

Abstract
Somatic spiking is known to regulate dendritic signaling and associative synaptic plasticity in many types of large neurons, but it is unclear whether somatic action potentials play similar roles in small neurons. Here we ask whether somatic action potentials can also influence dendritic signaling in an electrically compact neuron, the cerebellar stellate cell (SC). Experiments were conducted in rat brain slices using a combination of imaging and electrophysiology. We find that somatic action potentials elevate dendritic calcium levels in SCs. There was little attenuation of calcium signals with distance from the soma in SCs from postnatal day 17 (P17)–P19 rats, which had dendrites that averaged 60 μm in length, and in short SC dendrites from P30–P33 rats. Somatic action potentials evoke dendritic calcium increases that are not affected by blocking dendritic sodium channels. This indicates that dendritic signals in SCs do not rely on dendritic sodium channels, which differs from many types of large neurons, in which dendritic sodium channels and backpropagating action potentials allow somatic spikes to control dendritic calcium signaling. Despite the lack of active backpropagating action potentials, we find that trains of somatic action potentials elevate dendritic calcium sufficiently to release endocannabinoids and retrogradely suppress parallel fiber to SC synapses in P17–P19 rats. Prolonged SC firing at physiologically realistic frequencies produces retrograde suppression when combined with low-level group I metabotropic glutamate receptor activation. Somatic spiking also interacts with synaptic stimulation to promote associative plasticity. These findings indicate that in small neurons the passive spread of potential within dendrites can allow somatic spiking to regulate dendritic calcium signaling and synaptic plasticity.
Introduction
The manner in which neurons convey information between their somata and dendrites is a crucial determinant of how they process synaptic inputs and modify their synapses. The ability of action potentials to invade the dendrites of neurons is dependent on the electrotonic structure of the dendritic arbor and the density of dendritic sodium channels (Stuart et al., 2007). In large neurons, such as hippocampal and cortical pyramidal neurons, dendritic sodium channels permit action potentials to effectively backpropagate into their dendrites (Stuart and Sakmann, 1994; Spruston et al., 1995), which is essential to many forms of associative plasticity that require precisely timed presynaptic and postsynaptic activity (Magee and Johnston, 1997; Markram et al., 1997; Abbott and Nelson, 2000; Roberts and Bell, 2002; Sjöström et al., 2008). Backpropagating action potentials are also present in the dendrites of many other types of neurons, including large hippocampal and cortical interneurons (Häusser et al., 1995; Martina et al., 2000; Kaiser et al., 2001; Saraga et al., 2003; Goldberg and Yuste, 2005). Thus, the ability of neurons to convey spiking activity to their dendrites is important for postsynaptic regulation of synaptic plasticity.
For electrically compact neurons, it is not clear whether dendritic sodium channels are important for conveying somatic action potentials to dendrites. In most cases, little is known about whether the dendrites of such cells support action potentials, because they are thin and difficult to record from directly (Davie et al., 2006). Dendritic sodium channels promote the spread of somatic depolarizations into the dendrites of some small cortical interneurons (Goldberg et al., 2003). However, modeling studies of hippocampal parvalbumin-positive interneurons suggest that passive spread of somatic action potentials may be sufficient to depolarize dendrites (Emri et al., 2001). Thus, it remains an open question whether compact neurons require dendritic sodium channels to enable somatic potentials to influence dendritic signaling.
Here we investigate the manner in which somatic action potentials influence dendritic signaling in cerebellar stellate cells (SCs), which are small interneurons of the cerebellar cortex (Palay and Chan-Palay, 1974). We find that somatic action potentials evoke calcium entry into SC dendrites in a manner that does not rely on dendritic sodium channels. Trains of somatic action potentials elevate dendritic calcium to hundreds of nanomolar. Thus, in small neurons with short dendrites, active backpropagation of somatic action potentials is not required to convey somatic signals to the dendrites.
We went on to examine the functional consequences of such dendritic signaling. We found that high-frequency somatic spiking evoked the release of endocannabinoids (eCBs) from SC dendrites and resulted in synaptic suppression. Prolonged patterns of physiologically realistic firing frequencies promoted eCB signaling when group I metabotropic glutamate receptors (mGluR1/5) were activated. Finally, pairing somatic spiking with synaptic activation increased the magnitude of dendritic calcium signals and promoted eCB release. Thus, in the absence of active backpropagating action potentials, somatic spiking in SCs can elevate dendritic calcium sufficiently to promote eCB release and can interact with synaptic inputs to promote associative plasticity.
Materials and Methods
Tissue preparation.
Sprague Dawley rats [postnatal day 17 (P17)–P19 or P30–P33] were deeply anesthetized with halothane or isoflurane and decapitated. Brains were removed and placed in an ice-cold dissecting solution containing the following (in mm): 82.7 NaCl, 65 sucrose, 23.8 NaHCO3, 23.7 glucose, 6.8 MgCl2, 2.4 KCl, 1.4 NaH2PO4, and 0.5 CaCl2. Transverse (see Figs. 7, 8) or parasagittal (see Figs. 1–6, 9) slices (200–250 μm thick) were made from the cerebellum in ice-cold dissecting solution. Slices were incubated at 32°C for 30 min in the dissecting solution and 30 min in a saline solution containing the following (in mm): 125 NaCl, 26 NaHCO3, 25 glucose, 2.5 KCl, 2 CaCl2, 1.25 NaH2PO4, and 1 MgCl2, adjusted to 315 mOsm. Slices were perfused with saline bubbled with 95% O2/5% CO2 at 4 ml/min. All experiments were performed at 34 ± 1°C.
Figure 7.
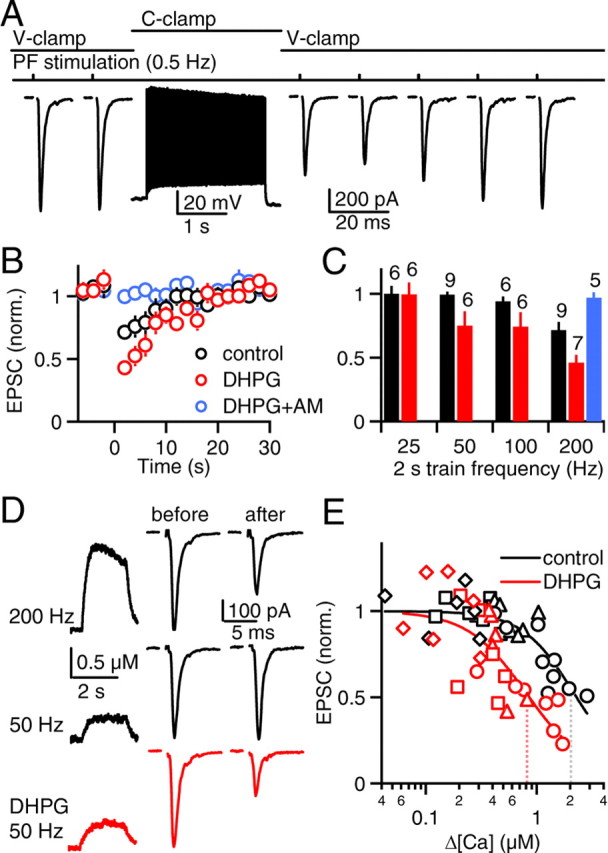
Brief, high-frequency action potential trains evoke retrograde eCB signaling in P17–P19 SCs, which is facilitated by mGluR1/5 activation. A, In a representative experiment, PFs were stimulated (0.5 Hz) in voltage clamp. The recording was switched to current clamp, and somatic action potentials were evoked (2 s, 200 Hz). The SC was returned to voltage clamp, and PF stimulation (0.5 Hz) was resumed. Such trials were repeated every minute for 4 min and then the EPSCs were normalized and averaged. B, The time courses of normalized PF EPSCs are shown for control conditions (black circles), in the presence of the mGluR1/5 receptor agonist DHPG (5 μm, red circles) and in the presence of DHPG and the CB1 receptor antagonist AM 251 (AM, 2 μm, blue circles). C, Normalized EPSC amplitudes are summarized for 2 s action potential trains of different frequencies. Numbers above the bars indicate the number of neurons recorded in each condition. D, Representative calcium transients (left) and synaptic responses (right) are shown for action potential trains of 200 Hz (top) and 50 Hz (middle) in control conditions and 50 Hz after wash-in of DHPG (bottom). Calcium transients are averages of four trials, each. E, Relative synaptic strength is plotted against peak calcium during the action potential trains and fit to the Hill equation (EPSC = 1 − 0.95[xn/(K0.5 + xn)], where 0.95 is the maximal suppression attained with CB1R activation, K0.5 is the half-maximal calcium, and n is the Hill coefficient). Dotted lines indicate the peak calcium required for half-maximal synaptic suppression (control: K0.5 = 2.2 ± 0.3 μm, n = 1.9 ± 0.4; DHPG: K0.5 = 0.8 ± 0.1 μm, n = 1.6 ± 0.4). Symbol shapes indicate the frequency of the action potential trains (diamonds: 25 Hz, squares: 50 Hz, triangles: 100 Hz, circles: 200 Hz).
Figure 8.
