

Abstract
DNA polymerase β (pol β) is the key gap-filling polymerase in base excision repair, the DNA repair pathway responsible for repairing up to 20,000 endogenous lesions per cell per day. Pol β is also widely used as a model polymerase for structure and function studies and several structural regions have been identified as being critical for the fidelity of the enzyme. One of these regions is the hydrophobic hinge, a network of hydrophobic residues located between the palm and fingers subdomains. Previous work by our lab has shown that hinge residues Y265, I260 and F272 are critical for polymerase fidelity by functioning in discrimination of the correct from incorrect dNTP during ground state binding. Our work aimed to elucidate the role of hinge residue I174 in polymerase fidelity. To study this residue, we conducted a genetic screen to identify mutants with a substitution at residue I174 that resulted in a mutator polymerase. We then chose the mutator mutant I174S for further study and found that it follows the same general kinetic pathway and has similar overall protein folding compared to the wild type (WT) pol β. Using single turnover kinetic analysis we found that I174S shows decreased fidelity when inserting opposite a template base G, and this loss of fidelity is due primarily to loss of discrimination during ground state dNTP binding. Molecular dynamics simulations show that mutation of residue I174 to serine results in an overall tightening of the hinge region, resulting in aberrant protein dynamics and fidelity. These results point to the hinge region as being critical in maintenance of the proper geometry of the dNTP binding pocket.
Maintenance of genomic integrity in the face of constant endogenous and exogenous DNA damaging agents is one of the keys to healthy cell survival. As evidence for the importance of genomic stability, cancer cells are characterized by a higher mutational burden than can be accounted for by the normal cellular mutation rate (reviewed in (1)). This mutator phenotype can be acquired due to loss of fidelity during DNA replication or faulty DNA repair (reviewed in (2)). One DNA repair pathway critical for the maintenance of genomic integrity is the base excision repair (BER) pathway, which is responsible for the repair of up to 20,000 spontaneous apurinic/apyrimidinic (AP) sites per cell per day (3).
BER is the DNA repair pathway primarily responsible for repairing lesions caused by reactive oxygen species and alkylating agents. DNA polymerase beta (pol β), an enzyme with both polymerase and deoxyribose phosphate (dRP) lyase activities, is a key enzyme in BER. It acts as the main gap-filling enzyme in both the short patch and in some cases the long patch subpathways (reviewed in (4)) (5–8), and the dRP lyase activity of pol β is necessary for certain subpathways as well (9). Pol β has been found to be mutated in approximately 30% of tumors studied to date (10). Several of these tumor-associated mutants have been shown to have aberrant polymerase or dRP lyase function (11–13). Additionally, some of these mutants can transform mouse cells in culture (13, 14). Based on these and other findings, it is thought that BER may function as a tumor suppressor mechanism (reviewed in (15)).
In addition to its role in BER, pol β is also useful as a model for the study of polymerase activity and fidelity (reviewed in (4)). It follows the standard two metal ion mechanism and is a small protein (39kD) that is easy to express and purify from E. coli. Several crystal structures are available(16–18), including those of the enzyme alone and in binary and ternary complexes with a variety of substrates and nucleotides. Finally, the enzyme has no known exonuclease activity, so the observed polymerase fidelity is due strictly to the accuracy of nucleotide incorporation.
Through the use of a genetic screen capable of identifying mutator mutants of pol β (19), the hinge region of pol β has been shown to contain residues critical for polymerase fidelity (19–22). The hinge is a hydrophobic region of the polymerase located between the palm and finger subdomains. It has an outside lining containing residues I174, T196 and Y265 and an inner lining of residues L194, I260 and F272. These residues likely help control the enzyme’s conformational change that occurs upon dNTP binding. Crystal structures show that when pol β is bound to gapped DNA it is present in an open structure and when it is bound to dNTP it is closed (16, 17). Upon closing, the fingers subdomain moves approximately 12 angstroms (18). Recent studies using single turnover kinetic analysis of mutator mutants with amino acid substitutions of hinge residues suggest that this region contributes to polymerase fidelity due to selectivity at the level of ground state dNTP binding (21, 23–25).
While many studies have shown the importance of hinge residues in polymerase fidelity (21–23, 26–29), the role of hinge residue I174 has not yet been investigated. Isoleucine 174 is located in the outside lining of the hinge and in the open conformation is in close proximity to residues Y265, T176 and K262. (Fig 1). These interactions could be interesting because Y265 has been shown to be critical for polymerase fidelity, both at the level of dNTP binding and in maintaining the substrate alignment necessary for efficient nucleotide insertion (25, 28, 29). Given its location in the hydrophobic hinge and its interaction with key hinge residues, it is important to determine what role residue I174 plays in pol β fidelity.
Figure 1. I174 and surrounding residues in the hydrophobic hinge region.

A close-up view of residue I174 (yellow sticks) and neighboring side-chains (green sticks) that are closely packed in this local network of interactions. The backbone cartoon is colored according to pol β sub-domains (palm, magenta; fingers, blue).
To further elucidate the role of the hinge in pol β fidelity, we have conducted an in-depth analysis of hinge residue I174. We began by mutating residue I174 to every other amino acid and the stop codon. We then used these variant polymerases in a genetic screen to identify mutator mutants. This screen identified the serine, threonine, aspartic acid and glycine substitution mutants as mutators in vivo. We then chose the I174S mutant for further study and found that it is an active polymerase that has similar presteady-state kinetics and folding patterns as wild type (WT) pol β. This mutant has decreased fidelity in vitro due to loss of discrimination at the level of ground state dNTP binding for a dTTP:G mispair and due to a combination of impaired ground state binding and chemistry for dATP:G and dGTP:G mispairs. Molecular dynamics simulations show that the observed loss of fidelity could be due to an overall tightening of the hinge region in the I174S mutant, resulting in aberrant enzyme dynamics. This tightening affects residues already known to be critical to pol β fidelity, such as Y265 and I260.
