

Abstract
About 70% of hepatocellular carcinomas are known to express α-fetoprotein, which is normally expressed in fetal but not in adult livers. To induce herpes simplex virus-thymidine kinase expression in these cancer cells, we constructed an adeno-associated viral vector containing the HSV-TK gene under the control of the α-fetoprotein enhancer and albumin promoter. We previously demonstrated in vitro that although this vector can transduce a variety of human cells, only transduced AFP and albumin-expressing hepatocellular carcinoma cell lines were sensitive to killing by ganciclovir (GCV). In the present study, we explored the effect of this vector on hepatocellular carcinoma cells in vivo. Subcutaneous tumors generated in nude mice by implanting hepatocellular carcinoma cells previously transduced with this vector shrank dramatically after treatment with GCV. Bystander effect was also observed on the tumors generated by mixing transduced and untransduced cells. To test whether the tumor cells can be transduced by the virus in vivo, we injected the recombinant adeno-associated virus into tumors generated by untransduced hepatocarcinoma cell line. Tumor growth were retarded after treatment with GCV. These experiments demonstrate the feasibility of in vivo transduction of tumor cell with rAAV.
Keywords: PLC/PRF/5 cell line, α-fetoprotein, nude mice, in vivo transduction
The most extensively used strategy for cancer gene therapy is to introduce a suicide gene, such as herpes simplex virus (HSV)-thymidine kinase (TK) gene followed by the antiviral drug, ganciclovir (GCV) treatment. GCV is a substrate for viral TK but a weak substrate for the mammalian enzyme. Viral TK phosphorylates GCV to the monophosphate form which is then converted to the triphosphate form by cytoplasmic enzymes. GCV triphosphate, when incorporated into replicating DNA, stops chain elongation and results in cell death (1, 2). In addition, GCV treatment also exerts a bystander effect as it causes not only the death the TK transduced cells but also the neighboring untransduced cells (3). These properties make the system attractive for cancer therapy.
Adeno-associated virus (AAV) is a potentially useful vector for cancer gene therapy as AAV is not known to cause any human disease. Unlike retrovirus, AAV can infect quiescent cells (4) and therefore facilitate the infection of slow growing tumor cells.
Random delivery of HSV-TK gene may harm normal proliferating cells, such as bone marrow and the epithelial cells of the intestine. HSV-TK delivered by adenovirus with nonspecific promoter has also been demonstrated to cause massive liver necrosis and death in animals after GCV treatment (5). One strategy to circumvent this problem is to deliver the TK gene in a tissue-specific manner as has been described with retroviral vectors (6, 7). Another is to express the gene specifically in the tissue to be treated. To achieve tissue-specific expression, several promoters and enhancers have been successfully used, including those for embryonic carcinoma antigen (8), tyrosinase (9) and α-fetoprotein (AFP) (10, 11). Since elevated levels of AFP have been observed in >70% of hepatocellular carcinomas (12), using AFP transcriptional sequence to achieve hepatocellular carcinoma specific gene expression is an ideal way. Because the package capacity of AAV is only 4.5 kb, it cannot accommodate the whole transcriptional sequence of AFP in the construct. To put TK gene with simian virus 40 polyA (2.2 kb) and a selectable marker, neoR gene (1.1 kb) together with a tissue specific promoter and enhancer within the virus, we used the minimum enhancer region of AFP (−3.7 to −3.3) and 0.3 kb minimum promoter of albumin. Our previous in vitro experiment (13) demonstrated that with this AAV vector, TK gene was specifically expressed in AFP and albumin positive hepatocellular carcinoma cells. Transgenic mice carrying this construct also showed predominant liver TK expression (13).
To study the in vivo function of this recombinant TK-AAV vector, we performed studies in athymic mice. Subcutaneous tumors generated by injection of TK-AAV transduced hepatocellular carcinoma cell line, PLC/PRF/5, shrank completely after 3 weeks GCV treatment. Bystander effect was also observed in vivo with GCV treatment when mice were inoculated with mixtures of different ratios of transduced and nontransduced cells. In addition, transduction of implanted untransduced tumors by direct injection of the TK-AAV also retarded tumor growth after treatment with GCV.
MATERIALS AND METHODS
Construction of Recombinant AAV Plasmids and Preparation of Viral Stocks.