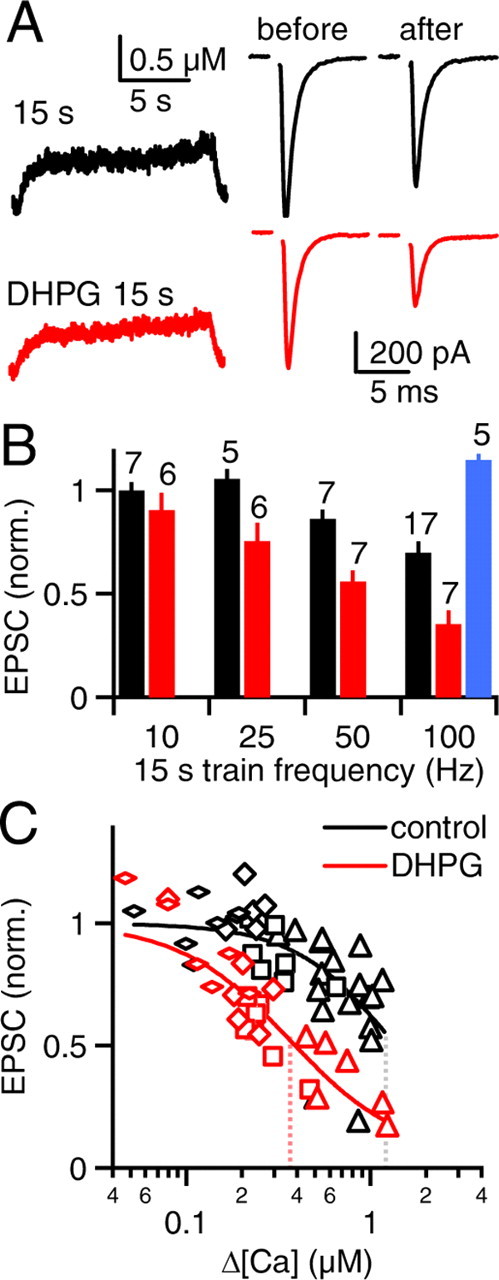
Prolonged somatic action potential trains promote retrograde eCB signaling. PFs were stimulated in voltage clamp and action potential trains were evoked in current clamp as in Figure 7. A, Representative calcium transients (left) and synaptic responses (right) are shown for an experiment in which somatic action potentials were evoked in SCs at 100 Hz for 15 s in control conditions (top) and in the presence of DHPG (bottom). B, Normalized EPSC amplitudes are summarized for 15 s action potential trains of different frequencies for control conditions (black), DHPG (red), and DHPG in the presence of AM 251 (AM, blue). Numbers above the bars indicate the number of neurons recorded in each condition. C, Relative synaptic strength is plotted against peak calcium during the action potential trains and fit to the Hill equation as in Figure 7E. Dotted lines indicate the peak calcium required for half-maximal synaptic suppression (control: K0.5 = 1.3 ± 0.2 μm, n = 1.9 ± 0.4; DHPG: K0.5 = 0.4 ± 0.1 μm, n = 1.4 ± 0.2). Symbol shapes indicate the frequency of the action potential trains (flat diamonds: 10 Hz, diamonds: 25 Hz, squares: 50 Hz, triangles: 100 Hz).
Figure 1.
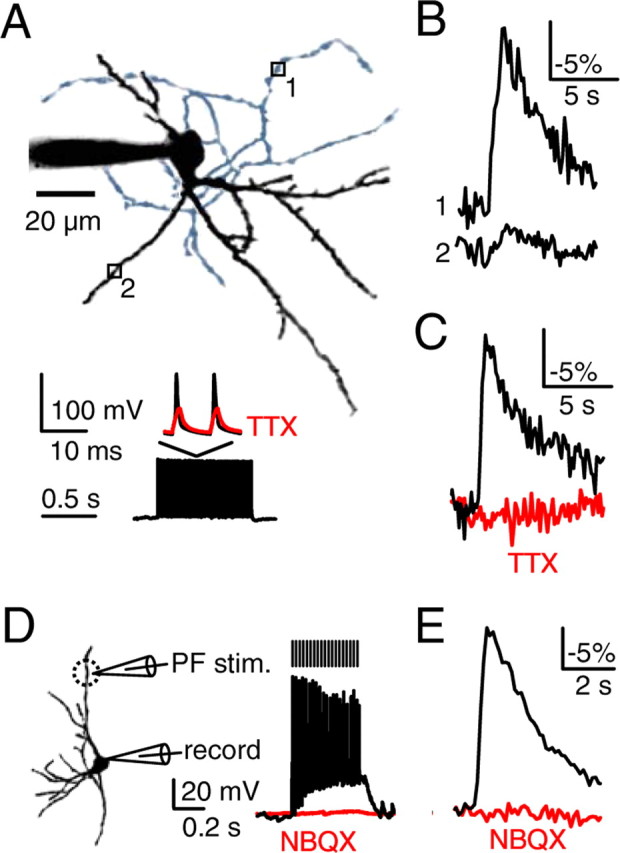
Somatic action potential trains elevate sodium levels in axons but not in dendrites. P17–P19 SCs were loaded with the sodium indicator SBFI (2 mm) through patch pipettes, and stimulus-evoked sodium transients were measured. Sodium elevations decrease SBFI fluorescence. Representative sodium transients (B, C, E) are averages of 3–5 trials (see Materials and Methods). In A and B, action potential trains were evoked in the soma (1 s, 100 Hz, A, bottom), whereas in D and E, PFs were activated (20 stimuli, 50 Hz). A, A representative SC is shown (top) with a black dendritic arbor and a blue axonal arbor. Sodium was measured at an axonal site (site 1) and a dendritic site (site 2). The resulting changes in SBFI fluorescence are indicated (B). C, Axonal sodium transients in control conditions and after wash-in of TTX (500 nm) are shown for a representative experiment (A, bottom, and C). D, Schematic (left) shows configuration for PF activation and somatic responses in control conditions (black trace) and after wash-in of NBQX (10 μm, red trace). The dashed circle indicates the area where dendritic sites were selected for imaging (see Materials and Methods). E, A representative experiment is shown in which PF stimulation evoked robust localized dendritic sodium transients that were blocked by NBQX.
Figure 2.
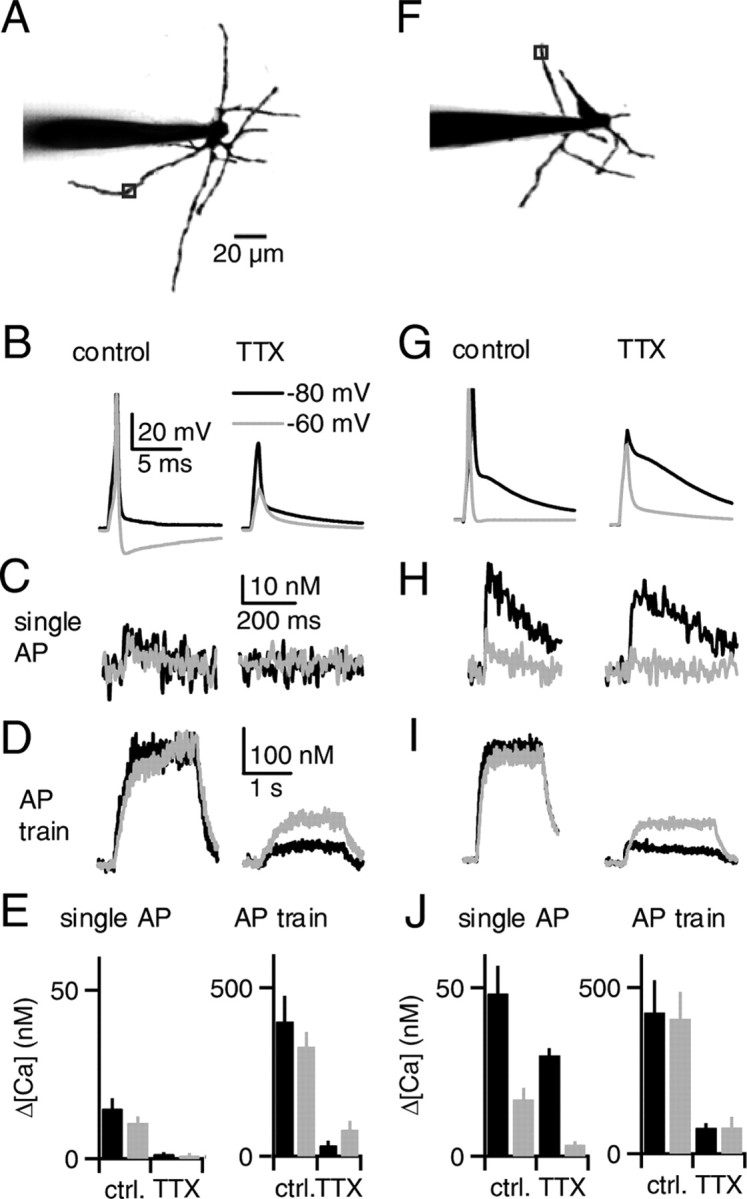
Action potentials in SCs elevate dendritic calcium. Recordings of P17–P19 SCs were made with pipettes containing a green calcium-sensitive indicator (fluo-5F, 50 μm) and a red calcium-insensitive dye (Alexa 594, 50 μm). Action potentials were evoked in current clamp from an initial potential of −80 mV (black traces) or −60 mV (gray traces), and calcium was measured at a dendritic site 50 μm from the soma. Representative calcium transients evoked by single action potentials (C, H) are averages of 12–16 trials, whereas transients evoked by 2 s 100 Hz trains (D, I) are averages of 3–5 trials. A–E and F–J represent ∼80% and 20% of neurons, respectively. In images of example neurons (A, F), axons were removed for clarity. In a cell representing the majority of neurons (A), a single action potential (B, left) elevated dendritic calcium by 10 nm, regardless of the initial potential (C, left). TTX eliminated the action potential (B, right) and substantially reduced the dendritic calcium elevations (C, right). D, Action potential trains (2 s, 100 Hz) elevated dendritic calcium to 300 nm (left). TTX reduced the calcium elevations evoked by trains (right). E, Recordings as in A–D are summarized (n = 5 neurons). In a cell representing the remaining neurons (F), a single action potential generated an afterdepolarization when evoked from −80 mV but not when evoked from −60 mV (G, left). Single action potentials elevated dendritic calcium to 40 and 10 nm when evoked from initial potentials of −80 mV and −60 mV, respectively (H, left). TTX eliminated the action potential but did not substantially reduce the afterdepolarization (G, right) or the calcium elevation (H, right) for an initial potential of −80 mV. However, calcium elevations were reduced when the initial potential was −60 mV. I, Action potential trains elevated dendritic calcium to 300 nm in control conditions, regardless of the initial potential (left). TTX reduced calcium elevations evoked by trains (right). J, Recordings as in F–I are summarized (n = 5 neurons).
Figure 3.