Materials & Methods
Bacterial Strains and Media
Bacterial strains and media for the tryptophan reversion assay and protein purification were used as described previously (22).
Chemicals and Reagents
All ultrapure deoxynucleoside triphosphates (dNTPs) were purchased from New England Biolabs. [γ-32P] ATP (5 mCi) and ATP were purchased from Amersham Biosciences and Sigma-Aldrich, respectively. All oligonucleotides used for changing the I174 codon and sequencing the mutagenesis products were purchased from Keck Biotechnology Research Center at Yale University and purified by denaturing polyacrylamide gel electrophoresis (20% acrylamide and 8% urea) as in (22).
Generation of I174 Mutants
I174 mutants were generated in the rat polB cDNA in the pAraBAD (pBADβ His,(22)) vector (Invitrogen) using the QuikChange Site-Directed Mutagenesis Protocol (Stratagene). For generation of mutants at position 174, primers with random nucleotides inserted at the position corresponding to the I174 codon were used (I174X-F and I174X-R, forward and reverse, respectively, Table 1). 50 μl PCR reactions were run according to the QuikChange Site-Directed Mutagenesis Kit protocol using 5ng template DNA. PCR reactions consisted of an initial denaturation step of 95°C for 30 seconds, followed by 16 cycles of denaturation at 95°C for 30 seconds, annealing at 55°C for 1 minute, and extension at 68°C for 10 minutes. To generate the I174 Y, N, K, and D mutants in the pBADβ His vector, the following sets of forward and reverse (F and R, respectively) primers were used: I174Y, I174N, I174K and I174D (Table 1). To generate the I174S mutation in the pol β cDNA in the pET28a vector, the I174S forward and reverse primers were used (Table 1). To verify the generated mutations, plasmids were purified using the Qiagen Miniprep kit and sequenced with the 432+b primer that binds within the pol β cDNA sequence (Table 1) for the pBADβ His vector and with the T7 forward and terminator primers for the pET28a constructs (Table 1).
Table 1.
Primer Sequences
| Name | Sequence(5′ → 3′) | Use |
|---|---|---|
| I174X-F | GCT GGA TCC CGA GTA CNN NGC TAC AGT CTG CGG CAG TTT CC | Generation of random I174 mutants in pBADβHis |
| I174X-R | GGA AAC TGC CGC AGA CTG TAG CNN NGT ACT CGG GAT CCA GC | |
| I174Y-F | GCT GGA TCC CGA GTA CTA TGC TAC AGT GTG CGG CAG TTT CC | Generation of Ile174Tyr mutant in pBADβHis |
| I174Y-R | GGA AAC TGC CGC AGA CTG TAG CAT AGT ACT CGG GAT CCA GC | |
| I174N-F | GCT GGA TCC CGA GTA CAA TGC TAC AGT CTG CGG CAG TTT CC | Generation of Ile174Asn mutant in pBADβHis |
| I174N-R | GGA AAC TGC CGC AGA CTG TAG CAT TGT ACT CGG GAT CCA GC | |
| I174K-F | GCT GGA TCC CGA GTA CAA AGC TAC AGT CTG CGG CAG TTT CC | Generation of Ile174Lys mutant in pBADβHis |
| I174K-R | GGA AAC TGC CGC AGA CTG TAG CAA AGT ACT CGG GAT CCA GC | |
| I174D-F | GCT GGA TCC CGA GTA CGA CGC TAC AGT CTG CGG CAG TTT CC | Generation of Ile174Asp mutant in pBADβHis |
| I174D-R | GGA AAC TGC CGC AGA CTG TAG CGT CGT ACT CGG GAT CCA GC | |
| I174S-F | GTT AAA AAG CTG GAT CCC GAG TAC AGC GCT ACA GTC TGC GGC AGT TTC CG | Generation of Ile174Ser mutant in pET2Ba |
| I174S-R | CGG AAA CTG CCG CAG ACT GTA GCG CTG TAC TCG GGA TCC AGC TTT TTA AC | |
| 432+b | GAA CCA CCA TCA GCG AAT TGG | Sequencing pBADβHis constructs |
| T7 forward | TAA TAC GAC TCA CTA TA | Sequencing pol beta pET28a+ constructs |
| T7 terminator | GCT AGT TAT TGC TCA GCG G | |
Forward (F) and reverse (R) sequences for primer used for site-directed mutagenesis and plasmid sequencing are listed. Underlined nucleotides represent sites of amino acid change. For nucleotides written as N, any of the four natural nucleotides were randomly inserted by the manufacturer.
Identification of Mutator Mutants in the Trp+ Reversion Screen
Mutator I174X mutants were identified using the Trp+ reversion genetic screen as previously described (22). In short, this screen uses a strain of E. coli that is incapable of making tryptophan due to an ochre mutation in the trpE gene, which encodes an essential enzyme in the tryptophan synthesis pathway. Therefore, these cells need to be grown in media supplemented with tryptophan. When grown on media lacking tryptophan, a Trp+ reversion phenotype can be observed as a result of 5 of 6 possible base substitution errors occurring by incorrect nucleotide incorporation into either the trpE gene or into the anticodon loop of suppressor tRNAs. These mutations can be induced by an error-prone polymerase in the cell, such as variant forms of pol β.
DNA Substrates
DNA substrates for use in kinetic studies were prepared as described previously (23). Sequences of DNA substrates are listed in Table 2.
Table 2. DNA Substrates.