Recombinant AAV plasmids, SSV9AFABTKneo, AAVTI2, and AAVAFABTI2 (Fig. 1) were constructed by standard plasmid subcloning technique. In SSV9AFABTKneo, the HSV-TK gene driven by a 0.3-kb human albumin promoter and 0.4-kb human AFP enhancer, as well as an upstream neomycin-resistance gene cassette, was inserted between the two inverted terminal repeats to replace the AAV genome. In AAVTI2 (provided by Avigen, Alameda, CA), a CMV promoter-driven HSV-TK gene was linked to the interleukin 2 (IL-2) gene by the encephalomyelitis virus internal ribosome entry site. To make AAVAFABTI2, the cytomegalovirus promoter in AAVTI2 was replaced by the albumin promoter and AFP enhancer. Preparation of virus stocks for infection of cells in vitro were performed with the procedure described by Samulski et al. (14). All the viruses used for in vivo experiment were prepared by Avigen.
Figure 1.
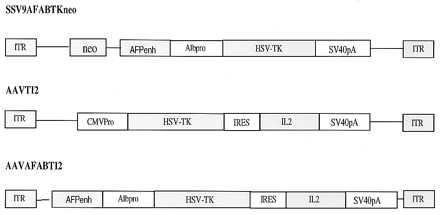
Structure of recombinant AAV viruses. In SSV9AFABTKneo, the HSV-TK with the albumin promoter (Alb pro) and AFP enhancer (AFP enh) was inserted downstream of the neoR gene and flanked by the two inverted terminal repeats. In AAVTI2, HSV-TK gene was linked to IL-2 gene by internal ribosome entry site, the bicistonic gene being driven by the cytomegalovirus promoter. AAVAFABTI2 virus was derived from AAVTI2 by replacing the cytomegalovirus promoter in AAVTI2 virus with albumin promoter and AFP enhancer. SSV9AFABTKneo and AAVAFABTI2 were used in experiments described in these studies.
Cell Line and Culture Conditions.
Human hepatocellular carcinoma cell line, PLC/PRF/5, was obtained from Yoshinor Murakami (National Cancer Research Institute, Tokyo) and was transduced by infecting with SSV9AFABTKneo virus as described (13). Both untransduced and transduced PLC/PRF/5 were maintained in DMEM with 10% fetal bovine serum. G418 (700 μg/ml) was added in above medium for culturing the transduced cells.
Generation of s.c. Tumors, in Vitro Transduction and GCV Treatment.
The transduced (TK+) or the untransduced (TK−) PLC/PRF/5 cells were trypsinized, washed with DMEM with 10% fetal calf serum to remove the enzyme, and harvested by centrifugation. The cell pellets were resuspended in serum-free DMEM medium. Cells (5 × 106) of TK+, TK−, or mixtures of different ratios of TK+ and TK− cells in 0.3 ml suspension were injected subcutaneously to the flank regions of adult nude mice (the Jackson Laboratory) to generate subcutaneous tumors. GCV (Roche Bioscience Products Development Unit, East Palo Alto, CA) treatment was started after the tumors grew to 0.5 to 1.3 cm in diameter. GCV (25 mg/kg) was injected i.p. twice daily for 21–40 days. The tumor size was monitored by measuring twice a week two diameters at right angles with a caliper and then converted to volume by the formula 4/3 X πr3 (r = sum of the 2 diameters divided by 4). In vivo transductions were conducted when the tumors generated by untransduced cell grew to 0.5–1.0 cm in diameter. About 1 × 1012 DNA copies of recombinant AAVs in 0.1 ml Hepes buffer were injected to each tumor in situ once or twice one day apart. GCV treatment was started 3 days after the initial inoculation.
Bystander Effect of TK+ PLC/PRF/5 Cells in Vitro and in Vivo.
To study the bystander effect in vitro, SSV9AFABTKneo transduced and untransduced PLC/PRF/5 cells were mixed in different ratios and plated on 60-mm tissue culture dishes at the density of 1 × 105 cells per dish. The cells were selected with 4 μM of GCV in DMEM with 10% fetal calf serum for 14 days. The resistant colonies were stained by Giemsa staining and counted. To test the in vivo bystander effect, tumors were generated by injecting 0.3 ml of cell suspension containing 5 × 106 of various ratios of transduced and untransduced PLC/PRF/5 cells to the flank regions of adult nude mice (The Jackson Laboratory). When the size of subcutaneous tumors reached 0.5–1.0 cm in diameter GCV at 25 mg/kg body weight was administered i.p. twice daily for 21 days and the tumor growth was monitored.
