

Abstract
Cytomegaloviruses (CMVs) are ubiquitous species-specific viruses that establish acute, persistent, and latent infections. Both human and mouse CMVs encode proteins that inhibit the activation of natural killer (NK) cells by downregulating cellular ligands for the NK cell activating receptor, NKG2D. The MCMV glycoprotein m152/gp40 downregulates the surface expression of RAE-1 in order to avoid NK cell control in vivo. So far it is unclear if there is a direct interaction between m152 and RAE-1, and if so, if m152 interacts differentially with the five identified RAE-1 isoforms, which are expressed as two groups in MCMV-susceptible or resistant mouse strains. To address these questions, we expressed and purified the extracellular domains of RAE-1 and m152, and performed size exclusion chromatography binding assays as well as analytical ultracentrifugation and isothermal titration calorimetry to characterize these interactions quantitatively. We further evaluated the role of full-length and naturally glycosylated m152 and RAE-1 in cotransfected HEK293T cells. Our results confirmed that m152 binds RAE-1 directly, relatively tightly (Kd < 5 μM), and with 1:1 stoichiometry. The binding is quantitatively different depending on particular RAE-1 isoforms, corresponding to the susceptibility to downregulation by m152. A PLWY motif found in RAE-1β, although contributing to its affinity for m152, does not influence the affinity of RAE-1 γ or δ, suggesting that other differences contribute to the RAE-1/m152 interaction. Molecular modeling of the different RAE-1 isoforms suggests a potential site for the m152 interaction.
Cytomegaloviruses (CMVs), ubiquitous species-specific large DNA viruses, possess a remarkable array of immunomodulatory mechanisms to permit evasion of the host's immune response (1, 2). The immunocompetent host employs both innate and adaptive arms of the immune system, exemplified by the natural killer (NK) cell and CD8+ T cell responses respectively, to control both acute infection and reactivation of latent infection (1, 3, 4). Accordingly, both mouse and human CMV (MCMV and HCMV) employ genes to avoid recognition of infected cells by NK and CD8+ T cells (5-7). NK cells effectively kill CMV-infected targets via NK activating receptors, among which NKG2D is one of the most potent and best studied. Ligands for NKG2D include, in the mouse, three groups of stress-induced MHC-I-like cell surface molecules known as MULT-1, RAE-1, and H60 (8), and in the human, the MHC class I-related chains A and B (MICA and MICB) and a family of UL16 binding and retinoic acid early transcript 1 proteins (ULBP1-ULBP4, RAET1E and RAET1G)(9). NKG2D is found on a variety of immune effector cells including NK cells, antigen-experienced CD8+ T cells, γδ T cells, and some activated CD4+ T cells (10-12). Contrasting with the NK cell response, the adaptive cytolytic T cell response to CMV infection is dependent on classical T cell receptor (TCR)-mediated recognition of viral peptides bound to host MHC-I-molecules (5), but recent evidence indicates that NKG2D may also function as a coreceptor for T cell recognition (13). As part of the ongoing co-evolution of virus and host, the CMVs have developed mechanisms to avoid the immune response. Two sets of MCMV genes contribute to viral immunoevasion: those that regulate the expression and recognition of NKG2D ligands and those that modulate recognition by CD8+ T cells. The former include MCMV genes m145, m152, and m155 that functionally impair the expression of MULT-1, RAE-1, and H60, respectively (m145 and m155 work cooperatively with m138, as well (14, 15)). The latter set of MCMV genes include m04 and m06 as well as m152 which perform similar functions with respect to the classical MHC-I antigen-presentation pathway (16-18).
Among the NKG2D ligands targeted by MCMV, of particular interest are the five murine RAE-1 isoforms (α,β,γ,δ, and ε), that are more than 90% identical in protein sequence, are expressed as glycophosphatidyl-inositol (GPI)-linked proteins, and are widely expressed in embryonic and tumor cells but not in adult tissues (19, 20). Of the viral immunoevasins, the m152 glycoprotein (also known as gp40) is remarkable because it counters both the NK cell and CD8+ T cell recognition pathways (17, 21-23), the first by lowering the expression of RAE-1, leading to decreased recognition by NKG2D-bearing NK cells, and the second by downregulation of some host MHC-I molecules, influencing T cell recognition of virus-infected cells (22). Despite efforts to understand the biochemical basis of the downregulation of RAE-1 by m152, direct interaction of m152 with RAE-1 has not been demonstrated (24). Although early studies based on cDNA transfection suggested that all five RAE-1 isoforms were efficiently downregulated at the cell surface by m152 (21), other experiments indicated that RAE-1δ was resistant to the effects of m152-mediated downregulation (24). An amino acid sequence motif in RAE-1α, β, and γ, PLWY, was shown to contribute to differences in the effect of m152 on the different RAE-1 isoforms (24).
To elucidate the biochemical basis of the proposed “cis” interaction of m152 with different RAE-1 isoforms, we have engineered RAE-1 and representative mutants for bacterial expression, and report here studies, both qualitative and quantitative, of the binding of recombinant m152 with this panel of RAE-1 molecules. Analysis by size exclusion chromatography (SEC) and analytical ultracentrifugation (AUC) reveals direct interaction of m152 with several isoforms of RAE-1, exposing quantitative differences. These studies explore the contribution of the PLWY motif of RAE-1 and suggest a competitive relationship between the m152 and NKG2D binding sites on RAE-1.
EXPERIMENTAL PROCEDURES
Plasmids
cDNA encoding RAE-1β was amplified by PCR using sense primer (5′ATTCATGTCGACATGGCCAAGGCAGCAGTGAC) and anti-sense primer (5′GACACGGTCGACTCACATCGCAAATGCAAATGC) from total RNA isolated from NIH3T3 cells (ATCC CRL-1658). RAE-1γ and δ cDNAs were synthesized (Ezbiolab) and cloned into the SalI restriction site of pB45-Neo plasmid (24). Deletion of the PLWY motif of RAE-1β and γ was carried out with the Qiagen XL-II mutagenesis kit according to the manufacturer's instructions using oligonucleotides 5′GATCCTACCCCAGCAGATGAAGCGAAGTGCTTCGTG for RAE-1β and 5′GCTCCTACCCCAGCAGATGAAGCGAAGTGCTTAGTG for RAE-1γ. Insertion of PLWY into RAE-1δ was done by gene synthesis (Ezbiolab). The pIRES-hr-GFP-II mammalian expression vector (Stratagene) encoding N-terminal FLAG-tagged versions of m152 and m153 were generated as described previously (25).
The portions of the cDNA encoding the ectodomains of m152 (residues 1-310) and m153 (residues 1-314) were cloned into the pMT-Bip-V5-His insect expression vector (Invitrogen) as described previously (25) for inducible, secreted expression of proteins in Drosophila S2 cells. The portion of the cDNA encoding the ectodomain of RAE-1 proteins (residues 28-205 of RAE-1β or γ, and residues 28-201 of RAE-1δ, see Fig. 4) were cloned into pET21b (Novagen) between NdeI and HindIII restriction sites for expression as inclusion bodies in E. coli. (The numbering convention for RAE-1 isoforms is based on the initiation methionine of the signal peptide as amino acid 1 (see Fig. 4). Numbering for the X-ray structure of RAE-1 differs by 30 (26)). All final constructs were verified by DNA sequencing.