DNA substrates used for each assay are listed. Substrates are shown as annealed product with the nucleotides in the gap underlined.
| DNA Substrate | Assay | Sequence |
|---|---|---|
| 45AG-U22-D22a | Presteady-state burst | 5′ GCCTCGCAGCCGTCCAACCAAC CAACCTCGATCCAATGCCGTCC 3′ CGGAGCGTCGGCAGGTTGGTTGAGTTGGAGCTAGGTTACGGCAGG |
| CIIb | Missing Base Primer Extension | 5′ TTGCGACTTATCAACGCCCACA AGITGTCTTCTCAGTCCT 3′AACGCTGAATAGTTGCGGGTGTAGTCATCAACAGAAGAGTCAGGA |
| CIIGc | One at a Time Primer Extension | 5′ TTGCGACTTATCAACGCCCACA AGITGTCTTCTCAGTCCT 3′ AACGCTGAATAGTTGCGGGTGTGGTCATCAACAGAAGAGTCAGGA |
| 45GG-U22-D22d | Single Turnover Misincorporation | 5′ GCCTCGCAGCCGTCCAACCAAC CAACCTCGATCCAATGCCGTCC 3′ CGGAGCGTCGGCAGGTTGGTTGGGTTGGAGCTAGGTTACGGCAGG |
The 45AG-U22-D22 substrate contains the 45 nucleotide 45AG template oligo with template A in the 1-bp gap annealed to the 22 nucleotidue U22 upstream oligo and the 22 nucleotide D22 downstream oligo.
The CII substrate contains the 45 nucleotide CIIT template oligo annealed to the 22 nucleotide CIIU upstream primer oligo and the 18 nucleotide CIID downstream oligo.
The CIIG substrate contains the same CIIU upstream primer and CIID downstream oligos used in the CII substrate annealed to the CIITG template oligo, which substitutes a template G for the first template A in the 5-bp gap found in the CII substrate.
The 45GG-U22-D22 substrate contains the U22 upstream primer oligo and the D22 downstream oligo used in the 45AG-U22-D22 substrate annealed to the 45GG template oligo, which substitutes a template G in the 1-bp gap compared to the template A in the 45AG-U22-D22 substrate.
Protein Expression and Purification
Rat WT and I174S pol β proteins were expressed in E. coli and purified using fast protein liquid chromatography as described previously (30).
Presteady-State Burst Assay
Presteady-state kinetic assays were conducted as described previously (30). In short, pol β bound to 1-bp gapped DNA substrate (100nM enzyme:300 nM DNA) is mixed with the correct dNTP and MgCl2 in a rapid quench apparatus at 37°C, followed by reaction quenching with EDTA on a millisecond timescale. The single base-pair extended product is then separated from unextended primer using denaturing 20% acrylamide gel electrophoresis, visualized and quantified using a Storm 860 Phosphorimager and ImageQuant software. Extended product at each time point is plotted and fit to the full burst equation where [E]App is the apparent enzyme concentration, which is proportional to the burst amplitude, k2 is the rate of product formation and k3 is the rate of product release.
Circular Dichroism
Circular dichroism wavelength scans were conducted with 8 μM wild type and I174S proteins in 10 mM K2HPO4 (pH 8.0). Scans were run in crystal cuvettes with 0.1 cm path length thermostated at 23°C in a Chirascan™ Circular Dichroism Spectrometer (Applied Photosystems). Ellipticity was measured in 0.5 nm steps from 190 – 260 nm. Three measurements were taken for each enzyme.
In Vitro Mutagenesis Assays
Primer extension assays were conducted as described previously (30). In short, an excess of enzyme (750 nM WT and 1500 nM I174S) bound to 50 nM 5-bp gapped DNA was mixed with various dNTP/MgCl2 mixes. Reactions were run for five minutes at 37°C and quenched by adding EDTA and 90% formamide gel loading buffer. Reaction products were separated on a sequencing gel, visualized and quantified using a Storm 860 Phosphorimager and ImageQuant software. For these assays, enzyme concentrations were based on the activity of the protein preparations derived from the presteady-state burst experiments, which is proportional to the apparent enzyme concentration of the burst equation. As the active fraction of I174S was about half that of WT, the standard concentration of enzyme was used for WT (750 nM, 15:1 enzyme:DNA ratio) and twice the amount of enzyme was used for I174S (1500 nM). The One at a Time Primer Extension assays used either 12.5 μM, 25 μM, or 50 μM of only one dNTP per reaction mixture.
Single Turnover Kinetic Assay
Single turnover misincorporation assays were conducted as described previously (23). Briefly, 1-bp gapped DNA (Table 2, 45GG-U22-D22) was mixed with 750 nM enzyme so >90% of DNA was bound. DNA binding was determined based on an apparent KD(DNA) of 2 nM for both WT and I174S obtained from gel electromobility shift assay results as in (30) (data not shown). Reactions were initiated by mixing various concentrations of dNTP with the other reaction components and incubated at 37°C. In cases where the incorrect dNTP was added, reactions were incubated up to 45 minutes and samples were collected and quenched at multiple time points. For reactions where the correct dNTP was provided, reactions were run using a KinTek rapid quench machine for times up to 10 seconds. All reactions were quenched by adding EDTA and products were analyzed as described for the Presteady-State Burst Assay. Concentration of extended product was then plotted against time for each reaction mixture and the data was fit to a single exponential curve yielding an observed rate constant (kobs): [product] = A(1 − ekobst). The kobs values for each dNTP/enzyme combination were then plotted against the [dNTP] and these data were fit to a hyperbolic equation: . The rate of polymerization (kpol) and the apparent dNTP binding constant (Kd) was determined for each enzyme (WT and I174S) for the insertion of each dNTP in the given 1-bp gapped sequence (Table 2, 45GG-U22-D22).