Southern Blot and RT-PCR Analyses.
The presence of TK gene in tumors that recurred was identified by Southern blot analysis of genomic DNA. The DNA was digested with PstI, separated on a 0.8% agarose gel, transferred onto a Hybond N+ nylon membrane (Amersham), and hybridized with a probe containing the full-length TK coding region and part of simian virus 40 Poly(A). Expression of TK gene in these tumors was analyzed by reverse transcription (RT)-PCR. RNAs were extracted from the tumors using RNAzol B (Biotecx Laboratories, Houston). Total RNA (1.5 μg) was used to synthesize cDNA in 20 μl reaction mixtures by standard protocol using oligo dT as primer and Moloney-murine leukemia virus reverse transcriptase (BRL). PCR reactions were carried out with 5 μl of RT products in 50 μl reactions using Amp Taq polymerase in PCR buffer containing 0.75 mM MgCl2 with annealing temperature of 63°C. Primers 5′-GGCATGCCTTATGCCGTGACCGAC and 5′-CCAGGTCGCATATCGTCGGTATGG, were used to amplify the 709-bp TK specific fragment. Human β-actin amplified with primers 5′-GACGACATGGAGAAAATCTGGCAC and 5′- GGCGACGTAGCACAGCTTCTCCTT served as internal control.
RESULTS
Transduced TK+ PLC/PRF/5 Cells Are Sensitive to GCV Treatment in Vivo.
To test the effect of GCV on transduced TK+ tumors in vivo, we used a TK+ PLC/PRF/5 cells previously isolated by infection with SSV9AFABTKneo virus and stably selected in vitro with G418. Transduced TK+ PLC/PRF/5 cells (5 × 106) were inoculated subcutaneously into adult nude mice on the right flank region. Twenty mice with tumors 0.8–1.3 cm in diameters were randomly assigned, 10 as the experimental group treated with GCV and 10 as the control group treated with PBS. GCV or PBS (25 mg/kg) were injected i.p. twice daily for 42 days. In the GCV-treated group, shrinkage of tumors was observed after 7 days’ treatment and, on the 21st day, 9 of 10 tumors shrank completely. One mouse had a residual 0.3-cm diameter tumor. In the control group, tumors grew continuously up to 2 cm in diameter by the end of 17th day when they were euthanized. The difference in tumor growth between the two groups are shown in (Figs. 2 and Fig. 3) and is statistically significant by Student’s t test.
Figure 2.
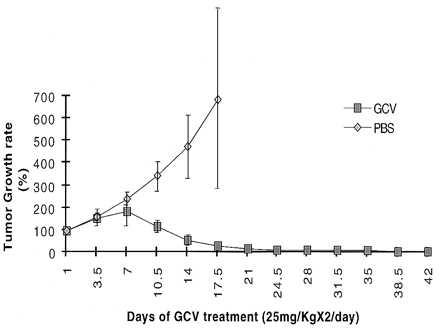
Effect of GCV on the growth of the transplanted TK+ PLC/PRF/5 cells in vivo. The tumor volumes prior to treatment were taken as 100%. The growth of tumor was calculated as the ratio of tumor volumes at any given time after the treatment to that before the treatment.
Figure 3.
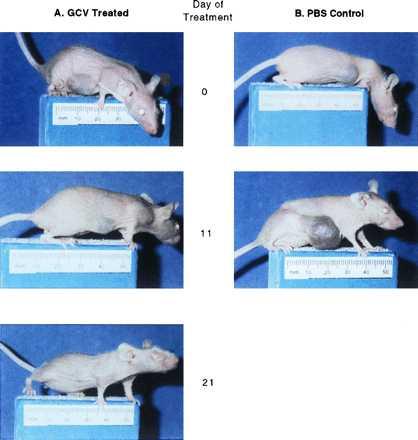
Illustration of the tumor sizes in a GCV-treated (Left) and a control mouse (Right). The tumors were ≈1 cm in diameter at the beginning of treatment (Day 0). The GCV treated mouse showed tumor decreasing in size after 11 days of treatment and grossly disappearing after 21 days of treatment. In the control mouse, the tumor continued to grow and reached 2 cm in diameter after 17.5 days when the mouse was euthanized.