FIGURE 4. RAE-1 isoforms differ in PLWY motif as well as in charged residues.

Amino acid sequence alignment of RAE-1 isoforms. The amino acid sequences of the extracellular domain of the five RAE-1 isoforms were aligned using ClustalW (45). Secondary structure elements of RAE-1β (PDB: 1JFM, (26)) are shown above the alignment. The PLWY motif is boxed in blue. Cysteine residues involved in the formation of disulfide bonds are shown as green dashed line. Charged residues involved in the formation of potential salt bridges are shown as blue dashed line. Differences in charged residues among RAE-1 isoforms are highlighted as black triangles. Predicted N-linked glycosylation sites are indicated as cyan ovals. The figure was generated with ESPRIPT 2.2 (46).
Protein Expression and Purification
Selection, expression and purification of the ectodomains of m152 and m153 encoded in pMT-Bip-V5-His from Drosophila S2 cells (Invitrogen) were described previously (25). Supernatant collected from S2 cells after induction with 1 mM Cu2+ for 4 to 6 days was dialyzed against PBS before applying to a Ni2+-NTA column (Qiagen) for initial purification, followed by further purification by SEC on a Superdex 75 column (GE Healthcare). (In the literature, the m152 protein is often referred to as m152/gp40. In this paper we will refer to the protein as m152, and the encoding gene as m152). The ectodomains of RAE-1 proteins encoded in pET21b (Novagen) were expressed as inclusion bodies in BL21Rosetta (pLysS) cells (Novagen) using the overnight express autoinduction system (Novagen). Inclusion bodies were subsequently solubilized in guanidine and refolded by dilution into an arginine buffer containing a glutathione redox couple (27). Refolded proteins were further purified on Superdex 75 (GE Healthcare). Protein concentration was determined by BCA protein assay with BSA as standard (Pierce). Similar protocols for E. coli expression, in vitro refolding and purification of RAE-1 isoforms have been used in other laboratories for studies of the interaction of RAE-1 with NKG2D both in binding studies (28) and for crystallization and X-ray structure determination (26). Following refolding, and purification on Superdex 75, all preparations were evaluated for purity by SDS polyacrylamide gel electrophoresis. All RAE-1 isoforms and mutants were also examined for reactivity with a pan RAE-1 monoclonal antibody (clone 186107, R&D Systems) by surface plasmon resonance, and for reactivity with NKG2D-Fc by ELISA confirming that these molecules were properly folded. In addition, data from sedimentation velocity (SV) experiments were analyzed to estimate the degree of purity and the frictional ratios (fr) of each of the preparations used for the AUC studies: m152 showed an fr of 1.35 and was of >90% purity; RAE-1β, fr =1.42, >95% purity; RAE-1β mutant, fr = 1.43, 90% homogeneous; RAE-1γ, fr=1.44, 98% homogeneous; RAE-1γ mutant, fr=1.42, 99% homogeneous; RAE-1δ, fr=1.39, 99% homogeneous; RAE-1δ mutant, fr=1.42, 99% homogeneous. If a fraction of the molecules were unfolded or aggregated, due to the increased translational friction, the sedimentation coefficients would be distinctly lower and the frictional ratio would be >2.0. Typical frictional ratios for most globular hydrated proteins are between 1.3 and 1.5. Such SV AUC analysis is particularly accurate with regard to the size homogeneity of proteins, and serves as a standard for detecting trace oligomeric impurities (29).
Cell lines and Transfectants
HEK293T (ATCC CRL-11268) cells were cultivated in DMEM supplemented with 10% FCS, 50 μM 2-mercaptoethanol, 1X non-essential amino acids (Bio Whittaker), and 50 μg/mL gentamicin. Drosophila S2 cells were cultured in Insect Express medium (Bio Whittaker) supplemented with 50 μg/mL gentamicin. Transient transfection of HEK293T cells was performed using Fugene 6 (Roche) according to the manufacturer's instruction. Briefly, 1.5 μg of pB45-neo encoding RAE-1γ or δ or their PLWY-deletion mutants were mixed with increasing amounts (6, 24, and 96 ng) of either pIRES-hr-GFP III (Invitrogen) alone or pIRES-hr-GFP III encoding m152. The mixture was then added to 5 × 105 HEK293T cells in 2ml growth medium along with Fugene 6 and incubated at 37°C for about 48 hours. Cells were then analyzed for surface RAE-1 expression by flow cytometry.
Flow Cytometry
Cells were incubated with PE-conjugated pan anti-RAE-1 mAb 186107 (R&D Systems), isotype control (eBioscience), or Cy3-conjugated anti-FLAG M2 antibody (Sigma). After three washes with PBS with 0.5% BSA and 0.03% NaN3, samples were examined on a FACSCalibur (Becton Dickinson) and analyzed with FlowJo software (Treestar).
Measurement of Binding by Chromatography, Analytical Ultracentrifugation, Isothermal Titration Calorimetry
Equimolar amounts (~1 nmol, 20 μL of each) of purified RAE-1 and m152 proteins were mixed in TBSE (25 mM Tris HCl, 150 mM NaCl, 10 mM EDTA, pH 7.5) and incubated on ice for 30 min before loading on a Shodex PROTEIN KW-803 HPLC gel filtration chromatography column (Thomson Instruments) equilibrated with TBSE. Elution was carried out at a flow rate of 0.75 mL/min and monitored by absorbance at 280 nm. The same amounts of RAE-1 and m152 proteins or gel filtration protein standards (670, 159, 44, 17, and 1.35 kDa, BioRad) were analyzed separately. Chromatographs were analyzed with 32 Karat software (Beckman-Coulter).
Analytical ultracentrifugation experiments were conducted in a Beckman-Coulter Optima XLI (Indianapolis, IN) with An50-Ti rotors, following the protocols previously described (30). A series of experiments was conducted with samples at different concentrations ranging from 1.2 to 45 μM and molar ratios of 1:1, 2:1, and 1:2 for the experiments with RAE-1β binding to m152, and at a 1:1 molar ratio for studying the interaction of the other molecules with m152. In brief, for sedimentation velocity (SV) experiments, 400 μL of sample was inserted in 12 mm charcoal-filled Epon centerpieces (100 μL was inserted in 3 mm pathlength centerpieces for samples at the highest concentrations), sedimented at 50,000 rpm and 20°C, and concentration profiles were recorded using the interference optical system. The data were analyzed by first calculating a diffusion-deconvoluted sedimentation coefficient distribution c(s) with the software SEDFIT (31), followed by integration of the peaks to determine the weight-average s-value of the mixture. For RAE-1β, additionally the amplitude and s-value of the reaction boundary and the undisturbed boundary were determined. The resulting isotherm data were globally fitted with models for mass action law and the boundary structure predicted by Gilbert-Jenkins theory, as implemented in SEDPHAT and described in detail elsewhere (32). In all analyses, the s-values of the separately studied individual components were used as prior knowledge (3.16 S and 2.08 S for m152 and RAE-1β, respectively). Error analysis was performed with the Monte-Carlo method.