Molecular Modeling and Molecular Dynamics Simulations
Models of native and mutant pol β were generated using a high-resolution DNA co-crystal structure (PDB code 2fms). Mutations at position 174 from Ile to Ser, Thr and Asp were introduced and modeled using the program VMD (31). The respective models were subjected to periodic-boundary molecular dynamics simulations performed for 10 ns using the Non-equilibrium Atomic Molecular Dynamics 2 (NAMD2) simulation package version 2.6 (32). The Charmm27 force field was used to parametrize intramolecular interactions with long-range non-bonding terms calculated up to a 12 Å cutoff for electrostatic and van der Waals interactions. All hydrogen bond lengths were held constant with the SHAKE-RATTLE-ROLL algorithm. The TIP3P model was employed for description of the water molecules. The time step for all simulations was set to 2 fs. The simulation temperature was maintained at 310 K using a temperature bath with a coupling constant of 5 ps-1. The lengths of the simulations were determined by the proper convergence of the monitored properties (typically for a period of 10 ns simulations). Trajectory analysis and molecular graphics images were generated using VMD. Root mean-square deviation analyses were performed to evaluate systems mobility and proper convergence (data not shown). Ramachandran maps, side-chain dihedral analysis, and inter-residue distance scatter plots were generated to monitor and document changes in the local structure (see Fig 1) around S174/T184/D174, T176, K62, Y265, Y266, V269, Q264 and I260 over time.
Results
I174S, T, D and G are Mutator Mutants
The tryptophan reversion assay is a genetic screen used to identify mutator mutants of pol β in vivo. Our study identified four amino acid substitution mutants at I174 that result in a mutator polymerase: serine, threonine, aspartic acid and glycine. All of these mutants showed an approximately 40-fold increase in mutation frequency compared to wild type (Fig 2). The threonine mutant showed inconsistent results, as exemplified by the large error bars, with the majority of replicates showing a mutator phenotype. This is probably due to a decrease in the stability of the I174T mutant protein. The serine mutant was chosen for further analysis due to its consistent results across multiple replicates.
Figure 2. I174S, T, D and G are mutator mutants.
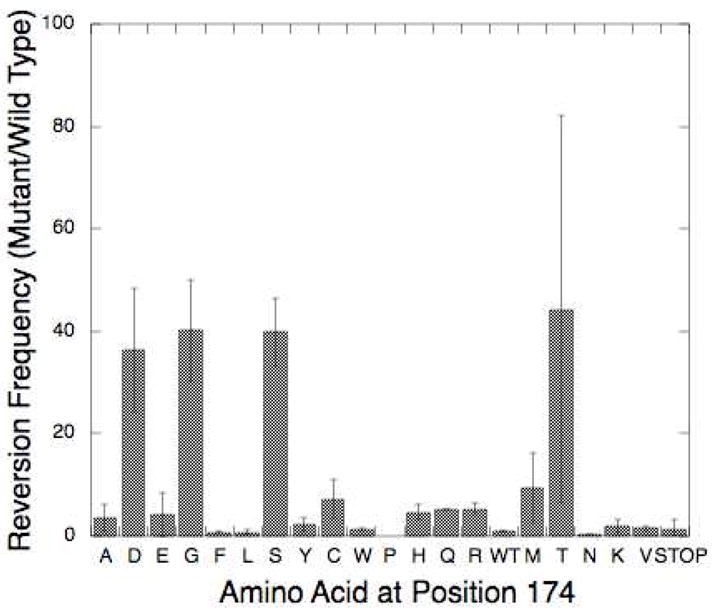
Results of the Trp+ Reversion Assay. I174 mutants are listed across the x-axis and mean mutant reversion frequency divided by the WT reversion frequency (± standard deviation) is graphed on the y-axis.
I174S and WT Share Similar Biochemical Characteristics and Protein Folding
To determine if the I174S mutant is active in vitro, we purified the His-tagged protein from E. coli and used the protein in a presteady-state kinetic assay, as described in Materials and Methods. This assay is used to determine both if the protein is active and if product release is slower than nucleotide incorporation, as it is for WT pol β. Our results show that the I174S mutant is an active polymerase (kobs = 11±3 s−1 and 8±2 s−1; kss = 2 s−1 and 1 s−1; WT and I174S respectively) that displays a biphasic burst of product formation like WT pol β (Fig 3A, inset), with a lower fraction of activity in the purified protein (Fig 3A, burst amplitude = 35% and 17%, WT and I174S respectively).
Figure 3. I174S and WT pol β have similar kinetic pathways and protein folding.

A. Results from a presteady-state kinetic burst assay. The inset shows a close-up of the results for I174S, illustrating that I174S has a biphasic burst of product formation similar to WT. The burst rates are 11±3 s−1 and 8±2 s−1 for WT and I174S, respectively. These results are representative of multiple trials using two different protein preparations conducted at 37°C.
B. Results from the circular dichroism assay.
To determine if the I174S mutant has the same overall folding pattern as WT pol β, we used a circular dichroism assay. This assay showed that there are no gross structural defects in the I174S protein when compared to wild type (Fig 3B).
I174S is a Mutator Mutant In Vitro
To confirm the mutator phenotype of I174S identified by the in vivo Trp+ reversion assay, we used an in vitro gap-filling primer extension assay utilizing one dNTP at a time to determine if the decreased fidelity of the I174S mutant was due primarily to misincorporation events, mispair extension, or a combination of the two. In this assay only a single nucleotide is provided for the polymerase reactions. If the nucleotide is incorporated opposite an incorrect templating base but the mispair is not extended, then a single band should be observed at the length of the misincorporation product (for example, Fig 4A lane 26). In this case the polymerase is likely a misincorporation mutator. If there are multiple bands past the band for the last correct incorporation, then the mutant is likely both a misincorporator and a mispair extender (for example, Fig 4A lane 14). If no bands are observed beyond the band representing the last correct product, then the polymerase mutant does not misincorporate. If all nucleotides are provided, pol β will conduct strand-displacement synthesis and continue extending the primer beyond the 5-bp gap (Fig 4A, lanes 3–8). Results from this assay show that I174S both misincorporates and extends mispairs when inserting a pyrimidine opposite a template G (dTTP) or T (dCTP) (Fig 4A, lanes 9–20) and misincorporates but does not extend mispairs when incorporating a purine opposite template G (Fig 4A, lanes 21–32). This misincorporation and mispair extension by I174S is greater than that observed for WT pol β. For instance, for the reactions using dTTP, 85% of the total I174S reaction product is a result of misinsertion or mispair extension, whereas only 25% of the WT reaction product is due to these events (Fig 4B, quantification of lanes 13 and 14). For the case of misinserting a purine opposite template G, 25% of the I174S reaction product is due to misinsertion compared to only 5% of the WT reaction product (Fig 4B, quantification of lanes 31 and 32). Given these results, we decided to investigate the mechanism underlying the I174S mutant’s ability to misincorporate opposite template G using single turnover kinetics.