Within 6 weeks after cessation of GCV treatment, six of the ten GCV treated mice, including the one with the residual tumor, had tumor recurrence. These recurred tumors would not respond to further GCV treatment. To test whether the recurrence was caused by gene deletion or silencing, genomic DNA was extracted from these tumors and analyzed by Southern blot, and total RNA was analyzed by RT-PCR. The TK gene was present and intact in the recurred tumors (Fig. 4A). However, the TK expression was not detectable in this tumors by RT-PCR analysis (see Fig. 4B). Therefore, recurrence is due to the silencing of the gene in a subpopulation of transduced cells.
Figure 4.
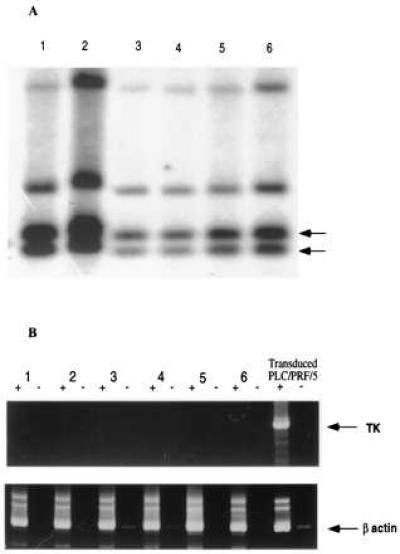
TK gene integrity and expression in recurred tumors. (A) Southern blot analysis showed the TK gene specific fragments of 1.0 and 0.8 kb in all six recurred tumors. (B) RT-PCR assay failed to show the TK-specific fragment detected in the transduced TK+ PLC/PRF/5 cells. RT-PCR for human β-actin serves as an internal control. PCR with or without reverse transcriptase is represented by + or −, respectively.
We observed that the tumors that recurred were those that had grown larger than 1 cm in diameter before GCV treatment. Hence, we conducted another experiment with eight mice where the GCV treatment was started when tumors were between 0.5 and 0.8 cm in diameter. After 21 days GCV treatment, all the tumors shrank completely. No tumor recurred after cessation of treatment up to a year (data not shown). Large tumors may allow gene silencing to occur as they took a longer time to shrink completely with GCV treatment. Hence, all the subsequent in vivo experiments were performed with tumor size of ≤1 cm in diameter.
Bystander Effect in Vivo and in Vitro.
To test if TK+ PLC/PRF/5 carcinoma cells has bystander effect, in vivo and in vitro experiments were conducted. Different ratios of TK+ and TK− PLC/PRF/5 cells were mixed and inoculated into nude mice as above. GCV treatment were started after the tumor grew to 0.5–1.0 cm in diameter and continued for 21 days. Tumors that contain 100, 90, and 50% TK+ cells shrank completely after GCV treatment. While tumors that contain 10% TK+ cells grew at the same rate as the 100% untransduced cells (Fig. 5). in Vitro experiments were also conducted with the same ratios of TK+ and TK− cells as in vivo experiments. Complete killing was never observed after GCV treatment even with as much as 90% TK+ cells (Fig. 6). Thus, with the PLC/PRF/5 cells, bystander effect was observed in vivo but not in vitro.
Figure 5.
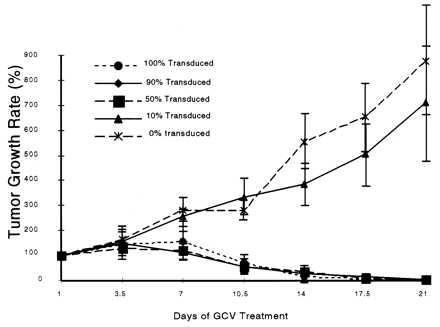
Bystander effect in vivo. Different ratios of SSV9AFABTKneo virus transduced and untransduced PLC/PRF/5 cells were inoculated to the right flank of the mice. GCV treatment started when tumor size was ≤1 cm in diameter. The tumor growth was calculated as described in Fig. 2. Complete shrinkage of tumors were obtained when the ratios of TK+: TK− were 10:0, 9:1, and 1:1.