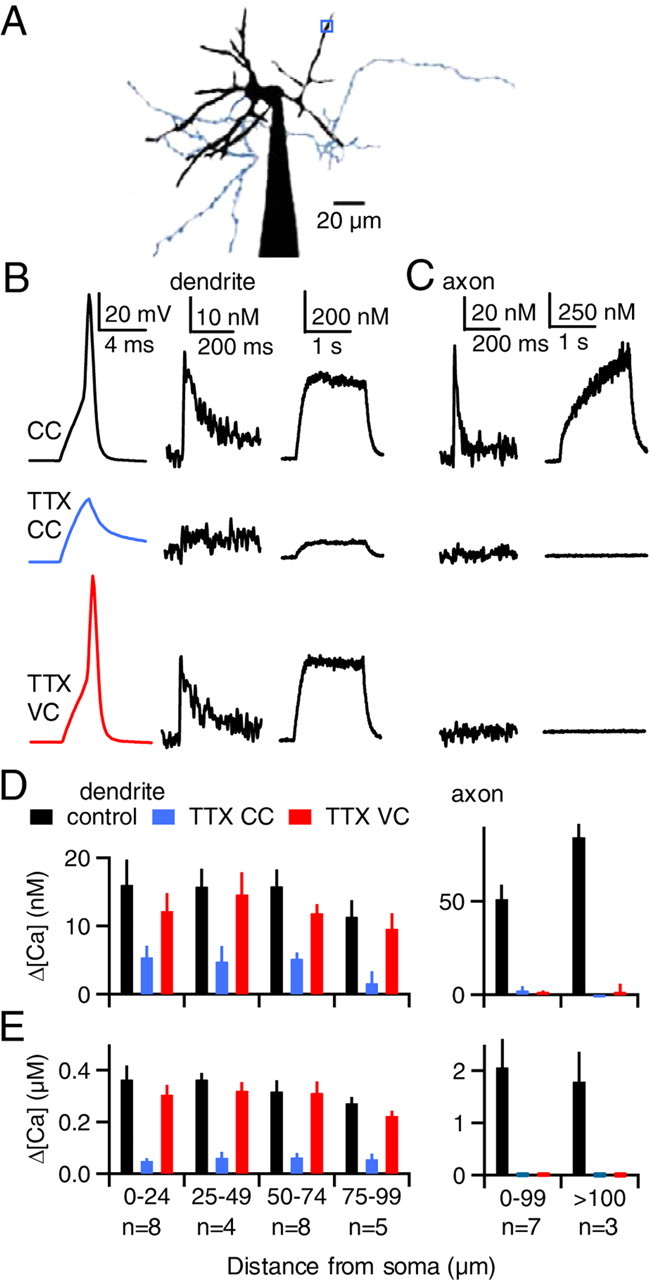
Somatic action potentials elevate dendritic calcium in a manner that does not rely on dendritic sodium channels. A, B, As shown for a representative experiment, action potentials were evoked in the soma of a P17–P19 SC, and the resulting calcium signals were measured at the site indicated in A (blue box). The axon is colored blue for clarity. B, Membrane voltage (left) and dendritic calcium signals following a single action potential (middle) and an action potential train (2 s, 100 Hz, right) are shown. Somatic depolarization evoked action potentials in current clamp (CC, top) but failed to do so in the presence of TTX (middle). In the presence of TTX, the cell was then voltage clamped with the action potential waveform (bottom). C, A second experiment is shown in which calcium was measured in an axon located 75 μm from the soma. Calcium signals evoked by either single action potentials (left) or trains of action potentials (right) are shown. D, E, Calcium elevations are summarized for dendritic sites (left) and axonal sites (right) for single action potentials (D) and 2 s action potential trains (E).
Figure 4.
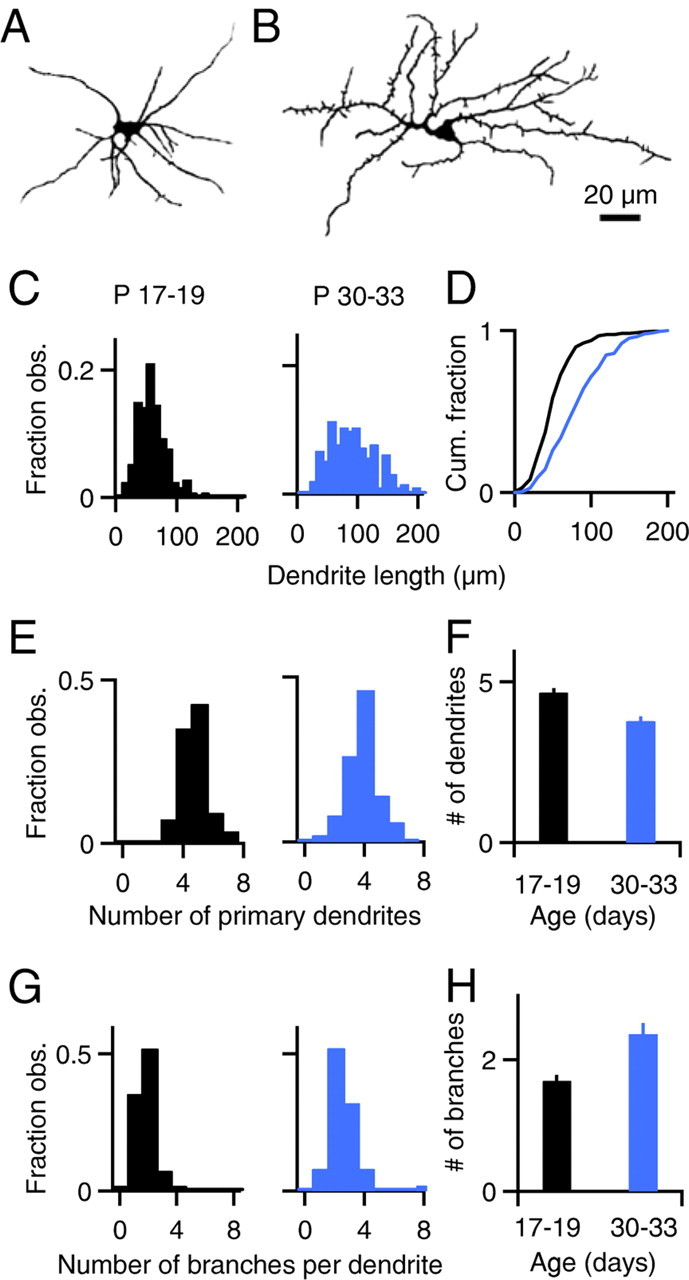
Morphology of stellate cell dendrites. SCs from P17–P19 or P30–P33 rats were loaded with Alexa 594, Z stacks were obtained, and dendrite morphology was analyzed using NeuronJ (Meijering et al., 2004). A, B, Shown are images of P18 (A) and P30 (B) SCs. Axons were removed for clarity. C, D, Histograms (C) and cumulative histograms (D) of SC dendrite lengths are shown for P17–P19 rats (C, left and D, black trace, n = 247 dendrites) and P30–P33 rats (C, right and D, blue trace, n = 193 dendrites). Histograms of the number of primary dendrites per SC (E) and the number of secondary branches per primary dendrite (G) are shown for P17–P19 rats (left, n = 54 neurons) and P30–P33 rats (right, n = 50 neurons). F, H, Experiments as in E and G are summarized.
Figure 5.
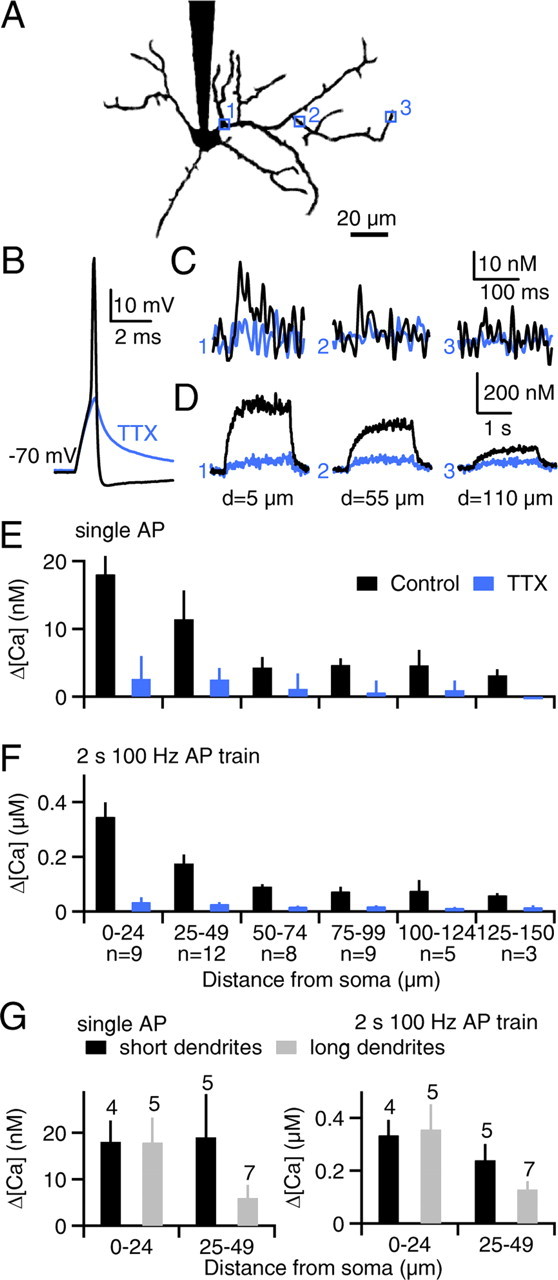
Action-potential-evoked calcium signals attenuate with distance from the soma in the dendrites of P30–P33 stellate cells. A–D, As shown for a representative SC obtained from a P30 rat, action potentials were evoked in the soma, and calcium levels were imaged at locations indicated by the numbered boxes (A, axon removed for clarity). Somatic membrane voltage in response to a single action potential (B) and dendritic calcium signals evoked by either single action potentials (C) or 2 s 100 Hz action potential trains (D) are shown for dendritic sites indicated in A. Blue traces indicate responses after application of TTX. Calcium elevations evoked by single action potentials (E) or action potential trains (F) are summarized. G, Data from the two most proximal regions in E and F are replotted for short dendrites (40–50 μm long, black bars) and long dendrites (>100 μm long, gray bars). Numbers above the bars indicate the number of dendrites analyzed.