Figure 4. I174S has a mutator phenotype in vitro.

A. Results from the One at a Time Primer Extension assay. Reactions were run for 5 minutes at 37°C. Odd numbered lanes show the product from reactions using WT pol β, and even numbered lanes show product from I174S reactions. Nucleotides included at varying concentrations are listed along the bottom of the gel image, while the templating bases corresponding to each band of extended product are shown along the side. P stands for the band corresponding to unextended primer. B. Quantitation of the One at at Time Primer Extension assay. Bands for reactions with the highest concentration of dNTP (50 μM) corresponding to the incorrect incorporation product from the one at a time primer extension assay (Panel A) were quantitated and presented as a fraction of total product for each reaction condition. For reactions in which dTTP, dATP, or dGTP were provided, all product longer than the primer was considered incorrect. For the reactions with dCTP incorporation, bands corresponding to insertion opposite the first two template Gs were considered correct incorporation product, and all additional bands were considered incorrect incorporation product.
I174S Lacks Discrimination at the Level of dNTP Binding
To determine which step is altered in I174S that permits it to misincorporate opposite template G more efficiently than WT, we used single turnover kinetics. This permits us to determine the rate of polymerization (kpol) and the dNTP binding affinity (Kd) for both WT and I174S when filling in a one-bp gap with G as a templating base (Fig 5).
Figure 5. Single turnover misincorporation opposite a template G.

Representative results of a single turnover misincorporation experiment, in this case the incorporation of dTTP opposite template G by WT. A. For each reaction run with varying concentrations of dNTP at 37°C, extended product was graphed against time and fit to a single exponential curve to obtain an observed rate constant. B. The observed rate constants were plotted against the corresponding concentration of dNTP ± standard error of the fit) and fit to a hyperbolic equation to yield the rates of polymerization and the dNTP dissociation constant for these conditions (see Table 3). Results shown are representative of the patterns of curves obtained for both WT and I174S incorporating all four dNTPs opposite a template G in a one base-pair gap.
Using this assay, we found that I174S has a 24-fold decrease in fidelity for misinsertion of dTTP (incorrect) opposite a template G when compared to WT pol β, as shown in Table 3. Although the rates of polymerization (kpol) are approximately the same for both enzymes under these conditions, I174S binds the correct nucleotide (dCTP) less tightly than wild type while binding the incorrect dTTP more tightly, leading to a greater than ten-fold loss in discrimination at the level of ground-state dNTP binding (Table 3).
Table 3.
Single turnover kinetic misincorporation constants opposite template G
| dNTP | Enzyme | kpol (s−1) | Kd (μM) | kpolc/kpoli | Kdi/Kdc | Efficiency(μM−1s−1) | Fidelity | Fold Change in Fidelity (WT/I174S) |
|---|---|---|---|---|---|---|---|---|
| dCTP | WT | 8.9 ± 0.5 | 9 ± 2 | 1.0 | ||||
| I174S | 6.7 ± 0.7 | 23 ± 6 | 2.9E-01 | |||||
| dTTP | WT | 0.1l5 ± 0.008 | 590 ± 70 | 78 | 66 | 1.9E-04 | 5,155 | 24 |
| I174S | 0.121 ± 0.007 | 90 ± 20 | 56 | 4 | 1.3E-03 | 217 | ||
| dATP | WT | 0.052 ± 0.002 | 72 ± 9 | 171 | 8 | 7.3E-04 | 1,376 | 13 |
| I174S | 0.093 ± 0.005 | 34 ± 6 | 73 | 1 | 2.8E-03 | 107 | ||
| dGTP | WT | 0.0094 ± 0.0004 | 120 ± 10 | 954 | 14 | 7.9E-05 | 12,879 | 11 |
| I174S | 0.022 ± 0.001 | 90 ± 10 | 313 | 4 | 2.5E-04 | 1,220 | ||
Kinetic constants obtained from single turnover misincorporation assays are listed for each enzyme (± standard error). Efficiency was calculated by dividing kpol by Kd for each condition. Fidelity was calculated as follows: [(kpol/Kd)c + (kpol|/Kd)i]/[(kpol/Kd)i], where c and i represent the correct and incorrect dNTPs, respectively.
In the case of misinserting a purine opposite a purine (in this case, dATP and dGTP opposite template G), most of the selectivity of both WT and I174S is due to discrimination at the level of kpol (Table 3). However, for the dATP opposite template G misinsertion, the discrimination at the level of kpol is approximately equal for both WT and I174S, and the 13-fold loss of fidelity by I174S likely results from the 8-fold loss of discrimination during dNTP binding. In the case of misinserting dGTP opposite G, the 11-fold loss of fidelity by I174S is likely due to loss of discrimination both at the level of kpol and during dNTP binding (Table 3). Overall, these results suggest that residue I174 is important for pol β fidelity, especially at the level of ground state dNTP binding.