To study the consistency with the results from isothermal titration calorimetry (ITC), a global analysis of the s-value isotherms and ITC titration was performed with SEDPHAT, and fractions of non-participating molecules of either m152 or RAE-1β were included in the model.
Sedimentation equilibrium (SE) experiments were conducted with 180 μL samples of RAE-1β and m152 at concentrations between 1 μM and 15 μM at molar ratios RAE-1β: m152 of 1:1, 1:2 and 2:1. Samples were inserted in Epon centerpieces with 12 mm optical pathlength. For detection, the absorbance optics at wavelengths of 280 nm and 250 nm were used to record concentration profiles sequentially at rotor speeds of 11,000 and 17,000 rpm, at 4°C. At each speed, equilibrium was established by assessing the difference in signal of scans at 6-hour intervals. Concentration gradients from different loading concentrations, molar ratios, and wavelengths were fitted with theoretical models for sedimentation equilibrium of bimolecular interacting systems forming reversible 1:1 complexes, as described (33). Statistical errors of the parameter estimates from the SE analysis were determined with F-statistics and the projection method. Molar extinction coefficients at 280 nm were determined from amino acid composition using the software SEDNTERP (courtesy of Dr. John Philo). Separate SE experiments were conducted in the same rotor and analyzed by global multi-speed and multi-signal analysis with a single-species model to determine the buoyant molar masses of the individual components (Mb = 10844 Da and Mb = 6855 Da for m152 and RAE-1β, respectively, consistent with the expectation based on amino acid composition). (Note that the buoyant molar mass is Mb = M(ν̄*ρ), where the buoyancy reduces the effective molar mass in sedimentation to approximately ¼ of the true molar mass). This also provided the ratios of extinction coefficients at 280 nm, 250 nm, and 230 nm.
m152 (10 μM) was titrated with RAE-1β (100 μM) over 23 injections (4× 2 μL, 19 × 15 μL) at 25°C using a VP-ITC microcalorimeter (GE Healthcare). Experimental samples were prepared by size exclusion chromatography (Superdex 75) and diluted to the indicated concentrations with filtered column running buffer (25 mM Tris, 500 mM NaCl, 10 mM EDTA, pH 7.5). Data analysis was performed using the global analysis software SEDPHAT (34) applying a 1:1 binary interaction model to obtain values for the apparent enthalpy change and binding constant. Error analysis was performed with the Monte-Carlo method. The global analysis of isotherms from SV and ITC was performed with the software SEDPHAT, assuming the absence of a significant temperature dependence of Kd between 20 °C and 25 °C. Global analysis of the isothermal titration calorimetry data along with the SV-AUC data of RAE-1β indicates that no more than 8% of the RAE-1β was incompetent monomer.
Molecular modeling of RAE-1 isoforms and mutants
Starting with the 2.85 Å structure of RAE-1β (PDB ID: 1JFM)(26) and the Clustal W alignment of the five RAE-1 isoforms shown in Fig. 4, residues of RAE-1β (in the first molecule of the asymmetric unit) were substituted by the corresponding residues of RAE-1γ, followed by idealization of geometry in Coot 0.4.1(35). Similar substitutions were made to generate the RAE-1δ model. To model the PLWY deletion of RAE-1δ, which lies between the β1 and β2 strands, the entire loop consisting of residues Lys12 through Tyr22 was deleted, rebuilt initially as poly-Ala, and then individual residues of RAE-1δ were introduced. Electrostatic surface calculations and molecular graphics illustrations were produced with PyMOL (http://www.pymol.org).
RESULTS
m152 binds to RAE-1 directly in vitro
Although several lines of evidence indicate that m152 can downregulate the cell surface expression of RAE-1 in BALB/c mice (21), it remains unclear whether this is due to a direct interaction of these molecules. Indeed, previous efforts in several labs have failed to demonstrate direct interaction by co-immunoprecipitation or immunoprecipitation and blotting (20, 21, 24). To examine whether the lumenal MHC-I-like domain of m152 interacts with the lumenal domain of RAE-1, we engineered and purified the ectodomains of these molecules and measured their interactions in a well-defined biochemical environment. As RAE-1 isoforms are expressed at the cell surface either as RAE-1α, β, or γ in BALB/c (but not C57BL/6) mice, or RAE-1δ and ε in C57BL/6 (but not BALB/c) mice (19, 20), we chose RAE-1β, γ, and δ as representatives of these two groups. By amino acid sequence (see Figure 4) RAE-1β and γ may be considered as representative of members of the family that contain the PLWY motif, and δ represents the deletion of the motif. Previous biosynthetic studies have focused on differences between RAE-1γ and δ (24). The ectodomains of RAE-1β, γ, and δ were expressed in bacteria as insoluble inclusion bodies, and subsequently solubilized, refolded, and purified. The ectodomain of m152 was expressed as a secreted protein and purified from insect cell supernatants. As shown in Figure 1A and B, both RAE-1 and m152 were purified as monomeric proteins.
Figure 1. m152 binds to RAE-1 directly.

A,B, Purification of RAE-1 and m152. The ectodomains of RAE-1 isoforms and of m152 were expressed and purified as described in Materials and Methods. Shown here are the chromatographs of RAE-1β (A, pink) and m152 (B, green) overlaid with the protein molecular weight standards (dashed line). C,D,E, Binding of m152 to RAE-1β, γ, and δ examined by a SEC binding assay. m152 and RAE-1 were mixed in equimolar amounts, incubated for 30 min on ice, analyzed chromatographically (blue), and compared to the profile of individual m152 (green) and RAE-1 proteins (pink) alone. Insets show enlargements of the peak regions. (Flow rate was 0.75 ml/min, so elution volumes may be calculated from the indicated time of elution.)