Figure 6.
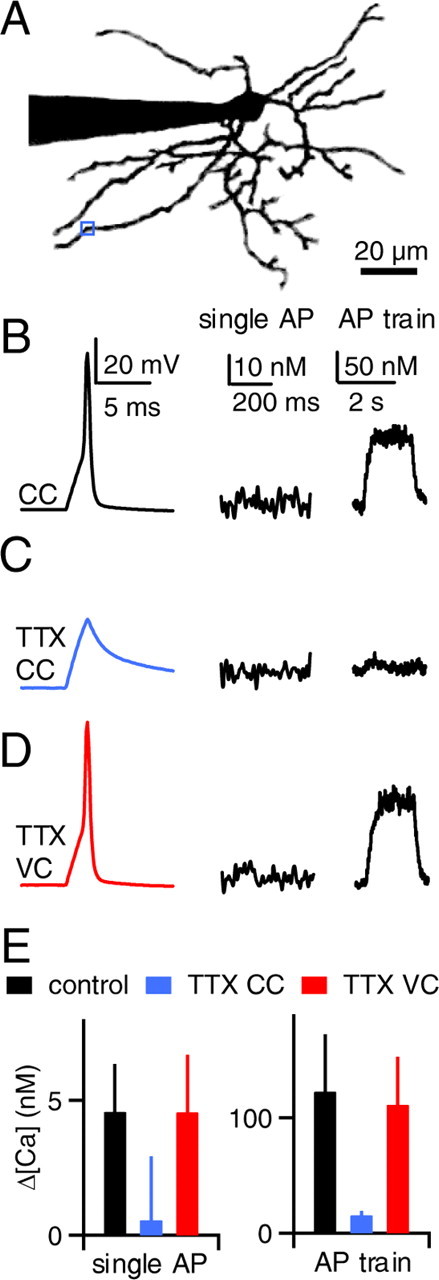
Somatic action potentials elevate dendritic calcium in a manner that does not rely on dendritic sodium channels in P30–P33 SCs. A–D, As shown for a representative experiment, action potentials were evoked in the soma and the resulting calcium signals were measured at the site indicated in A (blue box). The axon has been removed for clarity. B, Membrane voltage (left) and calcium elevations following a single action potential (middle) and an action potential train (2 s, 100 Hz, right) are shown. Calcium elevations were substantially smaller compared with SCs obtained from P17–P19 rats. Somatic depolarization evoked action potentials in current clamp (B) but failed to do so in the presence of TTX (C). In the presence of TTX, the cell was then voltage clamped with the action potential waveform (D). E, Calcium elevations are summarized for dendritic sites 70–100 μm from the soma (n = 6 neurons) for single action potentials (left) and action potential trains (right).
Figure 9.

SCs associate postsynaptic and presynaptic activity. A, In a representative experiment, membrane voltage (left), dendritic calcium transients (center), and relative synaptic strength before and after conditioning stimulation (right) are shown. Conditioning stimulation consisted of action potential trains (1 s, 100 Hz, top), PF bursts (5 stimuli, 50 Hz, middle), or PF bursts delivered in the middle of the action potential trains (bottom), with 7 trials averaged for each condition. The calculated sum of the calcium signal during the burst and train is superimposed on the measured calcium signal for combined stimulation (gray trace). Calcium elevation (B) and relative synaptic strength (C) are summarized (n = 6 neurons). Responses evoked by the PF burst alone and the action potential train paired with PF burst were significantly different (asterisk indicates p < 0.01, 1-way ANOVA).
Molecular layer interneurons are typically referred to as SCs or basket cells (BCs). SCs are located in the external part of the molecular layer and synapse primarily onto the dendrites of Purkinje cells (PCs), whereas BCs are located near the Purkinje cell layer and form distinctive synapses onto cell bodies and initial axon segments of PCs (Palay and Chan-Palay, 1974). In intermediate regions of the molecular layer the distinction between SCs and BCs is less clear. We therefore restricted our studies to the outer third of the molecular layer.
Electrophysiology.
Whole-cell recordings were obtained with borosilicate glass electrodes of 2–3.5 MΩ resistances. For depolarization-induced suppression of excitation (DSE) experiments (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), the internal solution contained the following (in mm): 145 Cs-MeSO3, 15 HEPES, 10 tris-phosphocreatine, 5 tetraethylammonium chloride (TEA-Cl), 2 Mg-ATP, 2 QX-314, 1 MgCl2, 0.3 Na-GTP, 0.2 EGTA, and 0.16 CaCl2, adjusted to 310 mOsm and pH 7.3. For all other experiments, the internal solution contained the following (in mm): 135 K-MeSO3, 10 HEPES, 2 Mg-ATP, 0.4 Na-GTP, 14 tris-phosphocreatine, adjusted to 310 mOsm and pH 7.3. Recordings were made with a Multiclamp 700B amplifier (Molecular Devices). For current-clamp experiments, series resistance was compensated completely by balancing the bridge on the amplifier. For voltage-clamp experiments, series resistance was compensated by 75%.
To evoke action potentials at the soma, 2 ms current pulses (1–2 nA) were delivered through the patch pipette. To stimulate parallel fibers (PFs), a borosilicate glass electrode filled with saline was placed in the molecular layer, and 0.2 ms shocks (25–100 μA) were delivered to the slice via a stimulus isolator (World Precision Instruments). In Figures 1, D and E, and 9, the stimulus electrode was placed above a dendrite as identified by two-photon imaging. In Figures 7 and 8 and supplemental Figure 1 (available at www.jneurosci.org as supplemental material), the stimulating electrode was placed in the molecular layer 100–250 μm away from the neuron to activate PFs that made synaptic contacts on dendrites ∼50 μm from the soma. Although we believe eCBs to be released throughout the dendritic arbor of SCs, eCBs do not affect synapses far from the release site (Kreitzer et al., 2002) and thus the eCBs that influence the PF synaptic inputs we are monitoring must be released from dendritic regions near the activated synapses. In Figures 1, D and E, and 9, the sites of PF synaptic inputs to SCs were determined by localizing the maximum PF-evoked dendritic sodium or calcium responses in the dendrite, respectively.
Experiments described in Figures 1–6 were performed in standard saline. Where indicated, tetrodotoxin (TTX, 500 nm) or 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX, 10 μm) was washed in. Experiments described in Figures 7–9 and supplemental Figure 1 (available at www.jneurosci.org as supplemental material) were performed in the presence of picrotoxin (20 μm) to block GABAA-mediated synaptic transmission and (2S)-3-[(15)-1-(3,4-dichlorophenyl)ethyl] amino-2-hydroxypropyl)(phenylmethyl) phosphinic acid (CGP 55845, 2 μm) to block GABAB receptors. Here, PFs were stimulated at 0.5 Hz to establish a 10 s baseline of EPSCs before conditioning stimuli were delivered to the neuron. After the conditioning stimulus, PFs were stimulated again at 0.5 Hz for 30 s. Trials were repeated every minute, and each experimental condition represents an average of 3–8 trials. Because the quantal size of PF to SC synapses is large, EPSCs onto SCs show considerable variability (Carter and Regehr, 2002). This variability was reduced by activating large numbers of inputs, and including 150 nm NBQX in the bath to reduce EPSC amplitudes by 60–75% to minimize series resistance errors (Beierlein and Regehr, 2006). Importantly, NBQX was not included in the experiments of Figure 9, which relied in part on synaptically evoked eCB release. As a result it was necessary to average many trials (6–8) in the experiments of Figure 9 to overcome the stochastic variability of the EPSC. Additionally, because experiments of Figure 9 compared three different stimulus conditions, for each repetition the three conditioning stimuli were delivered at random. To assess the effects of mGluR1/5 activation on eCB release, control data were collected, and then (S)-2-amino-2-(3,5-dihydroxyphenyl)acetic acid (DHPG, 5 μm) was washed in (Ohno-Shosaku et al., 2002). Experiments in the presence of DHPG were completed within 10 min after wash-in to minimize mGluR1/5-mediated synaptic suppression. All drugs were dissolved in water except for picrotoxin and CGP 55845, which were dissolved in dimethylsulfoxide. All drugs were purchased from Tocris, except for TTX, which was purchased from Sigma. All other chemicals were purchased from Sigma.
Imaging and data acquisition.
Imaging experiments were performed on a custom two-photon laser scanning microscope equipped with a pulsed Ti:Sapphire laser (Coherent) and 60× 0.9 numerical aperture objective (Olympus). Electrophysiology and imaging were controlled by custom software written in Matlab, which was generously provided by Bernardo Sabatini (Harvard Medical School, Boston, MA). For sodium-imaging experiments (Fig. 1), the fluorescent sodium indicator sodium-binding benzofuran isophthalate (SBFI; 2 mm) was added to the internal solution. Frame scans were performed at dendritic or axonal sites at 8 Hz. Relative sodium levels were determined by normalizing the average of 3–5 green fluorescence signals to blanked trials, which were interleaved with stimulus trials. For calcium-imaging experiments, a green fluorescent calcium indicator (50 μm fluo-5F for Figs. 2–6, 400 μm fluo-4FF for Figs. 7–9) and a red calcium-insensitive dye (Alexa 594, 50 μm) were added to the internal solution. Line scans across dendrites or axons were performed at 500 Hz to collect green and red fluorescence during single action potentials (repeated 12–16 times and averaged) or 2 s 100 Hz action potential trains (repeated 4–6 times and averaged). The ratio of green to red fluorescence (G/R) was converted to calcium concentration using published KD values for fluo-5F (Brenowitz and Regehr, 2007) (585 nm) and fluo-4FF (Yasuda et al., 2004) (8.1 μm) as well as minimum (Rmin) and maximum (Rmax) G/R values as described previously (Grynkiewicz et al., 1985; Brenowitz and Regehr, 2003). All fluorescent indicators and dyes were purchased from Invitrogen Detection Technologies.
Data analysis.
