cells were lysed in radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail. Protein-lysate (30 μg) was used for Western blot, and chemiluminescent detection of the Flag-Tag protein was carried out by using the Immun-Star Western C Kit.
Supplementary Material
Acknowledgments.
We thank D. Black for providing clones, reagents, and protocols for the cell biology experiments and for critically reading the manuscript. Support from the Swiss National Science Foundation National Center of Competence in Research in Structural Biology, the European Alternative Splicing Network (F.H.-T.A.), the National Institutes of Health (Grant GM085116 to D.R.), and the National Science Foundation (Grant MCB0747285 to D.R.) is acknowledged.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.L.B. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0907072107/DCSupplemental.
References
- 1.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black DL, Grabowski PJ. Alternative pre-mRNA splicing and neuronal function. Prog Mol Subcell Biol. 2003;31:187–216. doi: 10.1007/978-3-662-09728-1_7. [DOI] [PubMed] [Google Scholar]
- 4.Robinson F, Smith CW. A splicing repressor domain in polypyrimidine tract-binding protein. J Biol Chem. 2006;281:800–806. doi: 10.1074/jbc.M510578200. [DOI] [PubMed] [Google Scholar]
- 5.Spellman R, Smith CW. Novel modes of splicing repression by PTB. Trends Biochem Sci. 2006;31:73–76. doi: 10.1016/j.tibs.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Castelo-Branco P, et al. Polypyrimidine tract binding protein modulates efficiency of polyadenylation. Mol Cell Biol. 2004;24:4174–4183. doi: 10.1128/MCB.24.10.4174-4183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lou H, Helfman DM, Gagel RF, Berget SM. Polypyrimidine tract-binding protein positively regulates inclusion of an alternative 3′-terminal exon. Mol Cell Biol. 1999;19:78–85. doi: 10.1128/mcb.19.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knoch KP, et al. Polypyrimidine tract-binding protein promotes insulin secretory granule biogenesis. Nat Cell Biol. 2004;6:207–214. doi: 10.1038/ncb1099. [DOI] [PubMed] [Google Scholar]
- 9.Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 10.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 11.Vitali F, et al. Structure of the two most C-terminal RNA recognition motifs of PTB using segmental isotope labeling. EMBO J. 2006;25:150–162. doi: 10.1038/sj.emboj.7600911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez I, McAfee JG, Patton JG. Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry. 1997;36:11881–11890. doi: 10.1021/bi9711745. [DOI] [PubMed] [Google Scholar]
- 13.Singh R, Valcarcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 14.Oberstrass FC, et al. Structure of PTB bound to RNA: Specific binding and implications for splicing regulation. Science. 2005;309:2054–2057. doi: 10.1126/science.1114066. [DOI] [PubMed] [Google Scholar]
- 15.Izquierdo JM, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Rideau AP, et al. A peptide motif in Raver1 mediates splicing repression by interaction with the PTB RRM2 domain. Nat Struct Mol Biol. 2006;13:839–848. doi: 10.1038/nsmb1137. [DOI] [PubMed] [Google Scholar]
- 17.Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol. 2008;15:183–191. doi: 10.1038/nsmb.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rueda D, Wick K, McDowell SE, Walter NG. Diffusely bound Mg2+ ions slightly reorient stems I and II of the hammerhead ribozyme to increase the probability of formation of the catalytic core. Biochemistry. 2003;42:9924–9936. doi: 10.1021/bi0347757. [DOI] [PubMed] [Google Scholar]
- 19.Rueda D, Hsieh J, Day-Storms JJ, Fierke CA, Walter NG. The 5′ leader of precursor tRNAAsp bound to the Bacillus subtilis RNase P holoenzyme has an extended conformation. Biochemistry. 2005;44:16130–16139. doi: 10.1021/bi0519093. [DOI] [PubMed] [Google Scholar]
- 20.Karzai AW, Susskind MM, Sauer RT. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA) EMBO J. 1999;18:3793–3799. doi: 10.1093/emboj/18.13.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rueda D, Walter NG. Single molecule fluorescence control for nanotechnology. J Nanosci Nanotechnol. 2005;5:1990–2000. doi: 10.1166/jnn.2005.505. [DOI] [PubMed] [Google Scholar]
- 22.Klostermeier D, Millar DP. Time-resolved fluorescence resonance energy transfer: A versatile tool for the analysis of nucleic acids. Biopolymers. 2001;61:159–179. doi: 10.1002/bip.10146. [DOI] [PubMed] [Google Scholar]
- 23.Walter NG. Structural dynamics of catalytic RNA highlighted by fluorescence resonance energy transfer. Methods. 2001;25:19–30. doi: 10.1006/meth.2001.1212. [DOI] [PubMed] [Google Scholar]
- 24.Aleman EA, Lamichhane R, Rueda D. Exploring RNA folding one molecule at a time. Curr Opin Chem Biol. 2008;12:647–654. doi: 10.1016/j.cbpa.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Modafferi EF, Black DL. A complex intronic splicing enhancer from the c-src premRNA activates inclusion of a heterologous exon. Mol Cell Biol. 1997;17:6537–6545. doi: 10.1128/mcb.17.11.6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amir-Ahmady B, Boutz PL, Markovtsov V, Phillips ML, Black DL. Exon repression by polypyrimidine tract binding protein. RNA. 2005;11:699–716. doi: 10.1261/rna.2250405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Markovtsov V, et al. Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol Cell Biol. 2000;20:7463–7479. doi: 10.1128/mcb.20.20.7463-7479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson F, Jackson RJ, Smith CW. Expression of human nPTB is limited by extreme suboptimal codon content. PLoS One. 2008;3:e1801. doi: 10.1371/journal.pone.0001801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Auweter SD, Oberstrass FC, Allain FH. Solving the structure of PTB in complex with pyrimidine tracts: An NMR study of protein-RNA complexes of weak affinities. J Mol Biol. 2007;367:174–186. doi: 10.1016/j.jmb.2006.12.053. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Zhang W, Reed RB, Liu W, Grabowski PJ. Mutations in RRM4 uncouple the splicing repression and RNA-binding activities of polypyrimidine tract binding protein. RNA. 2002;8:137–149. doi: 10.1017/s1355838202015029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auweter SD, Allain FH. Structure-function relationships of the polypyrimidine tract binding protein. Cell Mol Life Sci. 2008;65:516–527. doi: 10.1007/s00018-007-7378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner EJ, Garcia-Blanco MA. Polypyrimidine tract binding protein antagonizes exon definition. Mol Cell Biol. 2001;21:3281–3288. doi: 10.1128/MCB.21.10.3281-3288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rueda D, Walter NG. Fluorescent energy transfer readout of an aptazyme-based biosensor. Methods Mol Biol. 2006;335:289–310. doi: 10.1385/1-59745-069-3:289. [DOI] [PubMed] [Google Scholar]
- 34.Ha T, et al. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature. 2002;419:638–641. doi: 10.1038/nature01083. [DOI] [PubMed] [Google Scholar]
- 35.Zhao R, Rueda D. RNA folding dynamics by single-molecule fluorescence resonance energy transfer. Methods. 2009 doi: 10.1016/j.ymeth.2009.04.017. In Press. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.