Molecular Dynamics Analysis Reveals a Tightening of the Hinge upon Mutation of I174
Two parallel molecular dynamics (MD) simulations were carried out each with two likely serine and aspartate rotamers in the context of position 174. In the first run, the serine hydroxyl or aspartate carboxylate were facing the solvent, and in a second simulation the side chain rotamers of the functional groups were chosen facing other residues in the hinge region (away from the bulk solvent). We analyzed all trajectories in detail but chose the second scenario as the far more convincing run as far as side chain interactions and conformational changes in the surrounding local structure of the protein were concerned. For reference, the wild type pol β structure (2fms) was subjected to the same protocol.
The key players involved in the local network surrounding position 174 are residues S174, T176, K62, Y265, Y266, V269, Q264 and I260 (see Fig 1). From distance analyses of these residues, it was observed that in the serine mutant, the hydroxyl group interacts with K262 and pulls the lysine toward 174. As compared to WT the 174–262 distance in the S174 simulation are significantly reduced (Fig 6A). This is accompanied by a decrease in distance between S174 and Y265, drawing them both closer. This is also supported by the fact that the distances between T176 and Y265 are considerably shorter compared to the WT (Fig 6A). A similar situation is found in the case of D174 (data not shown). Overall, this network of polar interactions seems to lead to a tightening of the entire hinge region.
Figure 6. Analysis of local structural changes in the hydrophobic hinge region during a 10 ns molecular dynamics simulation.

A. Scatter plot of inter-residue distances showing Ser174 and Lys262 on the x-axis and Ser174 and Tyr265 along the y-axis. Note the overall shortening of distances and the tightening of contacts in the I174S mutant (red) simulation as compared to the pol μ wild type trajectory (blue). The network of both polar and hydrophobic interactions around the Ser174 side chain actually involves residues T176, T196, I260, K262, Y265, Y266, and V269. B. The backbone torsion angles of residue Q264 show a bimodal distribution of Phi/Psi angles, a prominent difference compared to the 174S enzyme. Changes in the peptide backbone dihedrals of hydrophobic hinge residues could affect the specific flexibility of this important region and, possibly, change the geometry of the dNTP binding pocket. C. Distances between residues, Y271 and F272, and the catalytic aspartate residues, D192 and D256, plotted along the 10ns MD trajectory. The distances between Y272 and D256 (top lines) and F272 and D192 (bottom) indicate that the pol μ fingers domain in the wild type calculations (dashed blue lines) has a tendency to close more tightly towards the active site than in the I174S simulation (solid red lines). The tightening of side-chain/side-chain interactions in the I174S mutant (see Fig. 6A) appears to result in a slightly more open, perhaps less flexible fingers domain as compared to the wild type structure.
Upon mutation of I174 to serine, there is also a marked difference in the side chain and backbone dihedrals of T176 and V178 as compared to WT. Consequently, residues I260, Y266 and V269 show slight decreases in distance from Y265 as they move closer. Analysis of Ramachandran plots for residues 260–265, which are hinge residues adjacent to position 174 (See Fig 1), indicates that residue Q264 (and Y265 to some extent) show a bimodal distribution of Phi/Psi angles, a prominent difference compared to the WT (Fig 6B). Interestingly, however, residues 173–179 do not show any significant difference in their Ramachandran plots as compared to the WT (data not shown). Further analysis of residues lining the inside of the hinge (helix I and surrounding loops) shows that during the molecular dynamics calculations of wild type pol β, residues Y271 and F272 both move closer towards D192 and D256, while in the case of the mutants a subtle increase of the distances is observed (Fig 6C). The concerted shifts of these bulky side chains, which are directly adjacent to the incoming nucleotide, cause subtle changes in the shape and a small increase in the volume of the dNTP binding pocket of the mutant polymerase as compared to the wild type enzyme.
Discussion
Base excision repair is a DNA repair pathway critical to the maintenance of genomic integrity. DNA polymerase β acts as a key gap-filling enzyme in BER, and alterations in the polymerase fidelity could lead to increased mutagenesis and eventually cancer. Previous work by our lab has suggested that certain residues of the hydrophobic hinge region of pol β are critical for accurate nucleotide insertion (21–23, 26, 28, 29), thus implicating the hinge region in nucleotide discrimination. Because the hinge is a structural subdomain of Pol β, we wanted to study the roles of each of its residues in nucleotide discrimination. To study the role of hinge residue I174 in pol β fidelity, we began by altering this residue to every other amino acid and screening these mutants for in vivo mutator activity. This screen identified I174S, T, D and G amino acid substitution variants as mutator mutants. We then showed that the loss of fidelity in the I174S mutant is primarily due to loss of discrimination at the level of ground state dNTP binding, especially for misinsertion of dTTP opposite a template base G. These results, in combination with our previous work, suggest that the hinge plays an important role in nucleotide discrimination at the level of ground state binding.
It is not unusual for single amino acid substitution variants to affect pol β fidelity through altering dNTP binding. Previous studies have identified other residues that show a similar effect. These residues include Y271, which is located in the dNTP binding pocket, whose phenylalanine or serine mutants have aberrant dNTP binding (33). Studies involving the alanine mutant of R283, a residue that has been shown to stabilize the template DNA which is part of the dNTP binding pocket (16, 34), also show reduced fidelity caused by loss of discrimination during both ground state dNTP binding and the steps reflected by the single turnover kinetic constant kpol (35). Finally, two residues that bind the triphosphate moiety, R183 and R149, when mutated to alanine result in variants with decreased fidelity due to loss of selectivity during ground state dNTP binding (36).