SC dendrite morphologies obtained from two-photon z stacks were traced using the NeuronJ plugin for ImageJ (Abràmoff et al., 2004; Meijering et al., 2004). Dendrites were distinguished from axons by their larger diameter at the soma, which tapered with distance. Axons were thinner at the soma and did not taper. Primary dendrites were defined as the longest path of dendrite from the soma to the tip. Secondary branches were defined as protrusions from the primary dendrite that were longer than 10 μm.
Physiology and calcium-imaging data were analyzed off-line using Igor Pro (WaveMetrics). Averages are presented as mean ± SEM. Where indicated, one-way ANOVA or Student's t tests were performed to determine statistical significance of the data.
Results
Somatic action potential trains elevate sodium levels in axons but not in dendrites of stellate cells
Sodium action potentials elevate sodium within neurons (Jaffe et al., 1992; Regehr, 1997; Rose et al., 1999; Rose, 2003; Kole et al., 2008). We therefore measured sodium levels in SCs to provide an indirect measure of the relative sodium channel densities in axons and dendrites. We detected changes in intracellular sodium levels by recording from SCs with electrodes containing the sodium indicator SBFI (Minta and Tsien, 1989). Because individual action potentials only lead to small changes in intracellular sodium, this approach is not suited to measuring the sodium responses to single action potentials. Instead, we imaged dendritic and axonal regions 50 μm from the soma during trains of action potentials (1 s, 100 Hz) (Fig. 1A,B). Action potential trains produced robust decreases in SBFI fluorescence in axons (Fig. 1B, site 1) (−14 ± 2% ΔF/F, n = 8 neurons) corresponding to substantial increases in axonal sodium. TTX eliminated sodium action potentials (Fig. 1A, bottom) and nearly eliminated train-evoked axonal sodium increases (Fig. 1C) (ΔF/F was reduced from −17 ± 4% to −1.5 ± 1.5% in TTX, n = 4 neurons). In contrast to axons, action potential trains produced only small changes in dendritic fluorescence (Fig. 1B, site 2) (−2.3 ± 0.3% ΔF/F, n = 8 neurons). Thus, intense firing robustly elevates sodium levels in axons, but not in dendrites.
We next examined the sensitivity of our dendritic sodium measurements by activating parallel fibers (PFs) and measuring the resulting local sodium increases. In contrast to somatic action potentials, synaptic activation substantially elevated dendritic sodium (Fig. 1D,E). As shown for a representative experiment, PF activation (20 stimuli, 50 Hz) depolarized the cell (Fig. 1D, right) and elevated dendritic sodium near the site of stimulation (Fig. 1E). The AMPA receptor antagonist NBQX prevented both the somatic depolarization and the elevation of dendritic sodium (Fig. 1E) (ΔF/F was reduced from −19 ± 3% to −1.7 ± 1.3%, n = 4 neurons). This indicates that sodium entry through AMPA receptors can lead to dendritic sodium transients that are readily detected. These results demonstrate that sodium levels in dendrites can be robustly elevated by synaptic stimulation, but not by somatic spiking.
Somatic action potentials elevate dendritic calcium in stellate cells
We next measured dendritic calcium levels to determine the extent of action potential invasion into SC dendrites. Whole-cell recordings of SCs were obtained with electrodes containing the green fluorescent calcium indicator fluo-5F (Brenowitz and Regehr, 2007) (50 μm, KD = 585 nm) and the red calcium-insensitive fluorescent dye Alexa 594 (50 μm). Single action potentials or trains of action potentials (2 s, 100 Hz) were evoked in current clamp with brief current pulses (2 ms, 1–2 nA). Calcium levels were calculated from the green/red fluorescence obtained using line scans across dendritic sites 50 μm from the soma (Fig. 2A,F) (Yasuda et al., 2004).
The neurons we recorded from could be divided into two types of cells that differed with respect to the properties of single action potentials and associated calcium increases, but were similar in dendritic calcium signaling during action potential trains. Most of the neurons we recorded from (∼80%) (Fig. 2A–E) had action potentials with a strong afterhyperpolarization (Fig. 2B). In these cells, single action potentials elevated calcium to ∼10 nm, regardless of the initial potential of the cell (Fig. 2C, left). TTX eliminated the sodium action potential (Fig. 2B, right) and greatly reduced the dendritic calcium responses (Fig. 2C, right). Trains of action potentials (2 s, 100 Hz) elevated dendritic calcium to several hundred nanomolar (Fig. 2D, left). TTX greatly reduced the calcium elevations evoked by trains (Fig. 2D, right). Thus, in the majority of cells, TTX eliminated somatic action potentials, which in turn greatly reduced dendritic calcium elevations (Fig. 2E).
The remaining SCs (∼20%) (Fig. 2F) exhibited a slow afterdepolarization when a single action potential was evoked from −80 mV (Fig. 2G, left, black traces) but not −60 mV (Fig. 2G, left, gray traces). Single action potentials evoked larger dendritic calcium increases when evoked from −80 mV compared with −60 mV (Fig. 2H, left). This voltage dependence is consistent with the involvement of T-type calcium channels, which are present in SCs (Molineux et al., 2005). TTX did not reduce the slow afterdepolarization (Fig. 2G, right), only slightly reduced the magnitude of calcium increases evoked by single action potentials when the cell was initially at −80 mV and greatly reduced the magnitude of the calcium signals when the initial potential was −60 mV (Fig. 2H, right). Trains of action potentials (2 s, 100 Hz) produced similar calcium increases in control conditions, regardless of the initial potential (Fig. 2I, left). This is consistent with the observation that the slow afterdepolarization became much less prominent during trains (data not shown). TTX reduced the calcium elevations regardless of the initial potential (Fig. 2I, right). Thus, although for a holding potential of −80 mV the calcium increases evoked by single action potentials were only weakly TTX sensitive, calcium elevations evoked by trains of action potentials were greatly reduced in TTX (Fig. 2J).
These findings indicate that action potentials elevate dendritic calcium in SCs. This study is focused primarily on dendritic calcium signaling during trains of action potentials, and such trains elevated calcium in a similar TTX-sensitive manner in all SCs studied (Fig. 2E,J).
Action-potential-evoked dendritic calcium elevations do not require dendritic sodium channels
We next determined whether dendritic sodium channels are required for action-potential-evoked calcium elevations. As shown for a representative dendritic site (50 μm from the soma) (Fig. 3A), a single action potential and a train of action potentials elevated dendritic calcium by 20 and 300 nm, respectively (Fig. 3B, top). TTX prevented the generation of action potentials and greatly reduced the amplitude of calcium elevations (Fig. 3B, middle). In the presence of TTX, voltage clamping the soma with the action potential waveform recorded in control conditions (Stuart and Sakmann, 1994) elevated dendritic calcium to levels that were comparable to those measured in control conditions in current clamp (Fig. 3B, bottom). The similarity of the calcium signals evoked by action potentials in current clamp and by voltage-clamped action potentials in TTX indicates that dendritic sodium channels are not required for somatic action potentials to invade SC dendrites. Instead, passive spread of membrane depolarization is sufficient to convey somatic action potentials to the dendrites of SCs.
In another experiment, single action potentials and trains of action potentials also elevated axonal calcium levels (Fig. 3C, top). TTX eliminated axonal calcium elevations both in current clamp (Fig. 3C, middle) and when the soma was voltage clamped with the action potential waveform (Fig. 3C, bottom). The inability of voltage-clamped action potentials in TTX to evoke axonal calcium signals suggests that, in contrast to dendrites, the passive spread of depolarization associated with a somatic action potential is not effective at evoking calcium signals in axons.
Voltage-clamp rescue of calcium signals in the presence of TTX in dendrites but not axons was a consistent finding for single action potentials (Fig. 3D) and for trains of action potentials (Fig. 3E). For sites between 75 and 100 μm from the soma, the calcium signals evoked by single action potentials in current clamp, current clamp in TTX, and voltage-clamped action potentials in TTX were 11.3 ± 2.4 nm, 1.6 ± 1.8 nm, and 9.6 ± 2.2 nm, respectively (Fig. 3D, left) (n = 5 neurons). Action potential trains at the same sites elevated dendritic calcium by 272 ± 25 nm, 56 ± 22 nm, and 222 ± 20 nm, respectively (Fig. 3E, left). Dendritic calcium signals were slightly smaller with increasing distance from the soma (Fig. 3D,E, left). At axonal sites <100 μm from the soma, single action potentials and action potential trains evoked calcium signals (51 ± 8 nm and 2.06 ± 0.54 μm, respectively, n = 7 neurons) (Fig. 3D,E, right) that were larger than in dendrites (Fig. 3D,E, left). TTX reliably eliminated the axonal calcium elevations in current clamp and in action potential voltage clamp (Fig. 3D,E, right). Thus, somatic action potentials elevate dendritic calcium in a manner that does not rely on sodium channels in the dendrites. Despite the lack of dendritic sodium spikes, trains of somatic action potentials can elevate dendritic calcium to several hundred nanomolar.
The substantial calcium increases that can be evoked by somatic depolarization in the absence of sodium channels suggest that there is significant passive spread of potential within SC dendrites from P17–P19 rats. We therefore analyzed dendritic morphology, which is a major determinant of the passive spread of potential. In P17–P19 SCs, primary dendrites were short (Fig. 4A), with a mean length of 60 ± 2 μm (n = 247 dendrites) (Fig. 4C,D). There were 4.7 ± 0.1 primary dendrites per cell (n = 54 neurons) (Fig. 4E,F). The dendrites were not extensively branched, with only 1.7 ± 0.1 secondary branches per primary dendrite (Fig. 4G,H). The short dendrites and simple branching of P17–P19 SCs makes them well suited to support passive spread of somatic action potentials into the dendritic arbor.