To assess the interaction of m152 with different RAE-1 isoforms qualitatively, we analyzed mixtures of these molecules by SEC with columns that resolve differences in the required molecular weight range. SEC, although an inherently qualitative rather than quantitative method, permits rapid assessment of molecular interactions with Kd values in the micromolar range (36). RAE-1 and m152 at concentrations of about 50 μM were run on the SEC column either separately or after mixing. A shift of the eluted peak of the mixture to a shorter retention time compared to the peak of RAE-1 or m152 alone revealed the noncovalent direct interaction of the two proteins in solution. As shown in Figure 1C and D, when RAE-1β or γ was mixed with m152, a peak shift corresponding to the formation of a higher molecular weight species (i.e. protein complex) was observed. The peak shift was especially apparent with the mixture of RAE-1γ and m152 (Figure 1D), revealing a distinct peak for the complex, suggesting m152 might bind RAE-1γ with higher affinity. In contrast, when RAE-1δ was mixed with m152 (Figure 1E), the peaks superposed essentially with that of m152 alone, suggesting a significantly weaker interaction between RAE-1δ and m152 undetectable by this method. Together, these results suggested a direct interaction between the ectodomains of RAE-1β and γ and that of m152, prompting further analysis of their interactions (as well as that of RAE-1δ) by other definitive and quantitative means.
m152 binds to different RAE-1 isoforms with 1:1 stoichiometry but with differing affinity
Although SEC experiments suggested that m152 could interact with at least some isoforms of RAE-1, this is not a particularly quantitative technique. Therefore, to characterize quantitatively the interaction between RAE-1 and m152, SV and SE AUC experiments were performed for RAE-1β and m152 under conditions similar to those used in the binding experiment on the SEC column (Figure 2). A range of concentrations covering the expected binding isotherm, as well as different ratios of RAE-1β and m152 proteins, were examined at 20°C. Figure 2A shows the sedimentation coefficient distribution (c(s)) from the SV experiment. In the mixture of RAE-1β and m152, a peak with higher s-value can be discerned that is absent from samples of RAE-1β or m152 alone, indicating the formation of protein complexes (31, 37). The s-value of this peak (termed sfast) increases with higher protein concentration, consistent with the presence of a reaction boundary with relatively rapid reversible interactions (koff > 0.001/sec) (31, 32). The analysis of the isotherms of weight-average (sw) and reaction boundary (sfast) sedimentation coefficients as a function of protein concentration yielded a best-fit Kd value of 4.1 (3.1 – 5.1) μM for the RAE-1β/m152 interaction (Figure 2B), which was similar to the Kd derived from global non-linear least square fits of the data acquired from SE experiments conducted at 4°C (1.7 (1.6 – 4.9) μM) (Figure 2C) (30). Moreover, there was a close fit of all the centrifugation data to a simple 1:1 Langmuir binding model based on a single class of non-interacting binding sites, suggesting RAE-1 binds to m152 at 1:1 ratio. The SV analysis suggested the presence of 16% unreactive m152. (As noted in Experimental Procedures, analysis of data from the SV experiment independently substantiates that both m152 and RAE-1β were of high purity and were well-folded, globular proteins).
Figure 2. Biophysical experiments show m152 binds to RAE-1 at 1:1 ratio and with micromolar Kd.
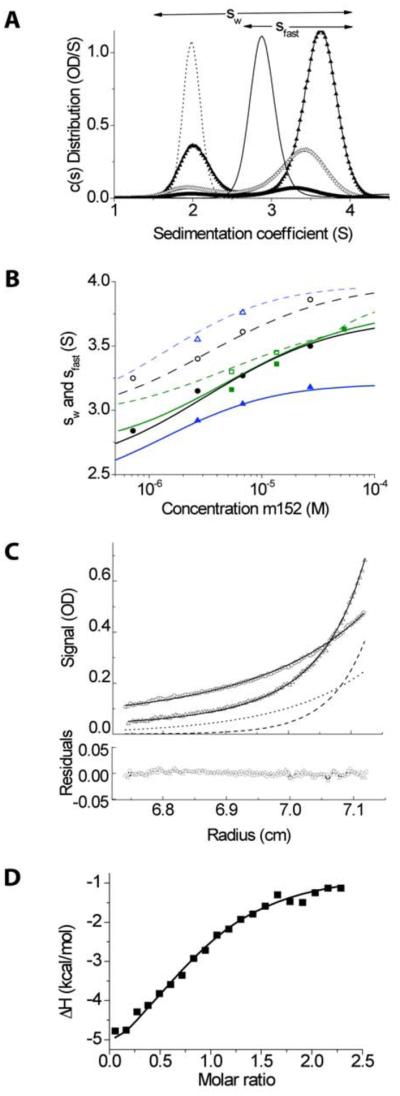
A, Sedimentation coefficient distribution c(s) measured in sedimentation velocity (SV) experiments for 11.3 μM RAE-1 alone (dotted line) and 11.3 μM m152 alone (thin line), and 0.63:1 mixtures of RAE-1 and m152 at 1.2 μM (thick line), 4.5 μM (open triangles), and 11.3 μM (filled triangles). Integration of c(s) traces over the range from ~1.5 to ~4.5 S or ~2.5 to ~4.5 S (indicated by horizontal arrows) yields values for the weighted-average s-value sw or the reaction boundary s-value sfast, respectively. B, Isotherms of sw (filled symbols) and sfast (open symbols) as a function of m152 concentration from dilution series at molar ratios RAE-1: m152 of 1.25:1 (black), 0.63:1 (green), and 2.5:1 (blue). The solid lines shown are from a global fit with a 1:1 binding model that incorporated additional data from the relative amplitude of the reaction boundary, as well as the data from isothermal titration calorimetry (ITC). The best-fit sw
value for the complex was 4.3 S, and the binding constant Kd = 4.1 μM. C, Equilibrium absorbance profiles at 280 nm of the mixture of 2.9 μM m152 and 10.2 μM RAE-1 sedimenting at 11,000 (circles) and 17,000 rpm (triangles), their best-fit distribution (solid lines) and the signal predicted for the complex (dotted and dashed lines). These data are taken from a global fit with a 1:1 binding model of 20 equilibrium gradients at different concentrations, rotor speeds, and absorbance wavelengths. D, Enthalpy isotherm from the titration of 100 μM RAE-1 into 10 μM m152 in ITC (symbols), based on a single titration series. The solid line shows the best-fit isotherm from a global fit with the SV isotherm data.
To confirm further the results from both SV and SE experiments and to gain insight into the thermodynamics of the RAE-1β/m152 interaction, isothermal titration calorimetry (ITC) was conducted (Fig. 2D) at 25 °C. The results revealed a Kd of 2.1 (1.7 – 2.7) μM, and a 1:1 stoichiometry with 7 (5 – 9) % unreacted m152 for the interaction between RAE-1β and m152. The thermodynamic parameters of ΔH and TΔS were determined to be −6 kCal/mol and 1.44 kCal/mol, respectively, suggesting the interaction is largely enthalpically driven. These results are in good agreement with the AUC measurements, and the results from a global analysis of the s-value isotherms from sedimentation velocity and the titration data from ITC indicate the absence of significant temperature dependence of the thermodynamic parameters between 20 °C and 25 °C.
To examine the differential interaction between RAE-1 isoforms and m152, SV experiments were carried out for RAE-1γ and δ with m152, and the isotherms of weight-average s-values were determined over a range of loading concentrations. Figure 3B shows a comparison of these data for RAE-1β, γ, and δ. Kd derived from the SV data demonstrated a significant difference in the affinity of m152 for different RAE-1 isoforms. RAE-1β and γ bind to m152 relatively tightly with Kd values of about 3 and 1 μM, respectively. In contrast, RAE-1δ binds to m152 more weakly, with a Kd of about 30 μM. These results are consistent with the qualitative binding analysis on SEC columns (Figure 1C, D, E, and Figure 3A), and support the view of the differential interaction between m152 and different RAE-1 isoforms. Although apparent differences in the elution time of the three different RAE-1 isoforms can be detected in the SEC profiles of Figure 1C, D, and E, the s-values determined from the SV experiments of all six molecules are between 1.92 and 2.05 S, essentially the same.