In addition to residues that occur in the dNTP binding pocket or that directly interact with the incoming dNTP, more distant residues of the hinge region have been shown to contribute to pol β fidelity by influencing dNTP binding. Mutator variants of two residues of the inner lining of the hinge were previously identified, I260Q and F272L (21, 22). The F272L variant displayed a moderate mutator phenotype, misincorporating a dGTP opposite a template base A more readily than WT due to a 3× loss of discrimination during dNTP binding (Table 4). The I260Q variant is a strong misincorporator, with misincorporations opposite templates A and C occurring due to 12–25 fold loss of discrimination during ground state dNTP binding compared to WT pol β, depending on the exact misincorporation event studied (Table 4). These results, together with our work analyzing the I174S variant, strongly suggest that the hydrophobic hinge contributes to polymerase fidelity through monitoring dNTP binding.
Table 4.
Comparison of hinge mutant fidelity
| Change in Discrimination(WT/Mutant) |
||||
|---|---|---|---|---|
| Hinge Mutant | Residue Location | Misincorporation Event | at kpol | at Kd(dNTP) |
| I260Q (23) | Inner Lining | A: dATP | 1.0 | 23 |
| A:dGTP | 1.2 | 16 | ||
| A:dCTP | 1.8 | 11 | ||
| C:dATP | 1.2 | 18 | ||
| C:dTTP | 1.3 | 12 | ||
| C:dCTP | 1.4 | 25 | ||
| F272L (21) | Inner Lining, dNTP Binding Pocket | A:dGTP | 1.1 | 3.0 |
Comparison of single turnover kinetic constants for hinge variants that misincorporate more than WT pol beta. Values for discrimination at kpol and Kd(dNTP) were taken from the referenced papers, and the ratio of discrimination for WT: Mutant was calculated.
The loss of discrimination during ground state binding observed for the I174S mutant suggests that the mutation of a bulky hydrophobic amino acid to a smaller polar residue results in the alteration of the shape or the size of the dNTP binding pocket. The fact that a small charged residue, aspartic acid, resulted in a mutator polymerase while substitution with a large polar residue, glutamic acid, did not suggests that the size of the side chain at position 174 is important for fidelity. Molecular dynamics simulations provide evidence for a mechanism by which the mutation of a hinge residue could impact the configuration of the dNTP binding pocket by showing that mutating I174 to serine or aspartate in silico coincides with a marked increase mainly in polar but also van der Waals interactions. These interactions cause an overall tightening of the entire network of residues consisting of S174, T176, K62, Y265, Y266, and V269.
The hydrophobic hinge is a dynamic structure that undergoes a conformational change in the course of binding dNTP (16). Molecular dynamics simulations reveal that along with the tightening, or altered positioning, of the surface hinge residues upon mutation of I174 to serine or aspartate, the movement, or dynamics, of interior residues relative to the palm subdomain is altered as well. Namely, the side chains of Y271 and F272, which both line the DNA binding cleft and interact with the incoming nucleotide, have decreased mobility in the 174S and D simulations relative to that observed in the WT simulation. This alteration of the subtle movements of the Y271 and F272 side-chains caused by the tightening of the surface residues could directly alter the size and the shape of the dNTP binding pocket and may, therefore, allow non-cognate dNTPs to bind more readily to the mutant active site.
A similar situation was suggested to occur in the case of the I260Q and I260M mutator hinge mutants (12, 23). A small cavity adjacent to I260 was shown to disappear upon mutation to glutamine or methionine. This small cavity is thought to accommodate local motions that occur upon complete closure of the fingers subdomain, and without this pocket the hinge region may no longer be capable of completely closing the fingers subdomain around the nascent base pair. Consequently, the size and shape of the dNTP pocket would be altered, presumably leading to a change in the ground state binding of non-cognate nucleotides. The combined analysis of aberrant fidelity in hinge mutants suggests a common underlying structural principle that lowered fidelity in pol β stems from small but significant restrictions in the movement of the fingers subdomain. Despite the differences in cause (mutators I174S or I260Q), the net effect is the same: lowered discrimination during ground state binding and accommodation of non-Watson-Crick base pairs in the active site of mutator mutants.
Understanding the impact of residues distant from the polymerase active site on polymerase fidelity is crucial for determining how polymerases maintain genomic stability on a regular basis. Additionally, this structural knowledge can provide insight into the role of tumor-associated mutants with substitutions found in well-characterized regions. For instance, the I260M prostate cancer associated hinge mutant was shown to be a sequence-specific mutator (12), a finding consistent with the role of the hydrophobic hinge in the maintenance of pol β fidelity. With the knowledge of how each structure in a polymerase contributes to its activity and fidelity, we can understand how a normal polymerase maintains genomic stability and how a mutated polymerase contributes to a cancer phenotype.
Acknowledgments
This work was supported by NIH grant CA80830 (to J.B.S.).