Dendritic signaling in stellate cells of P30–P33 rats
It is possible that developmental changes in dendritic morphology or sodium channel density alter dendritic signaling in the SCs of older animals. We therefore extended our studies of dendritic signaling to SCs from older rats (P30–P33). The dendrites were more variable in length (Fig. 4B,C) and the average length of primary dendrites was longer than in young animals (90 ± 3 μm, n = 193 dendrites) (Fig. 4C,D). SCs from P30–P33 rats had 3.8 ± 0.2 primary dendrites (n = 50 neurons) (Fig. 4E,F) and maintained a simple branching pattern, with 2.4 ± 0.2 secondary branches per primary dendrite (Fig. 4G,H). We also observed numerous short protrusions from the dendrites (Fig. 4B), but as these did not appear to be full-fledged dendrites, they were excluded from analysis (see Materials and Methods). Thus, in P30–P33 SCs some of the dendrites have short, simple morphologies that favor the spread of passive potentials, as in P17–P19 SCs, but the rest of the dendrites are longer and more attenuation is possible with distance from the soma.
We characterized action potential-evoked dendritic calcium increases in P30–P33 SCs (Fig. 5). Action potentials were evoked in the soma and calcium levels were measured at various dendritic locations, as in Figure 3. As shown for a representative experiment (Fig. 5A–D), single action potentials and 2 s 100 Hz action potential trains in control conditions elevated dendritic calcium near the soma comparably to younger SCs (Fig. 5C,D, site 1, black traces). However, calcium signals strongly attenuated with distance from the soma (Fig. 5C,D, sites 2 and 3). On average, dendritic calcium elevations at 75–100 μm from the soma were ∼25% of those observed in dendrites near the soma (Fig. 5E,F, black bars). TTX eliminated the action potential (Fig. 5B, blue trace) and substantially reduced dendritic calcium elevations (Fig. 5C–F, blue). The large TTX-insensitive calcium signals observed in some P17–P19 SCs (Fig. 2F–J) were not observed in any P30–P33 SCs. Thus, action potentials elevate dendritic calcium in older SCs, but on average somatic action potentials are ineffective at elevating calcium in distal compartments.
To gain insight into whether dendritic length influences calcium signaling, we divided the data from Figure 5, E and F, into groups based on the length of the primary dendrite (Fig. 5G). In dendrites longer than 100 μm, calcium elevations between 25 and 50 μm from the soma were only 20% of those observed at sites <25 μm from the soma (Fig. 5G, gray bars). In contrast, dendrites between 40 and 50 μm long exhibited little attenuation (Fig. 5G, black bars). These findings suggest that calcium signaling is similar in short dendrites of P30–P33 SCs and in dendrites of P17–P19 SCs.
Finally, we determined whether dendritic sodium channels contribute to action-potential-evoked calcium elevations in P30–P33 SCs. We performed voltage-clamp rescue experiments in the presence of TTX at sites between 70 and 100 μm from the soma (Fig. 6). In the presence of TTX, voltage-clamped action potentials elevated dendritic calcium to levels similar to control conditions in current clamp, as shown in a representative experiment (Fig. 6A–D). This finding was consistent among six neurons tested for both single action potentials and 2 s 100 Hz action potential trains (Fig. 6E). Thus, dendritic sodium channels do not contribute to action-potential-evoked dendritic calcium increases in P30–P33 SCs.
Action potential trains and retrograde endocannabinoid signaling
Previous studies have shown that SCs can regulate the strength of their synaptic inputs by releasing eCBs from their dendrites in a calcium-dependent manner (Beierlein and Regehr, 2006; Soler-Llavina and Sabatini, 2006). We tested the role of somatic action potentials in regulating the release of eCBs from SC dendrites. We focused on the dendrites of SCs from P17–P19 rats in these studies (Figs. 7–9) because they had consistently short dendrites and large evoked calcium increases.
We initially tested whether high-frequency spike trains elevate calcium sufficiently to evoke eCB release and suppress synaptic transmission. As shown in a representative experiment (Fig. 7A), PF EPSCs were first measured in voltage clamp. The recording was then switched to current clamp and action potentials were evoked in the neuron (2 s, 200 Hz). Following the train, the recording was returned to voltage clamp and PF EPSCs were measured again. For 2-s-long action potential trains in control conditions, 200 Hz trains transiently reduced EPSC amplitude to 71 ± 6% of the initial value (Fig. 7B, black circles) (n = 9 neurons), but lower frequency trains did not result in suppression (Fig. 7C, black bars). This suggests that for 2 s trains, SCs must fire at frequencies greater than the 10–60 Hz firing frequencies observed in vivo (Ekerot and Jörntell, 2003).
The activation of mGluR1/5 can reduce the calcium levels necessary to evoke eCB release by activating postsynaptic the β isoform of phospholipase C (PLCβ) (Varma et al., 2001; Galante and Diana, 2004; Brenowitz and Regehr, 2005; Hashimotodani et al., 2005; Maejima et al., 2005). We tested whether SC spiking would more effectively promote eCB release when paired with activation of mGluR1/5 receptors. Although strong activation of mGluR1/5 can drive eCB release on its own (Maejima et al., 2001), low-level activation can reduce the calcium dependence of eCB release without affecting synaptic transmission (Ohno-Shosaku et al., 2002). We therefore bath applied the mGluR1/5 agonist DHPG at a concentration (5 μm) that alone did not substantially affect synaptic strength (94 ± 10% of control, n = 29). In the presence of DHPG, 200 Hz trains reduced normalized EPSC amplitudes to 46 ± 6% of baseline amplitude (Fig. 7B, red circles) (n = 7 neurons). The strong suppression in the presence of DHPG was prevented by the CB1 receptor antagonist AM 251 (2 μm) (Fig. 7B,C, blue). In the presence of DHPG, 100 Hz and 50 Hz trains also suppressed synaptic strength, but 25 Hz trains did not (Fig. 7C) (p < 0.05, unpaired t tests between control and DHPG for 50, 100, and 200 Hz). These findings indicate that action potential trains in SCs can retrogradely suppress their synaptic inputs by promoting eCB release from their dendrites. mGluR1/5 activation increases the magnitude of suppression and lowers the firing frequencies required for suppression.
To gain insight into how mGluR1/5 activation enhances eCB release, we determined the calcium dependence of spiking-evoked suppression (Fig. 7D,E) (Wang and Zucker, 2001; Brenowitz and Regehr, 2003; Brenowitz et al., 2006). Experiments were performed as in Figure 7A, except that dendritic calcium was also measured during the trains. In control conditions, a 2 s 200 Hz action potential train elevated dendritic calcium to 1.5 μm and reduced synaptic strength by 30% (Fig. 7D, top), and a 50 Hz train elevated calcium to 0.5 μm and did not reduce synaptic strength (Fig. 7D, middle). After wash-in of DHPG, the 50 Hz train reduced synaptic strength by 50% even though it did not alter the calcium signal (Fig. 7D, bottom).
We performed a series of such experiments to determine the calcium dependence of spike-evoked suppression (Fig. 7E). We plotted normalized EPSC amplitudes against peak calcium elevation for 25, 50, 100, and 200 Hz trains and fit the data with the Hill equation. In control conditions, half-maximal suppression occurred at 2.15 ± 0.26 μm calcium, and in DHPG, half-maximal suppression occurred at 0.80 ± 0.11 μm calcium. Thus, mGluR1/5 activation facilitates spiking-evoked eCB signaling by increasing the calcium sensitivity of eCB release.
These results suggest that SCs require lower levels of calcium to evoke eCB release compared with other cell types, where 4–7 μm calcium is required for half-maximal synaptic suppression (Wang and Zucker, 2001, Brenowitz and Regehr, 2003). Importantly, in such studies dendritic calcium was elevated by depolarizing the neuron in voltage clamp with a Cs-based internal solution and is widely termed depolarization-induced suppression of inhibitory (DSI) and excitatory (DSE) synapses (Lenz and Alger, 1999; Kreitzer and Regehr, 2001; Wilson and Nicoll, 2001). Photolysis of caged-calcium has also been used to determine the calcium dependence of eCB release and requires 4 μm calcium to evoke half-maximal synaptic suppression (Wang and Zucker, 2001). The similar calcium sensitivities reported for studies in which depolarization or photolysis of caged-calcium were used to evoke eCB release suggest that bulk calcium rather than local calcium triggers release.
To gain insight into the apparent difference between the calcium dependence of eCB signaling in SCs and other neurons, we determined the calcium dependence of DSE at PF to SC synapses. Here, we performed experiments similar to those of Figure 7, but used a Cs-based internal solution and elevated dendritic calcium by depolarizing the cell from a holding potential of −70 mV to potentials between −50 and 0 mV for 2 s (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Half-maximal DSE occurred at 4.5 ± 0.4 μm. Thus, when eCB signaling at PF to SC synapses is evoked by depolarization in a Cs-based internal solution, similar levels of calcium are required for eCB release compared with other neurons. One possible explanation for the differences in the calcium dependence of eCB signaling for K-based and Cs-based internal solutions is that high local calcium signals (which are not readily detected with our imaging methodology) could be more prominent when eCB release is driven by action potentials using a K-based internal solution, which would cause us to underestimate the calcium levels that trigger eCB release (as opposed to a smaller steady calcium influx in experiments in which a Cs-based internal solution was used).
Prolonged action potential trains and mGluR1/5 activation promote spiking-evoked endocannabinoid signaling
Prolonged calcium elevations can be more effective than brief calcium increases at evoking eCB release (Brenowitz et al., 2006). We therefore examined the calcium dependence and mGluR1/5 dependence of eCB signaling evoked by 15 s action potential trains.
A 15 s 100 Hz train elevated calcium to 0.5 μm and reduced synaptic strength by 20% (Fig. 8A, top). After wash-in of DHPG, the 100 Hz train reduced synaptic strength by 60% without affecting the dendritic calcium signal (Fig. 8A, bottom). Although 2 s 100 Hz trains did not reduce synaptic strength, 15 s 100 Hz trains reduced synaptic strength by 30 ± 5% on average (Fig. 8B, black bars) (n = 17 neurons). DHPG facilitated synaptic suppression evoked by 15 s trains, permitting frequencies as low as 25 Hz to evoke synaptic suppression (Fig. 8B, red bars) (p < 0.01, unpaired t tests between control and DHPG for 25, 50, and 100 Hz trains). At the highest frequency tested (100 Hz), AM 251 blocked all synaptic suppression (Fig. 8B, blue bar).