Figure 3. The PLWY motif contributes to the binding affinity of RAE-1β to m152.

A, SEC binding assay. The PLWY motif was deleted from RAE-1β and γ and inserted into RAE-1δ by site-directed mutagenesis. The binding of wild type and mutant RAE-1 proteins to m152 was examined by SEC as described in Materials and Methods. The results from wild type RAE-1 proteins are the same as shown in Figure 1C, D, E and shown here for ease of comparison. B, Sedimentation velocity ultracentrifugation. Isotherms of weighted-average sedimentation coefficients sw (in experimental units) obtained from SV experiments of equimolar mixtures of RAE-1 and m152 (symbols) and best-fit isotherm with a 1:1 binding model (solid lines) using the pre-determined s-value of RAE-1 and m152, and a complex s-value fixed to that determined in the extensive characterization of RAE-1β/ m152 interaction of Figure 2. The insets show the sedimentation coefficient distributions c(s) underlying the sw data points, for approximately equimolar loading concentrations of 1 (cyan), 2 (magenta), 7 (blue), 14 (green), and 32 (red) μM. Precise concentrations were measured from the interference optical signal and varied slightly.
The PLWY motif contributes to the affinity of RAE-1β to m152
Recent analysis of the comparative function of RAE-1 isoforms noted the deletion of a PLWY motif in RAE-1δ as compared with RAE-1γ (24). Since this sequence lies in an exposed loop connecting the β1 and β2 strands (see Figure 6), we hypothesized that it might contribute to the molecular interactions of RAE-1 with m152. To gain insight into the site of interaction and establish the molecular basis for the differential interaction of RAE-1 isoforms with m152, we aligned the amino acid sequences of the five identified RAE-1 isoforms (Figure 4). RAE-1 proteins share more than 90% sequence identity (19). However, among the several sequence variants, the PLWY motif (aa 49-52) is present in RAE-1α, β, γ, but is deleted in RAE-1δ and substituted with LPWC in RAE-1ε. The presence or absence of the PLWY motif seems to correlate with RAE-1 function, as RAE-1 proteins are expressed exclusively as two groups (RAE-1α, β, and γ or RAE1δ and ε) in different mouse strains. We hypothesized that differences in the PLWY motif may contribute to the differential interaction of RAE-1 isoforms with m152, and that the differential interactions might be reflected in the relative control of cell surface expression of the different RAE-1 isoforms under the influence of m152. To test this idea, we constructed PLWY-deletion mutants of RAE-1β and γ, as well as a PLWY-addition mutant of RAE-1δ, and expressed, refolded, and purified the ectodomains of these RAE-1 mutants. We then examined the binding interaction between RAE-1 mutants and m152 by SEC and AUC using the same methods we used for wild type RAE-1 proteins (Figure 3). As shown in Figure 3A, deletion of the PLWY motif from RAE-1β (i.e. the RAE-1β mutant) caused loss of the more rapidly eluting “mixture” peak detected chromatographically, suggesting decreased binding. However, deletion of PLWY from RAE-1γ or insertion of the motif into RAE-1δ (i.e. RAE-1γ and δ mutant) caused little effect on the apparent binding between RAE-1 and m152. These qualitative SEC results were consistent with the observations made in the AUC experiments (Figure 3B), as deletion of PLWY in RAE-1β led to a 10-fold decrease in affinity (Kd changing from 3.1 μM to 31 μM) whereas deletion of the motif from RAE-1γ resulted in no significant change in Kd (1μM as compared with 1.4 μM). Insertion of the motif into RAE-1δ did not cause any measurable change in the Kd as well. Thus, these results suggested that the PLWY motif may be involved, but is not crucial in mediating the RAE-1γ or δ and m152 interaction.
Figure 6. RAE-1 isoforms differ in surface electrostatic charge.

A, Models of RAE-1γ and δ were constructed in Coot based on the crystal structure of RAE-1β (PDB 1JFM chain A (26)). RAE-1β shows 92 and 94% amino acid sequence identity with RAE-1γ and δ, respectively. Residues in RAE-1β were replaced with the corresponding residues of RAE-1γ and δ followed by geometry idealization as described in Material and Methods. The molecular surfaces are colored by electrostatic potential, with positively charged areas in blue and negatively charged areas in red. B, Ribbon diagrams of RAE-1β, γ, and δ. Positions of some of the polymorphic charged residues are shown as stick representations superposed on the ribbons. For RAE-1β, residues Lys89 (blue), Glu178 (red), and Lys181 (blue) are indicated; for RAE-1γ, Glu89 (red), Asp97 (red), and Asp178 (red); and for RAE-1δ, Glu89 (red), Asp97 (red), Glu178(red), and Lys181(blue) are shown. (Numbering is based on the alignment in Figure 4).