Figure 6.
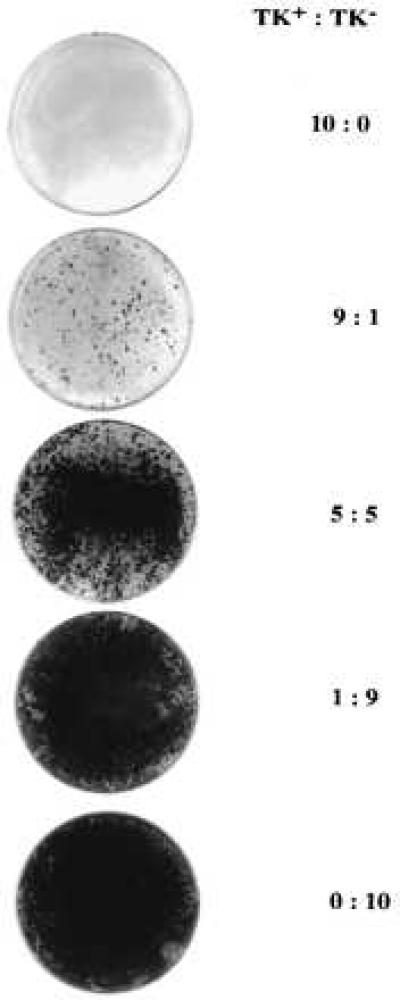
Bystander effect in vitro. Different ratios of TK+ and TK− PLC/PRF/5 cells as indicated were plated on 60-mm tissue culture dishes at the density of 1 × 105 cell per dish and cultured in medium containing 4 μM/ml GCV for 14 days. Colonies were stained with Giemsa.
In Vivo Transduction of the Tumor Cells with Recombinant TK-AAV Virus.
To test the feasibility of direct infection of the tumor cells by the recombinant AAV virus in vivo, untransduced PLC/PRF/5 cells were inoculated in both right and left flanks of the nude mice. Three weeks later, eight mice with similar tumor size of 0.5–1.0 cm in diameter on both flanks were chosen. SSV9AFABTKneo virus (1 × 1012 copies) in 0.1 ml Hepes buffer was injected into the tumors on right flank of the mice, while the tumor on the left flank remained uninjected. GCV treatment was started 3 days after the inoculation. Growth retardation was noticed in the inoculated tumors. The differences of the means of the tumor size between the right and left site were statistically significant at day 10 and day 14 (Student’s t test) (Figs. 7A and 8).
Figure 7.
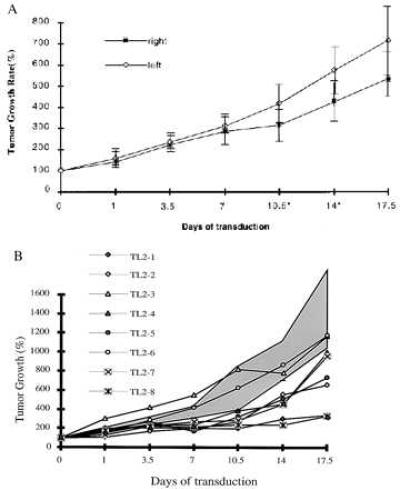
in Vivo transduction of PLC/PRF/5 tumors by direct injecting rAAV virus. (A) Subcutaneous tumors were generated by inoculating untransduced TK− PLC/PRF/5 cells into both flanks of mouse. TK-AAV (1012 copies) (derived from the plasmid SSV9AFABTKneo) in 0.1 ml Hepes buffer was injected at the right tumors when they were 0.5–1.0 cm in diameter (Day 0). GCV treatment was started on day 3 and continued for 17.5 days. Tumor growth was monitored as described in Materials and Methods. The difference in tumor growth rate between left and right side are statistically significant (P < 0.05) between day 10.5 and 14 (*) by Student’s t test. (B) Tumors were generated by injecting untransduced PLC/PRF/5 cells in both flanks in eight mice as described in A. Virus derived from AAVAFABTI2 containing TK and IL-2 were injected into the left tumors on day 0 and day 2 and AAVLacZ virus into the right tumors. GCV treatment started at day 3 and continued to day 17.5. The means and SDs of the growth rate for eight LacZ injected tumors is presented as shaded area. The growth rate of the eight TK-IL-2 virus injected tumors was plotted individually. Tumor retardation were observed on six tumors, two of which stopped growing for 10 days after the treatment.