References
- 1.Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–3239. [PubMed] [Google Scholar]
- 2.Venkatesan RN, Bielas JH, Loeb LA. Generation of mutator mutants during carcinogenesis. DNA Repair (Amst) 2006;5:294–302. doi: 10.1016/j.dnarep.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 3.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 4.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase Beta. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 5.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) Embo J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 7.Dianov GL, Prasad R, Wilson SH, Bohr VA. Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J Biol Chem. 1999;274:13741–13743. doi: 10.1074/jbc.274.20.13741. [DOI] [PubMed] [Google Scholar]
- 8.Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000;275:4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- 9.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 11.Lang T, Maitra M, Starcevic D, Li SX, Sweasy JB. A DNA polymerase beta mutant from colon cancer cells induces mutations. Proc Natl Acad Sci U S A. 2004;101:6074–6079. doi: 10.1073/pnas.0308571101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalal S, Hile S, Eckert KA, Sun KW, Starcevic D, Sweasy JB. Prostate-cancer-associated I260M variant of DNA polymerase beta is a sequence-specific mutator. Biochemistry. 2005;44:15664–15673. doi: 10.1021/bi051179z. [DOI] [PubMed] [Google Scholar]
- 13.Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweasy JB, Lang T, Starcevic D, Sun KW, Lai CC, Dimaio D, Dalal S. Expression of DNA polymerase {beta} cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci U S A. 2005;102:14350–14355. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell Cycle. 2006;5:250–259. doi: 10.4161/cc.5.3.2414. [DOI] [PubMed] [Google Scholar]
- 16.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 17.Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J. Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP. Science. 1994;264:1891–1903. [PubMed] [Google Scholar]
- 18.Sawaya MR, Pelletier H, Kumar A, Wilson SH, Kraut J. Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism. Science. 1994;264:1930–1935. doi: 10.1126/science.7516581. [DOI] [PubMed] [Google Scholar]
- 19.Sweasy JB, Yoon MS. Characterization of DNA polymerase beta mutants with amino acid substitutions located in the C-terminal portion of the enzyme. Mol Gen Genet. 1995;248:217–224. doi: 10.1007/BF02190803. [DOI] [PubMed] [Google Scholar]
- 20.Washington SL, Yoon MS, Chagovetz AM, Li SX, Clairmont CA, Preston BD, Eckert KA, Sweasy JB. A genetic system to identify DNA polymerase beta mutator mutants. Proc Natl Acad Sci U S A. 1997;94:1321–1326. doi: 10.1073/pnas.94.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li SX, Vaccaro JA, Sweasy JB. Involvement of phenylalanine 272 of DNA polymerase beta in discriminating between correct and incorrect deoxynucleoside triphosphates. Biochemistry. 1999;38:4800–4808. doi: 10.1021/bi9827058. [DOI] [PubMed] [Google Scholar]
- 22.Starcevic D, Dalal S, Sweasy J. Hinge residue Ile260 of DNA polymerase beta is important for enzyme activity and fidelity. Biochemistry. 2005;44:3775–3784. doi: 10.1021/bi047956x. [DOI] [PubMed] [Google Scholar]
- 23.Starcevic D, Dalal S, Jaeger J, Sweasy JB. The hydrophobic hinge region of rat DNA polymerase beta is critical for substrate binding pocket geometry. J Biol Chem. 2005;280:28388–28393. doi: 10.1074/jbc.M502178200. [DOI] [PubMed] [Google Scholar]
- 24.Dalal S, Starcevic D, Jaeger J, Sweasy JB. The I260Q variant of DNA polymerase beta extends mispaired primer termini due to its increased affinity for deoxynucleotide triphosphate substrates. Biochemistry. 2008;47:12118–12125. doi: 10.1021/bi8011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah AM, Maitra M, Sweasy JB. Variants of DNA polymerase Beta extend mispaired DNA due to increased affinity for nucleotide substrate. Biochemistry. 2003;42:10709–10717. doi: 10.1021/bi034885d. [DOI] [PubMed] [Google Scholar]
- 26.Clairmont CA, Narayanan L, Sun KW, Glazer PM, Sweasy JB. The Tyr-265-to-Cys mutator mutant of DNA polymerase beta induces a mutator phenotype in mouse LN12 cells. Proc Natl Acad Sci U S A. 1999;96:9580–9585. doi: 10.1073/pnas.96.17.9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Opresko PL, Shiman R, Eckert KA. Hydrophobic interactions in the hinge domain of DNA polymerase beta are important but not sufficient for maintaining fidelity of DNA synthesis. Biochemistry. 2000;39:11399–11407. doi: 10.1021/bi000698t. [DOI] [PubMed] [Google Scholar]
- 28.Opresko PL, Sweasy JB, Eckert KA. The mutator form of polymerase beta with amino acid substitution at tyrosine 265 in the hinge region displays an increase in both base substitution and frame shift errors. Biochemistry. 1998;37:2111–2119. doi: 10.1021/bi9722711. [DOI] [PubMed] [Google Scholar]
- 29.Shah AM, Li SX, Anderson KS, Sweasy JB. Y265H mutator mutant of DNA polymerase beta. Proper teometric alignment is critical for fidelity. J Biol Chem. 2001;276:10824–10831. doi: 10.1074/jbc.M008680200. [DOI] [PubMed] [Google Scholar]
- 30.Murphy DL, Kosa J, Jaeger J, Sweasy JB. The Asp285 variant of DNA polymerase beta extends mispaired primer termini via increased nucleotide binding. Biochemistry. 2008;47:8048–8057. doi: 10.1021/bi702104y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 32.Kale L, Bhandarkar RSM, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: Greater scalability for parallel molecular dynamics. Journal of Computational Physics. 1999;151:283–312. [Google Scholar]
- 33.Kraynov VS, Werneburg BG, Zhong X, Lee H, Ahn J, Tsai MD. DNA polymerase beta: analysis of the contributions of tyrosine-271 and asparagine-279 to substrate specificity and fidelity of DNA replication by pre-steady-state kinetics. Biochem J. 1997;323:103–111. doi: 10.1042/bj3230103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Werneburg BG, Ahn J, Zhong X, Hondal RJ, Kraynov VS, Tsai MD. DNA polymerase beta: pre-steady-state kinetic analysis and roles of arginine-283 in catalysis and fidelity. Biochemistry. 1996;35:7041–7050. doi: 10.1021/bi9527202. [DOI] [PubMed] [Google Scholar]
- 35.Ahn J, Werneburg BG, Tsai MD. DNA polymerase beta: structure-fidelity relationship from Pre-steady-state kinetic analyses of all possible correct and incorrect base pairs for wild type and R283A mutant. Biochemistry. 1997;36:1100–1107. doi: 10.1021/bi961653o. [DOI] [PubMed] [Google Scholar]
- 36.Kraynov VS, Showalter AK, Liu J, Zhong X, Tsai MD. DNA polymerase beta: contributions of template-positioning and dNTP triphosphate-binding residues to catalysis and fidelity. Biochemistry. 2000;39:16008–16015. doi: 10.1021/bi0008480. [DOI] [PubMed] [Google Scholar]