m152 downregulates surface RAE-1 expression differently in transfected HEK293T cells
Our in vitro characterization of the RAE-1 and m152 interaction revealed that RAE-1 binds to m152 with a Kd as low as 1 μM and at a 1:1 ratio. The magnitude and stoichiometry of the interaction between RAE-1 and m152 suggest that such direct interaction may contribute to the downregulation of RAE-1 surface expression by m152. To test this idea and evaluate the role of full length, naturally glycosylated proteins in a cellular system, we transiently transfected human HEK293T cells with either m152 cDNA or a control vector, along with either of the RAE-1γ or δ cDNAs as well as their corresponding PLWY-deletion or PLWY-insertion mutants, respectively. A similar experiment has been described previously by Lodoen et al (21). In those experiments equal amounts of the RAE-1 and m152 cDNA were cotransfected into HEK293T cells. They showed that all the five RAE-1 isoforms were susceptible to the downregulation by m152. To explore the relative effects of m152 on the cell surface expression of different RAE-1 isoforms, we transfected HEK293T cells with a constant amount of RAE-1 vector and varied the amount of a cotransfected m152-encoding vector that carried an IRES-GFP (see Figure 5 and Experimental Procedures). Forty-eight hours following transfection, cells were examined by flow cytometry for the expression of GFP as an indication of the transfection efficiency and for surface expression of RAE-1. In Figure 5A the two-dimensional dot-plot of GFP vs. RAE-1 for the RAE-1γ wild type transfection is shown. The level of GFP expression from the IRES-GFP increases steadily with increasing amounts of the IRES-GFP-only vector. RAE-1γ surface expression in the absence of m152 (center column) remains approximately constant, varying from 77% (66.3 + 11%) of total cells for the lowest dose of the empty vector to about 60% (9.83 + 50.6%) with the highest, reflecting some variability apparently due to differences in total efficiency of transfection. Remarkably, when increasing amounts of the m152-encoding, IRES-GFP vector are included, a progressively smaller proportion of the cells are positive for RAE-1, ranging from 52% (42.5 + 9.74%) of the cells RAE-1 positive at the lowest dose of m152 vector to 16% (8.07 + 8.39%) at the highest dose. This result is further emphasized if one only considers the GFP+ cells, where RAE-1 positive cells are 62% (9.74/15.5%) at the lowest dose of m152, to 49% (9.41/19.21%) at the intermediate dose, to 21.2% (8.39/38.89%) at the highest dose. Similar two-dimensional analyses were used to generate the histograms for both RAE-1γ and RAE-1δ as well as their respective PLWY deletion and insertion mutants, respectively (Figure 5B). The uninhibited surface expression of RAE-1 is shown by the blue lines and the m152 downregulated levels are shown in green. Thus it is clear that both RAE-1γ and the RAE-1γ mutant are similarly downregulated by m152 coexpression (Figure 5B, first and second columns). RAE-1δ wild type is detectably but less effectively downregulated by m152 than RAE-1γ (Figure 5B, third column). (Downregulation of RAE-1δ is not detectable at all at the lowest dose of m152, while for RAE-1γ there is clearly an effect at that dose). Finally, introduction of the PLWY motif into RAE-1δ, as the RAE-1δ mutant, shows no major difference in m152 responsiveness as compared to the wild type. It is crucial to note that “downregulation” of cell surface expressed RAE-1 isoforms as measured by cell surface immunofluorescent staining formally tells us only that the serological epitopes of RAE-1 are no longer available to the detecting antibody when the RAE-1 is coexpressed with m152. The molecular basis for this may be the intracellular interaction of m152 with RAE-1 and subsequent diversion of the normal trafficking of RAE-1 due to the m152 interaction. Alternatively, the m152/RAE-1 complex may go directly to the cell surface at the same rate and with the same steady state level of RAE-1, but with the epitopes of RAE-1 obscured or masked by the binding of m152. However, pulse-chase labeling studies indicated that RAE-1γ, when expressed coordinately with m152, accumulated as lower molecular weight forms in the ERGIC, consistent with the interpretation that m152/RAE-1γ interaction reduces the cell surface expression of the RAE-1γ molecule (24).
Figure5. m152 differently downregulates surface expression of RAE-1 isoforms.

A, A constant amount of wild type and the PLWY mutant of RAE-1γ or δ cDNAs were cotransfected into human HEK293T cells along with an empty vector carrying an IRES-GFP (left and middle column) or the corresponding m152-encoding vector (right column) at increasing amounts as indicated. 48 hours after transfection, cells were harvested and stained with either an isotype control Ig (left column) or rat pan anti-RAE-1 mAb (186107, middle and right column) and analyzed for surface RAE-1 expression by flow cytometry. Results are representative of three independent experiments. Shown here is the two-dimensional dot plot of RAE-1γ and m152 cotransfection experiment. B, Summary of the above described cotransfection experiments in histogram. Red, isotype control; blue, cotransfected RAE-1 with empty vector and stained with 186107; green, cotransfected RAE-1 with m152 and stained with 186107.
DISCUSSION
In this study, we hypothesized that the physical interaction between the extracellular domains of the viral protein m152 and the host stress-induced proteins RAE-1 contribute to the regulation of surface expression of RAE-1. Previous efforts to demonstrate the interaction directly by coprecipitation have not been fruitful. Using recombinant RAE-1 isoforms and mutants expressed in E. coli and m152 expressed in insect cells, we have demonstrated by several different methods the direct physical interaction of the viral and host-encoded molecules. We have shown that m152 binds RAE-1 directly and at a 1:1 ratio. The binding affinities differ detectably among RAE-1 isoforms. RAE-1β and γ, which are expressed on MCMV-susceptible mouse strains such as BALB/c, bind m152 tightly (Kd =1-3 μM), a level that can be compared to the affinity of the NKG2D/RAE-1 interaction (Kd =350-730 nM (28)). In contrast, RAE-1δ, which is expressed on MCMV-resistant mouse strains such as C57BL/6, CBA and C3H, binds m152 weakly (Kd = 34 μM), which is about 50-fold lower than its affinity for NKG2D (Kd =726 nM). The difference in affinity of RAE-1β or RAE-1γ for m152 as compared with RAE-1δ suggests that the less effective downregulation of RAE-1δ by m152 as reported recently by Arapovic et al (24) may be a result of this lower affinity. It is interesting to note that the SV analysis of the m152/RAE-1β interaction revealed a concentration-dependent increase in sfast, indicative of relatively rapid reversible interaction (Figure 2A). This result may explain the experimental difficulties in detecting m152/RAE-1 interaction by coimmunoprecipitation “pull-down” experiments. Similarly, it was observed that the luminal domain of m152 is involved in the retention of MHC class I molecules in the ERGIC region in the cells, but without biochemical evidence for their direct interaction (23, 38). This suggests the interactions of m152/RAE-1 and m152/MHC-I may share similar intracellular regulatory pathways.
The differing affinities of RAE-1 isoforms for the MCMV m152 immunoevasin suggest a scenario for the ongoing evolution of MCMV immunoevasins. Genetic resistance of different mouse strains to MCMV infection is determined to some degree by loci that control NK activating receptors such as Ly49H (expressed in the MCMV resistant strain C57BL/6) (39). In MCMV sensitive strains such as BALB/c, NKG2D, as an ITAM-associated activating receptor, plays an important role in control of viral titre following infection, and NKG2D ligands, particularly the stress-induced MULT-1, H60, and RAE-1 molecules, serve as the stimulus for NK cell activation. The virus gains a counter advantage by evolving evasins that antagonize the expression or function of the NKG2D ligands, particularly important in the setting of Ly49H negative mouse strains. These “sensitive” mouse strains may then gain advantage over the virus by the expression of more potent NKG2D ligands such as RAE-1α, β, and γ, while the resistant strains, because they have an additional NK activating receptor, Ly49H, have less need for the expression of better NKG2D ligands. In response to the expression of “good” NKG2D ligands, the virus refines the function of molecules to downregulate not only MULT-1 and H60, but also RAE-1α, β, and γ, accomplishing this with m152/gp40. The strong direct interaction of the m152 lumenal domain with RAE-1β and γ as we have measured here results in the effective downregulation of these NKG2D ligands, and the quantitative difference between RAE-1β and γ interaction with m152 as compared with RAE-1δ reflects the greater need for effective control of RAE-1β and γ (and presumably RAE-1α as well) to counter the NKG2D susceptibility of MCMV infected cells by NK “sensitive” strains.