Figure 8.
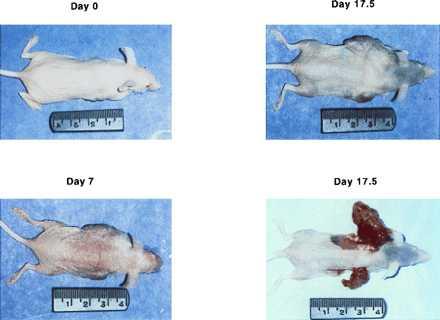
Photomicrographs comparing the size of the AAV-treated (Right) or -untreated (Left) tumors in one of the mice. On day 0, the tumors were about the same size when the GCV was started. The virus-injected tumor was appreciably smaller in day 7. On day 17.5, the mouse was euthanized and the tumors exposed for comparison of their sizes.
One problem encountered with this experiment was that only 0.1 ml of fluid could be injected into the small tumors with each injection. This may not provide sufficient virus to attain an adequate multiplicity of infection. Hence, two injections were used in the next experiment. AAVAFABTI2 virus, which is a recombinant AAV virus constructed by linking HSV-TK and IL-2 with internal ribosome entry site was used. Both genes share the same albumin promoter and AFP enhancer. To rule out the possibility that the AAV vector itself somehow inhibited tumor growth, we used AAVLacZ, a recombinant AAV virus containing LacZ gene driven by cytomegalovirus promoter as control. Since the experiment was done in nude mice, the IL-2 in AAVAFABTI2 should have no influence on the tumor growth. Untransduced PLC/PRF/5 cells were injected to both flanks of the nude mice as above. When the tumors grew to 0.5–1.0 cm in diameter, 1 × 1012 copies of virus were injected into the tumors directly on day 0 and day 2, AAVAFABTI2 virus into the tumors on the left flank and AAVLacZ virus into the tumors on the right flank. GCV treatment was started on day 3. Six out of the eight tumors injected with AAVAFABTI2 virus showed a variable inhibition of growth during the treatment. Tumors infected with AAVLacZ were not affected by the GCV treatment. The difference of tumor size between AAVAFABTI2 tumors and AAVLacZ tumors was statistically significant at day 10 (Student’s t test) (Fig. 7B). The variability in results was due to the difficulty in injecting fluid into solid tumors. Variable amounts of the vector containing fluid extravasated into the surrounding tissue in some cases. Since the AAVAFABTI2 contains the IL-2 gene, the virus was also inoculated the same way to another eight tumors, and the animals were left without GCV treatment to rule out any influence of the IL-2 on tumor growth in nude mice. The growth of the tumors did not show any change (data not shown). These results demonstrate that the effect on tumor growth was due to TK gene expression and GCV treatment.
DISCUSSION
PLC/PRF/5 is a human hepatocellular carcinoma cell line that was derived from malignant liver tissue. This cell line expresses both albumin and AFP (13, 15). We previously demonstrated (13) that the HSV-TK gene that was driven by AFP enhancer and albumin promoter and delivered via AAV vector, could express in these cells and render them sensitive to GCV treatment in vitro. To test if the AAV virus carrying this TK construct can control tumor growth in vivo, we performed experiments in nude mice. Mice bearing tumors derived from AAV-TK-transduced PLC/PRF/5 were treated with GCV. Tumors ≤1 cm in diameter were eradicated after GCV treatment. However, tumors >1 cm in diameter at the onset of GCV treatment either did not completely shrink or recurred after the treatment had ceased. Resistance to GCV has been observed in other HSV-TK+ tumor cell lines and attributed to HSV-TK gene loss or methylation, inadequate GCV concentrations, or presence of noncycling HSV-TK+ cells during the period of GCV administration (16–18). The tumor recurrence in this study was caused by gene silencing, as no TK RNA was detected by RT-PCR. It appears that large tumors tend to recur perhaps because of the presence of some noncycling HSV-TK+ cells during the GCV treatment. Alternatively, inadequate GCV concentrations may allow some HSV-TK+ cells to survive for a longer period and have the chance to silence the gene by methylation or other mechanisms. A recent report by Chen et al. (19) indicated that hyperacetylation of histones is involved in the silencing of virally transduced genes and administration of sodium butyrate or trichostatin A can reactive the gene in vitro. Whether or not this treatment is effective or feasible in vivo requires additional investigation.