We determined the calcium dependence of eCB signaling for prolonged trains by plotting normalized EPSC amplitude against peak calcium elevation during 15 s action potential trains (Fig. 8C). In control conditions for 15 s trains half-maximal suppression occurred at 1.29 ± 0.23 μm calcium, which is approximately a factor of 2 lower than for 2 s trains. In DHPG half-maximal suppression occurred at 0.36 ± 0.03 μm calcium for 15 s trains, which is also about a factor of 2 lower than for 2 s trains.
Thus, 15 s action potential trains are more effective at evoking eCB signaling, compared with 2 s trains. Pairing mGluR1/5 activation with prolonged SC firing is particularly effective at evoking eCB release. Here, physiologically relevant firing frequencies in SCs (Ekerot and Jörntell, 2003) (25–60 Hz) are sufficient to regulate the strength of their PF synaptic inputs.
Stellate cells associate presynaptic and postsynaptic activity
It has been previously shown that presynaptic activity can evoke retrograde eCB signaling in SCs (Beierlein and Regehr, 2006; Soler-Llavina and Sabatini, 2006). Our present results indicate that somatic spiking in SCs can also evoke retrograde eCB signaling. We therefore determined whether presynaptic activity and postsynaptic activity interact to promote eCB signaling at PF to SC synapses.
We measured dendritic calcium and synaptic strength for experiments using three conditioning stimuli: an action potential train (1 s, 100 Hz), a PF burst (5 stimuli, 50 Hz), and coincident stimulation where the PF burst was delivered in the middle of the action potential train. Although more prolonged PF activation can produce large retrograde inhibition (Brown et al., 2003), brief PF activation produced little or no synaptic suppression. In a representative experiment, the spike train elevated dendritic calcium to 0.5 μm and did not change synaptic strength (Fig. 9A, top). The PF burst locally elevated dendritic calcium to 2 μm and reduced EPSC amplitude by 20% (Fig. 9A, middle). Combining the train and burst elevated calcium to 2.5 μm and reduced EPSC amplitude by 40% (Fig. 9A, bottom). For combined stimulation, the calcium increase was well approximated by the linear summation of the calcium elevations for the train alone and the burst alone (Fig. 9A, bottom, gray trace). The linear summation of calcium elevation evoked by presynaptic and postsynaptic activity was consistent among six neurons tested (Fig. 9B). The train and burst generated significantly larger calcium increases compared with the burst alone (p < 0.01, one-way ANOVA).
Action potential trains also consistently promoted synaptic suppression when paired with PF bursts (Fig. 9C). The trains did not substantially reduce synaptic strength on their own (0.89 ± 0.05 normalized EPSC), but pairing them with the PF burst reduced the EPSC amplitude to a greater extent than the PF burst alone (reduced to 0.50 ± 0.08 fraction of baseline, compared with 0.74 ± 0.06, p < 0.01, one-way ANOVA). This indicates that postsynaptic spiking activity facilitates synaptically evoked retrograde eCB signaling. Here, passive spread of somatic action potentials influences dendritic calcium signaling to promote eCB release from SC dendrites during brief bursts of synaptic activity.
Discussion
We have shown that in SCs, somatic action potentials effectively depolarize short dendrites without requiring dendritic sodium channels. Despite the lack of active backpropagating action potentials, trains of action potentials in the dendrites of P17–P19 SCs elevated dendritic calcium sufficiently to evoke dendritic eCB release that retrogradely suppressed PF synaptic inputs. When mGluR1/5 was activated, prolonged SC firing at physiologically relevant rates produced retrograde suppression. SC spiking also enhanced synaptically evoked eCB signaling and promoted associative plasticity at PF to SC synapses. These findings suggest that in electrically compact neurons, passive spread of potential changes in the soma can allow somatic spiking to regulate dendritic signaling, even in the absence of backpropagating action potentials.
Spread of somatic potentials within stellate cells
Our findings indicate that in SCs, dendritic sodium channels do not play an important role in conveying potentials from the soma to the dendrites. Trains of somatic action potentials were ineffective at elevating dendritic sodium, suggesting that SC dendrites express sodium channels at low density, if at all. In most SCs somatic depolarization evoked a large rapid sodium action potential that was eliminated by TTX. In most P17–P19 SCs, and in all P30–P33 SCs, a single spike evoked small increases in dendritic calcium that were largely eliminated by TTX. Because SCs fire spontaneously in vivo (Ekerot and Jörntell, 2003), we investigated dendritic calcium elevations evoked by trains of action potentials and found that high-frequency trains elevated calcium to several hundred nanomolar. Regardless of whether slow afterdepolarizations were observed, TTX greatly reduced the magnitude of the calcium signals evoked by such trains, and voltage clamp of the soma in the presence of TTX rescued dendritic calcium increases. This indicates that in SCs, somatic action potentials depolarize dendrites sufficiently to open voltage-gated calcium channels without relying on sodium channels in the dendrites.
There are several differences in dendritic signaling between P17–P19 and P30–P33 SCs. The most striking difference is that in the longer dendrites of P30–P33 SCs calcium increases evoked by single somatic action potentials or trains of action potentials exhibit a pronounced attenuation with distance from the soma, whereas in the dendrites of P17–P19 SCs very little attenuation is apparent. It is likely that the more pronounced attenuation is a consequence of dendritic length, as calcium signals do not attenuate significantly in the short dendrites of P30–P33 SCs. Thus it seems that in P17–P19 SCs and in the shorter dendrites of P30–P33 SCs, the dendrites are sufficiently compact that passive propagation of somatic action potentials provides enough depolarization to open voltage-gated calcium channels. It remains unclear whether such dendritic signaling is important for conveying somatic potentials into short dendrites of other neurons as well (Kampa and Stuart, 2006; Nevian et al., 2007; Zhou et al., 2008; Acker and Antic, 2009).
The more pronounced attenuation of calcium signaling in the long dendrites of P30–P33 dendrites may reflect passive attenuation of voltage. A-type potassium channels, which are thought to limit the spread of depolarization in other types of cells (Magee and Johnston, 1997; Goldberg et al., 2003; Goldberg and Yuste, 2005), are also present in SCs and may limit the spread of somatic depolarization (Molineux et al., 2005). Another difference is that in a small fraction of P17–P19 cells, somatic depolarization evoked a sodium spike and a slow TTX-insensitive afterdepolarization lasting >10 ms, whereas in P30–P33 SCs such afterdepolarizations were never observed. A sodium spike accompanied by a slow afterdepolarization increased dendritic calcium by ∼50 nm, and TTX did not eliminate these calcium signals, suggesting that slow afterdepolarizations are produced by low-threshold calcium channels.
Sodium and calcium signaling are both markedly different in the dendrites and axons of SCs. Somatic spike trains evoked large TTX-sensitive sodium signals in the axons but not in the dendrites. This suggests that there is a much higher density of sodium channels in axons than in dendrites, although more reliable invasion of action potentials during spike trains could also contribute to the disparity in sodium signals. Another striking difference is that in the presence of TTX, voltage clamp of the soma rescued calcium signals in dendrites, but not axons. This is consistent with passive attenuation of somatic potentials being more pronounced in SC axons, which are much thinner than dendrites (Rall, 1969; Palay and Chan-Palay, 1974). Although prolonged somatic depolarization in the presence of TTX can elevate axonal calcium levels (Christie and Jahr, 2008), we find that somatic voltage clamp with trains of action potentials in the presence of TTX does not significantly elevate axonal calcium. This likely reflects the fact that transient depolarizations are strongly filtered by the passive properties of the axons (Rall and Segev, 1985; Spruston et al., 1993) so that compared with longer lasting changes in somatic potential, trains of action potentials are less effective at depolarizing the axon.
Stellate cell spiking and endocannabinoid signaling
We found that in P17–P19 SCs, somatic action potentials evoked eCB release from dendrites and suppressed PF synaptic inputs. For brief (2 s) action potential trains in control conditions, extreme frequencies (200 Hz) were needed to suppress synaptic strength. Two factors lowered the firing frequency required for retrograde signaling. First, mGluR1/5 activation promoted retrograde suppression, permitting 2 s 50 Hz trains to reduce synaptic strength. Second, prolonging the duration of firing to 15 s lowered the firing frequency required to produce retrograde signaling (100 Hz). When mGluR1/5 activation was combined with prolonged trains, firing frequencies of just 25 Hz were needed for retrograde signaling. Both mGluR1/5 activation and prolongation of firing appear to promote retrograde signaling by lowering the calcium levels required to evoke eCB release (Brenowitz and Regehr, 2005; Hashimotodani et al., 2005; Maejima et al., 2005). mGluR1/5 leads to eCB release through a pathway involving PLCβ, which has a relatively low calcium requirement. Both mGluR1/5-dependent and -independent retrograde signaling are dependent on the duration of SC firing, but the molecular mechanism underlying this enhancement of retrograde signaling is not understood.
The robust suppression observed when mGluR1/5 activation was paired with prolonged SC firing suggests that SC spiking could lead to retrograde suppression in vivo. Previous studies have shown that molecular layer interneurons can fire for many seconds at 10–60 Hz (Ekerot and Jörntell, 2003). Although glutamate signaling and mGluR1/5 activation that occurs within the cerebellar cortex in vivo has not been well characterized, mGluR1 activation in vivo is thought to be important because mice lacking mGluR1 exhibit profound ataxia (Aiba et al., 1994). It is likely that glutamate signals in the slice are very different from those that occur in vivo, because granule cells within the slice are not spontaneously active and stimulation activates only a small fraction of the granule cells within the slice. Therefore, low doses of DHPG provided a means of mimicking the activation of mGluR1/5 that might occur in vivo.
In P30–P33 SC dendritic regions near the soma, or throughout dendrites that are <100 μm long, it is possible that the eCB release and retrograde suppression could be evoked by somatic action potentials. In the distal regions of the long dendrites of P30–P33 SCs, it seems unlikely that high-frequency trains of somatic action potentials alone would evoke sufficient dendritic calcium signals to trigger eCB release. However, it remains a possibility that, when paired with mGluR1/5 activation, somatic action potentials could regulate eCB release in the long dendrites of SCs in older animals.