This discussion is based on our knowledge of the genetics and relative resistance of several well-characterized inbred mouse strains, and a more complete analysis would require an extensive understanding of the expression patterns and RAE-1 isoform differences among wild outbred mice. To our knowledge such data are not yet available. However, the role of the MCMV genes is beginning to be assessed by the analysis of DNA of a number of independent isolates of virus from free-living mice (40-42) Remarkably, the m152 gene has been highly conserved, suggesting that its evolutionary advantage is either to encode a protein that interacts with a molecule with conserved structure, or to serve multiple functions, such as the dual function of m152 interaction both with RAE-1 and host MHC-I. Needless to say, these molecules as well as others play a complex cooperative role in the mutual adaptation of virus and host, and our studies of one set of these interactions offer only a hint of the greater complexity. It should be emphasized, however, that the preservation of a gene that contributes to the fitness of the virus may result from a rather small advantage that may elude detection by laboratory evaluation of viral titre resulting from controlled doses and routes of infection.
To understand the molecular mechanisms that determine the differential m152/RAE-1 interaction, we explored whether the PLWY motif, which is present in RAE-1α, β, γ whereas absent from RAE-1δ and mutated to LPWC in RAE-1ε, may play a role in the m152/RAE-1 interaction. Our results confirmed that deletion of PLWY in RAE-1β indeed caused a 10-fold decrease in its binding affinity to m152, suggesting that the PLWY motif has a key role in the m152/RAE-1β interaction. However, the binding affinity of RAE-1γ or δ for m152 was not influenced by the presence or absence of PLWY, indicating that other differences among RAE-1 isoforms contribute to the interaction and to the resulting downregulation of m152. We further found that aside from the PLWY motif, RAE-1 isoforms differ at a number of positions in charged amino acid residues (Figure 4). To gain insight into the possible effects of these amino acid differences, we have examined the surface electrostatic distribution of the RAE-1β X-ray structure, and have generated molecular models of RAE-1γ and RAE-1δ as well (Figure 6).
Our first consideration was whether the predicted N-linked glycosylation sites of RAE-1 (Asn38, 70, 83, 143, and 156 --- all conserved, with the exception of Asn156 which is absent from RAE-1δ) might impinge on the m152 interaction. All of these are distant from the NKG2D/RAE-1 interface (26) and are located either beneath the platform domain (Asn38 and 143) or at various peripheral positions (Asn83 at the N-terminus of the α1 helix, and Asn70 and 156 in loops connecting β strands). Thus, we focused on the “top” of the RAE-1 molecule, which serves as the NKG2D binding interface (26).
Looking at a top view of the RAE-1β structure in comparison with the models, there are three major regions that seem to be influenced by the substitutions that distinguish the three isoforms. The first and most obvious region is that where PLWY is present in RAE-1β and γ and where PLWY is deleted in RAE-1δ. This region flanks the α1 helix, focused on the loop connecting the β1 and β2 strands. The second major area involves the third β-strand of the α1 domain as well as the left hand side of the α1 helix (see left box in Figure 6A and highlighted residues of the α1 helix in Figure 6B). This region is basic in RAE-1β, acidic in RAE-1γ, and partly acidic and partly basic in RAE-1δ. The third region spans the right hand side of both the α1 and α2 helices, is basic in RAE-1β, acidic in RAE-1γ, and has subregions of both acidic and basic character in RAE-1δ. A more detailed understanding clarifying the complexities of the interaction of m152 with the various RAE-1 isoforms must await structure determination of a complex of RAE-1 with m152. Whether the downregulation of host MHC-I molecules by m152 expression is due to a direct interaction of m152 with MHC-I, and whether such a direct interaction is mediated by a surface of MHC-I structurally homologous to that of RAE-1 are questions that demand further detailed investigation.
MCMV infection provides a valid model system for understanding not only available mechanisms for NK and T cell mediated immunity to infection and virus resistance to host immunity, but also for gaining insight into the ongoing coevolution of virus and host. Our studies of the relative differences in the interaction of the viral immunoevasin m152 with host NKG2D ligands provide a glimpse of the ongoing adaptations of virus and host. The recently identified H60 isoforms (H60a, b, c) exhibit some features similar to the RAE-1 isoforms (43, 44). The relative interactions of m155 for the different H60 isoforms have yet to be addressed. Thus, differential expression and regulation of host ligands for NKG2D, as influenced by the virus, seem to be common features of the ongoing interplay and evolution of host response and viral immunoevasion. The likelihood that mouse and human viruses may share similar immune evasion strategies raises the prospect for identifying targets for novel anti-viral therapeutics based on disrupting the interaction between immunoevasins and their specific ligands in the host.
ACKNOWLEDGMENTS
We thank Andrea Balbo for help with the ultracentrifugation experiments, and Dr. John Coligan for critical comments on the manuscript.
ABBREVIATIONS AND TEXTUAL FOOTNOTES
- AUC
analytical ultracentrifugation
- ITC
isothermal titration calorimetry
- NK
natural killer
- PAGE
polyacrylamide gel electrophoresis
- sw
weight average coefficient
- sfast
reaction boundary coefficient
- SE
sedimentation equilibrium
- SV
sedimentation velocity
- SEC
size exclusion chromatography
Footnotes
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Lenac T, Arapovic J, Traven L, Krmpotic A, Jonjic S. Med Microbiol Immunol. 2008;197:159–166. doi: 10.1007/s00430-008-0080-7. [DOI] [PubMed] [Google Scholar]
- 2.>Miller-Kittrell M, Sparer TE. Feeling manipulated: cytomegalovirus immune manipulation. Virol J. 2009;6:4. doi: 10.1186/1743-422X-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powers C, DeFilippis V, Malouli D, Fruh K. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol. 2008;325:333–359. doi: 10.1007/978-3-540-77349-8_19. [DOI] [PubMed] [Google Scholar]
- 4.French AR, Yokoyama WM. Natural killer cells and viral infections. Curr Opin Immunol. 2003;15:45–51. doi: 10.1016/s095279150200002x. [DOI] [PubMed] [Google Scholar]
- 5.Doom CM, Hill AB. MHC class I immune evasion in MCMV infection. Med Microbiol Immunol. 2008;197:191–204. doi: 10.1007/s00430-008-0089-y. [DOI] [PubMed] [Google Scholar]
- 6.Wagner M, Gutermann A, Podlech J, Reddehase MJ, Koszinowski UH. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med. 2002;196:805–816. doi: 10.1084/jem.20020811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu Rev Immunol. 2000;18:861–926. doi: 10.1146/annurev.immunol.18.1.861. [DOI] [PubMed] [Google Scholar]
- 8.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez S, Groh V, Spies T. Immunobiology of human NKG2D and its ligands. Curr Top Microbiol Immunol. 2006;298:121–138. doi: 10.1007/3-540-27743-9_6. [DOI] [PubMed] [Google Scholar]