Previous experiments have shown that TK+ cells can cause killing of neighboring TK− cells through bystander effect upon GCV treatment (20). The mechanism responsible for this antitumor effect is believed to be the transfer of phosphorylated GCV to TK− tumor cells, an effect reported in vitro by Moolten (17) and called “metabolic cooperation.” Since this phenomenon is observed only when cells are plated at high density, it has been attributed to metabolic cooperation mediated through gap junctions (21–23). The bystander effect also occurs in vivo during the treatment of gliomas, sarcomas, or carcinomas (20, 24). With our cell line, we observed minimal, if any, bystander effects in vitro, even with densely plated cells at high ratios of transduced cell to untransduced cells. In contrast in vivo experiments showed tumoricidal effect with a 50–50 mixture of transduced and untransduced cells on GCV treatment. One possible explanation is that PLC/PRF/5 cell may form gap junctions efficiently in vivo but not in vitro. Results obtained with U-87 gliomas indicated that bystander effect is not sufficient to restrain the tumor growth in immunodeficient animals (23). However, our experiments demonstrated that with hepatocellular carcinoma cell line, PLC/PRF/5, the bystander effect was efficient in destroying the tumor completely when half of the cells were transduced. Hence, the efficiency of bystander effect may differ with different tumors.
in Vivo transduction has also been carried out in this experiment to test the feasibility of direct injection of this recombinant TK-AAV in vivo. With one time injection, the tumor growth was retarded as a group (Fig. 7A). In second experiment, we infected the tumors with the virus twice to increase the multiplicity of infection, six out of eight tumors responded. Although, we did not achieve complete eradication of tumors in these experiments, partial response was seen when the injections were adequate.
These studies suggest that to use this approach for the treatment of hepatocellular carcinoma in humans, several conditions are necessary. First, the tumor must be small in size, as large tumors may be conducive to gene silencing. Secondly, because the virus is of relatively low titer, it must be delivered efficiently and directly into the tumor. An opportunity of accomplishing these two goals may be illustrated by the procedure of chemical embolization of hepatoma, where a catheter is introduced through the hepatic artery to embolize the tumor when it is small. The viral vector could conceivably be introduced in a similar manner. In addition to tumor killing by TK and GCV, immunotherapy will be necessary to help control the tumor (25). Because our experiments were performed in nude mice, we cannot test the effect of the IL-2 in our viral construct. An immunocompetent animal model is needed to test this or other cytokines.
Acknowledgments
We thank Dr. Peter Colosi of Avigen, Inc., for preparing the AAV vector and Dr. Jefferson Chan for reviewing the manuscript, and Lisa Woldin for editing of the manuscript. This work was supported in part by National Institutes of Health Grant DK16666. The transgenic facility used in this study is supported by a grant from the Lucille P. Markey Charitable Trust to the University of California, San Francisco, Program in Biological Sciences.