Stellate cell spiking and associative plasticity
We also found that the association of SC spiking and synaptic activation promoted the release of eCBs. The calcium increases arising from SC spiking summated linearly with the local dendritic calcium produced by PF activation. As a result there was a significant enhancement of retrograde eCB signaling at active synapses during periods of postsynaptic spiking. The properties of the associative plasticity at PF to SC synapses differ from forms of associative plasticity observed in other cells. For some cells, associative plasticity is highly timing dependent and involves pairing a single sodium spike with synaptic activation (Abbott and Nelson, 2000; Roberts and Bell, 2002; Sjöström et al., 2008). In contrast, it is necessary to fire SCs many times to elevate dendritic calcium levels sufficiently to produce retrograde signaling because the dendritic calcium increases arising from single action potentials are small. At other synapses, a supralinear calcium signal is observed, in which paired stimulation leads to calcium signals that are larger than the linear summation of the signals resulting from each stimulus alone (Magee and Johnston, 1997; Schiller et al., 1998; Wang et al., 2000; Waters et al., 2003; Brenowitz and Regehr, 2005; Canepari et al., 2007; Gasparini et al., 2007). Consequently, the magnitude of plasticity can be very large for paired stimulation, even if it is small for each stimulus alone. In contrast, there is not a pronounced supralinear calcium increase in SC dendrites, which explains the rather modest associative effect seen when pairing synaptic activation and postsynaptic spiking.
Footnotes
This work was supported by National Institutes of Health Grants DA024090 to W.G.R. and DA025450 to M.H.M. We thank M. Antal, A. Best, M. Carey, J. Crowley, D. Fioravante, C. Hull, and T. Pressler for comments on this manuscript.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Abràmoff MD, Magalhães PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Acker CD, Antic SD. Quantitative assessment of the distributions of membrane conductances involved in action potential backpropagation along Basal dendrites. J Neurophysiol. 2009;101:1524–1541. doi: 10.1152/jn.00651.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Local interneurons regulate synaptic strength by retrograde release of endocannabinoids. J Neurosci. 2006;26:9935–9943. doi: 10.1523/JNEUROSCI.0958-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Calcium dependence of retrograde inhibition by endocannabinoids at synapses onto Purkinje cells. J Neurosci. 2003;23:6373–6384. doi: 10.1523/JNEUROSCI.23-15-06373.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron. 2005;45:419–431. doi: 10.1016/j.neuron.2004.12.045. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Reliability and heterogeneity of calcium signaling at single presynaptic boutons of cerebellar granule cells. J Neurosci. 2007;27:7888–7898. doi: 10.1523/JNEUROSCI.1064-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Best AR, Regehr WG. Sustained elevation of dendritic calcium evokes widespread endocannabinoid release and suppression of synapses onto cerebellar Purkinje cells. J Neurosci. 2006;26:6841–6850. doi: 10.1523/JNEUROSCI.1280-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SP, Brenowitz SD, Regehr WG. Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci. 2003;6:1048–1057. doi: 10.1038/nn1126. [DOI] [PubMed] [Google Scholar]
- Canepari M, Djurisic M, Zecevic D. Dendritic signals from rat hippocampal CA1 pyramidal neurons during coincident pre- and post-synaptic activity: a combined voltage- and calcium-imaging study. J Physiol. 2007;580:463–484. doi: 10.1113/jphysiol.2006.125005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Dendritic NMDA receptors activate axonal calcium channels. Neuron. 2008;60:298–307. doi: 10.1016/j.neuron.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie JT, Kole MH, Letzkus JJ, Rancz EA, Spruston N, Stuart GJ, Häusser M. Dendritic patch-clamp recording. Nat Protoc. 2006;1:1235–1247. doi: 10.1038/nprot.2006.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekerot CF, Jörntell H. Parallel fiber receptive fields: a key to understanding cerebellar operation and learning. Cerebellum. 2003;2:101–109. doi: 10.1080/14734220309411. [DOI] [PubMed] [Google Scholar]
- Emri Z, Antal K, Gulyás AI, Megías M, Freund TF. Electrotonic profile and passive propagation of synaptic potentials in three subpopulations of hippocampal CA1 interneurons. Neuroscience. 2001;104:1013–1026. doi: 10.1016/s0306-4522(01)00136-1. [DOI] [PubMed] [Google Scholar]
- Galante M, Diana MA. Group I metabotropic glutamate receptors inhibit GABA release at interneuron–Purkinje cell synapses through endocannabinoid production. J Neurosci. 2004;24:4865–4874. doi: 10.1523/JNEUROSCI.0403-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, Losonczy A, Chen X, Johnston D, Magee JC. Associative pairing enhances action potential back-propagation in radial oblique branches of CA1 pyramidal neurons. J Physiol. 2007;580:787–800. doi: 10.1113/jphysiol.2006.121343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JH, Yuste R. Space matters: local and global dendritic Ca2+ compartmentalization in cortical interneurons. Trends Neurosci. 2005;28:158–167. doi: 10.1016/j.tins.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Goldberg JH, Tamas G, Yuste R. Ca2+ imaging of mouse neocortical interneurone dendrites: Ia-type K+ channels control action potential backpropagation. J Physiol. 2003;551:49–65. doi: 10.1113/jphysiol.2003.042580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Häusser M, Stuart G, Racca C, Sakmann B. Axonal initiation and active dendritic propagation of action potentials in substantia nigra neurons. Neuron. 1995;15:637–647. doi: 10.1016/0896-6273(95)90152-3. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- Kaiser KM, Zilberter Y, Sakmann B. Back-propagating action potentials mediate calcium signalling in dendrites of bitufted interneurons in layer 2/3 of rat somatosensory cortex. J Physiol. 2001;535:17–31. doi: 10.1111/j.1469-7793.2001.t01-1-00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Stuart GJ. Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J Neurosci. 2006;26:7424–7432. doi: 10.1523/JNEUROSCI.3062-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–186. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Carter AG, Regehr WG. Inhibition of interneuron firing extends the spread of endocannabinoid signaling in the cerebellum. Neuron. 2002;34:787–796. doi: 10.1016/s0896-6273(02)00695-5. [DOI] [PubMed] [Google Scholar]
- Lenz RA, Alger BE. Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol. 1999;521:147–157. doi: 10.1111/j.1469-7793.1999.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cβ4 signaling cascade in the cerebellum. J Neurosci. 2005;25:6826–6835. doi: 10.1523/JNEUROSCI.0945-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Martina M, Vida I, Jonas P. Distal initiation and active propagation of action potentials in interneuron dendrites. Science. 2000;287:295–300. doi: 10.1126/science.287.5451.295. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Minta A, Tsien RY. Fluorescent indicators for cytosolic sodium. J Biol Chem. 1989;264:19449–19457. [PubMed] [Google Scholar]
- Molineux ML, Fernandez FR, Mehaffey WH, Turner RW. A-type and T-type currents interact to produce a novel spike latency-voltage relationship in cerebellar stellate cells. J Neurosci. 2005;25:10863–10873. doi: 10.1523/JNEUROSCI.3436-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Larkum ME, Polsky A, Schiller J. Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nat Neurosci. 2007;10:206–214. doi: 10.1038/nn1826. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. New York: Springer-Verlag; 1974. [Google Scholar]
- Rall W. Time constants and electrotonic length of membrane cylinders and neurons. Biophys J. 1969;9:1483–1508. doi: 10.1016/S0006-3495(69)86467-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W, Segev I. I. Space-clamp problems when voltage-clamping branched neurons with intracellular microelectrodes. In: Smith TGJ, Lecar H, Redman SJ, editors. Voltage and patch-clamping with microelectrodes. Bethesda, MD: American Physiological Society; 1985. pp. 191–215. [Google Scholar]
- Regehr WG. Interplay between sodium and calcium dynamics in granule cell presynaptic terminals. Biophys J. 1997;73:2476–2488. doi: 10.1016/S0006-3495(97)78276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PD, Bell CC. Spike timing dependent synaptic plasticity in biological systems. Biol Cybern. 2002;87:392–403. doi: 10.1007/s00422-002-0361-y. [DOI] [PubMed] [Google Scholar]
- Rose CR. High-resolution Na+ imaging in dendrites and spines. Pflugers Arch. 2003;446:317–321. doi: 10.1007/s00424-003-1036-9. [DOI] [PubMed] [Google Scholar]
- Rose CR, Kovalchuk Y, Eilers J, Konnerth A. Two-photon Na+ imaging in spines and fine dendrites of central neurons. Pflugers Arch. 1999;439:201–207. doi: 10.1007/s004249900123. [DOI] [PubMed] [Google Scholar]
- Saraga F, Wu CP, Zhang L, Skinner FK. Active dendrites and spike propagation in multi-compartment models of oriens-lacunosum/moleculare hippocampal interneurons. J Physiol. 2003;552:673–689. doi: 10.1113/jphysiol.2003.046177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham DE. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci. 1998;1:114–118. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Sabatini BL. Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat Neurosci. 2006;9:798–806. doi: 10.1038/nn1698. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Williams SH, Johnston D. Voltage- and space-clamp errors associated with the measurement of electrotonically remote synaptic events. J Neurophysiol. 1993;70:781–802. doi: 10.1152/jn.1993.70.2.781. [DOI] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995;268:297–300. doi: 10.1126/science.7716524. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Sakmann B. Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature. 1994;367:69–72. doi: 10.1038/367069a0. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Spruston N, Häusser M. Dendrites. Ed 2. Oxford: Oxford UP; 2007. [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21:RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zucker RS. Photolysis-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol. 2001;533:757–763. doi: 10.1111/j.1469-7793.2001.t01-1-00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Denk W, Häusser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]
- Waters J, Larkum M, Sakmann B, Helmchen F. Supralinear Ca2+ influx into dendritic tufts of layer 2/3 neocortical pyramidal neurons in vitro and in vivo. J Neurosci. 2003;23:8558–8567. doi: 10.1523/JNEUROSCI.23-24-08558.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Zhou WL, Yan P, Wuskell JP, Loew LM, Antic SD. Dynamics of action potential backpropagation in basal dendrites of prefrontal cortical pyramidal neurons. Eur J Neurosci. 2008;27:923–936. doi: 10.1111/j.1460-9568.2008.06075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]