- 10.Lanier LL. On guard--activating NK cell receptors. Nat Immunol. 2001;2:23–27. doi: 10.1038/83130. [DOI] [PubMed] [Google Scholar]
- 11.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 12.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strid J, Roberts SJ, Filler RB, Lewis JM, Kwong BY, Schpero W, Kaplan DH, Hayday AC, Girardi M. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat Immunol. 2008;9:146–154. doi: 10.1038/ni1556. [DOI] [PubMed] [Google Scholar]
- 14.Arapovic J, Lenac Rovis T, Reddy AB, Krmpotic A, Jonjic S. Promiscuity of MCMV immunoevasin of NKG2D: m138/fcr-1 down-modulates RAE-1varepsilon in addition to MULT-1 and H60. Mol Immunol. 2009 doi: 10.1016/j.molimm.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Arapovic J, Lenac Rovis T, Reddy AB, Krmpotic A, Jonjic S. Promiscuity of MCMV immunoevasin of NKG2D: m138/fcr-1 down-modulates RAE-1varepsilon in addition to MULT-1 and H60. Mol Immunol. 2009 doi: 10.1016/j.molimm.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Gutermann A, Bubeck A, Wagner M, Reusch U, Menard C, Koszinowski UH. Strategies for the identification and analysis of viral immune-evasive genes-cytomegalovirus as an example. Curr Top Microbiol Immunol. 2002;269:1–22. doi: 10.1007/978-3-642-59421-2_1. [DOI] [PubMed] [Google Scholar]
- 17.Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, Jonjic S. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol. 2002;3:529–535. doi: 10.1038/ni799. [DOI] [PubMed] [Google Scholar]
- 18.Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol. 2002;3:1019–1025. doi: 10.1038/ni1102-1019. [DOI] [PubMed] [Google Scholar]
- 19.Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 20.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 21.Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med. 2003;197:1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinto AK, Jamieson AM, Raulet DH, Hill AB. The role of NKG2D signaling in inhibition of cytotoxic T-lymphocyte lysis by the Murine cytomegalovirus immunoevasin m152/gp40. J Virol. 2007;81:12564–12571. doi: 10.1128/JVI.01328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziegler H, Muranyi W, Burgert HG, Kremmer E, Koszinowski UH. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 2000;19:870–881. doi: 10.1093/emboj/19.5.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arapovic J, Lenac T, Antulov R, Polic B, Ruzsics Z, Carayannopoulos LN, Koszinowski UH, Krmpotic A, Jonjic S. Differential susceptibility of RAE-1 isoforms to mouse cytomegalovirus. J Virol. 2009;83:8198–8207. doi: 10.1128/JVI.02549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mans J, Natarajan K, Balbo A, Schuck P, Eikel D, Hess S, Robinson H, Simic H, Jonjic S, Tiemessen CT, Margulies DH. Cellular expression and crystal structure of the murine cytomegalovirus major histocompatibility complex class I-like glycoprotein, m153. J Biol Chem. 2007;282:35247–35258. doi: 10.1074/jbc.M706782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li P, McDermott G, Strong RK. Crystal structures of RAE-1beta and its complex with the activating immunoreceptor NKG2D. Immunity. 2002;16:77–86. doi: 10.1016/s1074-7613(02)00258-3. [DOI] [PubMed] [Google Scholar]
- 27.Natarajan K, Hicks A, Mans J, Robinson H, Guan R, Mariuzza RA, Margulies DH. Crystal structure of the murine cytomegalovirus MHC-I homolog m144. J Mol Biol. 2006;358:157–171. doi: 10.1016/j.jmb.2006.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Callaghan CA, Cerwenka A, Willcox BE, Lanier LL, Bjorkman PJ. Molecular competition for NKG2D: H60 and RAE1 compete unequally for NKG2D with dominance of H60. Immunity. 2001;15:201–211. doi: 10.1016/s1074-7613(01)00187-x. [DOI] [PubMed] [Google Scholar]
- 29.Brown PH, Balbo A, Schuck P. A bayesian approach for quantifying trace amounts of antibody aggregates by sedimentation velocity analytical ultracentrifugation. AAPS J. 2008;10:481–493. doi: 10.1208/s12248-008-9058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balbo A, Brown PH, Braswell EH, Schuck P. Measuring protein-protein interactions by equilibrium sedimentation. Curr Protoc Immunol Chapter. 2007;18 doi: 10.1002/0471142735.im1808s79. Unit 18 18. [DOI] [PubMed] [Google Scholar]
- 31.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dam J, Schuck P. Sedimentation velocity analysis of heterogeneous protein-protein interactions: sedimentation coefficient distributions c(s) and asymptotic boundary profiles from Gilbert-Jenkins theory. Biophys J. 2005;89:651–666. doi: 10.1529/biophysj.105.059584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, Schuck P. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem. 2004;326:234–256. doi: 10.1016/j.ab.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 34.Houtman JC, Brown PH, Bowden B, Yamaguchi H, Appella E, Samelson LE, Schuck P. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 2007;16:30–42. doi: 10.1110/ps.062558507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Mayer CL, Snyder WK, Swietlicka MA, Vanschoiack AD, Austin CR, McFarland BJ. Size-exclusion chromatography can identify faster-associating protein complexes and evaluate design strategies. BMC Res Notes. 2009;2:135. doi: 10.1186/1756-0500-2-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown PH, Balbo A, Schuck P. Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Curr Protoc Immunol Chapter. 2008;18 doi: 10.1002/0471142735.im1815s81. Unit 18 15. [DOI] [PubMed] [Google Scholar]
- 38.Ziegler H, Thale R, Lucin P, Muranyi W, Flohr T, Hengel H, Farrell H, Rawlinson W, Koszinowski UH. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 1997;6:57–66. doi: 10.1016/s1074-7613(00)80242-3. [DOI] [PubMed] [Google Scholar]
- 39.Scalzo AA, Yokoyama WM. Cmv1 and natural killer cell responses to murine cytomegalovirus infection. Curr Top Microbiol Immunol. 2008;321:101–122. doi: 10.1007/978-3-540-75203-5_5. [DOI] [PubMed] [Google Scholar]
- 40.Smith LM, McWhorter AR, Masters LL, Shellam GR, Redwood AJ. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J Virol. 2008;82:6689–6696. doi: 10.1128/JVI.00160-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith LM, Shellam GR, Redwood AJ. Genes of murine cytomegalovirus exist as a number of distinct genotypes. Virology. 2006;352:450–465. doi: 10.1016/j.virol.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 42.Mans J, Zhi L, Revilleza MJ, Smith L, Redwood A, Natarajan K, Margulies DH. Structure and function of murine cytomegalovirus MHC-I-like molecules: how the virus turned the host defense to its advantage. Immunol Res. 2009;43:264–279. doi: 10.1007/s12026-008-8081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takada A, Yoshida S, Kajikawa M, Miyatake Y, Tomaru U, Sakai M, Chiba H, Maenaka K, Kohda D, Fugo K, Kasahara M. Two novel NKG2D ligands of the mouse H60 family with differential expression patterns and binding affinities to NKG2D. J Immunol. 2008;180:1678–1685. doi: 10.4049/jimmunol.180.3.1678. [DOI] [PubMed] [Google Scholar]
- 44.Whang MI, Guerra N, Raulet DH. Costimulation of dendritic epidermal gammadelta T cells by a new NKG2D ligand expressed specifically in the skin. J Immunol. 2009;182:4557–4564. doi: 10.4049/jimmunol.0802439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]