ABBREVIATIONS
- AFPp
α-fetoprotein
- AAV
adeno-associated virus
- HSV
herpes simplex virus
- TK
thymidine kinase
- GCV
ganciclovir
- IL-2
interleukin 2
- RT
reverse transcription
References
- 1.Ezzeddine Z D, Martuza R L, Platika D, Short M P, Malick A, Choi B, Breakefield X O. New Biol. 1991;3:608–614. [PubMed] [Google Scholar]
- 2.Heyman R A, Borrelli E, Lesley J, Anderson D, Richman D D, Baird S M, Hyman R, Evans R M. Proc Natl Acad Sci USA. 1989;86:2698–2702. doi: 10.1073/pnas.86.8.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takamiya Y, Short M P, Ezzeddine Z D, Moolten F L, Breakefield X O, Martuza R L. J Neurosci Res. 1992;33:493–503. doi: 10.1002/jnr.490330316. [DOI] [PubMed] [Google Scholar]
- 4.Flotte T R, Afione S A, Zeitlin P L. Am J Respir Cell Mol Biol. 1994;11:517–521. doi: 10.1165/ajrcmb.11.5.7946381. [DOI] [PubMed] [Google Scholar]
- 5.Brand K, Arnold W, Bartels T, Lieber A, Kay M A, Strauss M, Dorken B. Cancer Gene Ther. 1997;4:9–16. [PubMed] [Google Scholar]
- 6.Kasahara N, Dozy A M, Kan Y W. Science. 1994;266:1373–1376. doi: 10.1126/science.7973726. [DOI] [PubMed] [Google Scholar]
- 7.Somia N V, Zoppe M, Verma I M. Proc Natl Acad Sci USA. 1995;92:7570–7574. doi: 10.1073/pnas.92.16.7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osaki T, Tanio Y, Tachibana I, Hosoe S, Kumagai T, Kawase I, Oikawa S, Kishimoto T. Cancer Res. 1994;54:5258–5261. [PubMed] [Google Scholar]
- 9.Vile R G, Hart I R. Cancer Res. 1993;53:3860–3864. [PubMed] [Google Scholar]
- 10.Kaneko S, Hallenbeck P, Kotani T, Nakabayashi H, McGarrity G, Tamaoki T, Anderson W F, Chiang Y L. Cancer Res. 1995;55:5283–5287. [PubMed] [Google Scholar]
- 11.Huber B E, Richards C A, Krenitsky T A. Proc Natl Acad Sci USA. 1991;88:8039–8043. doi: 10.1073/pnas.88.18.8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakabayashi H, Hashimoto T, Miyao Y, Tjong K K, Chan J, Tamaoki T. Mol Cell Biol. 1991;11:5885–5893. doi: 10.1128/mcb.11.12.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su H, Chang J C, Xu S M, Kan Y W. Hum Gene Ther. 1996;7:463–470. doi: 10.1089/hum.1996.7.4-463. [DOI] [PubMed] [Google Scholar]
- 14.Samulski R J, Chang L S, Shenk T. J Virol. 1989;63:3822–3828. doi: 10.1128/jvi.63.9.3822-3828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander J J, Bey E M, Geddes E W, Lecatsas G. S Afr Med J. 1976;50:2124–2128. [PubMed] [Google Scholar]
- 16.Vrionis F D, Wu J K, Qi P, Cano W G, Cherington V. J Neurosurg. 1996;84:250–257. doi: 10.3171/jns.1996.84.2.0250. [DOI] [PubMed] [Google Scholar]
- 17.Moolten F L. Cancer Res. 1986;46:5276–5281. [PubMed] [Google Scholar]
- 18.Moolten F L, Wells J M. J Natl Cancer Inst. 1990;82:297–300. doi: 10.1093/jnci/82.4.297. [DOI] [PubMed] [Google Scholar]
- 19.Chen W Y, Bailey E C, McCune S L, Dong J Y, Townes T M. Proc Natl Acad Sci USA. 1997;94:5798–5803. doi: 10.1073/pnas.94.11.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Culver K W, Ram Z, Wallbridge S, Ishii H, Oldfield E H, Blaese R M. Science. 1992;256:1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 21.Bi W L, Parysek L M, Warnick R, Stambrook P J. Hum Gene Ther. 1993;4:725–731. doi: 10.1089/hum.1993.4.6-725. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg G, Bertram J S. Cancer Res. 1994;54:3947–3948. [PubMed] [Google Scholar]
- 23.Colombo B M, Benedetti S, Ottolenghi S, Mora M, Pollo B, Poli G, Finocchiaro G. Hum Gene Ther. 1995;6:763–772. doi: 10.1089/hum.1995.6.6-763. [DOI] [PubMed] [Google Scholar]
- 24.Kaneko Y, Tsukamoto A. Cancer Lett. 1995;96:105–110. doi: 10.1016/0304-3835(95)03919-n. [DOI] [PubMed] [Google Scholar]
- 25.Freeman S M, Ramesh R, Shastri M, Munshi A, Jensen A K, Marrogi A J. Cancer Lett. 1995;92:167–174. doi: 10.1016/0304-3835(95)03771-n. [DOI] [PubMed] [Google Scholar]