

Abstract
Tyrosine autophosphorylation of receptor tyrosine kinases plays a critical role in regulation of kinase activity and in recruitment and activation of intracellular signaling pathways. Autophosphorylation is mediated by a sequential and precisely ordered intermolecular (trans) reaction. In this report we present structural and biochemical experiments demonstrating that formation of an asymmetric dimer between activated FGFR1 kinase domains is required for transphosphorylation of FGFR1 in FGF-stimulated cells. Transphosphorylation is mediated by specific asymmetric contacts between the N-lobe of one kinase molecule, which serves as an active enzyme, and specific docking sites on the C-lobe of a second kinase molecule, which serves a substrate. Pathological loss-of-function mutations or oncogenic activating mutations in this interface may hinder or facilitate asymmetric dimer formation and transphosphorylation, respectively. The experiments presented in this report provide the molecular basis underlying the control of transphosphorylation of FGF receptors and other receptor tyrosine kinases.
Keywords: cell signaling, phosphorylation, protein kinases, protein–protein interactions, surface receptors
Ligand-induced tyrosine autophosphorylation plays an important role in the control of activation and cell signaling by receptor tyrosine kinases (1–6). Structural and biochemical studies have shown that autophosphorylation of fibroblast growth factor receptor 1 (FGFR1) (7, 8) and FGFR2 (9) are mediated by a sequential and precisely ordered intermolecular reaction that can be divided into three phases. The first phase involves transphosphorylation of a tyrosine located in the activation loop (Y653 in FGFR1) of the catalytic core resulting in 50–100-fold stimulation of kinase activity (7). In the second phase, tyrosine residues that serve as docking sites for signaling proteins are phosphorylated including tyrosines in the kinase insert region (Y583, Y585), the juxtamembrane region (Y463), and in the C-terminal tail (Y766) of FGFR1. In the final and third phase, Y654, a second tyrosine located in the activation loop is phosphorylated, resulting in an additional 10-fold increase in FGFR1 kinase activity (7). Interestingly, tyrosines that are adjacent to one another (e.g. Y653, Y654 and Y583, Y585) are not phosphorylated sequentially, suggesting that both sequence and structural specificities dictate the order of phosphorylation. Although tyrosine phosphorylation plays a major role in cell signaling, it is not yet clear what the structural basis for transautophosphorylation is. In other words, the molecular mechanism underlying how one kinase (the enzyme) within the dimerized receptor specifically and sequentially catalyzes phosphorylation of tyrosine(s) of the other kinase (the substrate) is not yet resolved.
We previously determined the crystal structure of activated FGFR1 kinase domain bound to a phospholipase Cγ (PLCγ) fragment composed of two SH2 domains and a tyrosine phosphorylation site (Fig. 1) (PDB code 3GQI) (10). In this structure we found that the substrate-binding pocket of the kinase molecule (the enzyme molecule, termed molecule E) is occupied by the residue equivalent to Y583 of a symmetry-related molecule (the substrate molecule, termed molecule S). This tyrosine (substituted by an phenylalanine residue) is located in the kinase insert and is the second FGFR1 tyrosine that becomes phosphorylated in vitro (7). On reexamination of the crystal structure, 3GQI, we found that there is a substantial crystallographic interface between the N-lobe of the molecule that serves as an enzyme and the C-lobe of molecule that functions as a substrate. In this interface there are direct interactions between R577′ and D519 (Fig. S1A). Inherited mutations have been documented that result in D519N, a loss-of-function mutation causing lacrimo-auriculo-dento-digital syndrome (11), and in R576W, a somatic gain of function mutation found in glioblastoma (12). In the current study we used structural and biochemical tools to show that R577 is involved in creating, in vivo, an asymmetric FGFR1 dimer that allows transphosphorylation of Y583 and other tyrosine autophosphorylation sites in FGF-stimulated cells. This study provides the basis for understanding molecular-level specificity in FGFR1 transphosphorylation and cell signaling.
Fig. 1.

The overall structure of asymmetric activated FGFR1 kinase dimer and detailed views of inter receptor contacts. (A) Asymmetric dimer of active phosphorylated FGFR1 is shown in ribbon diagram. Molecules E and S of the asymmetric dimer are colored in cyan and green, respectively. (B) A detailed view of the interface formed between kinases in the asymmetric dimer. ATP analog (AMP-PCP) and interacting residues are shown in stick representation and the magnesium ion is shown as a blue sphere. Residues from molecule S are labeled with primes. The color scheme applied in this figure is used for all figures. Secondary structures are labeled in blue. (C) Surface representation of molecule E is depicted in cyan with interacting residues of the molecule S in stick and ribbon representation. Representative residues from molecule S are labeled. (D) Surface representation of molecule S is shown in green with interacting residues of molecule E (Pale Cyan) in stick and ribbon representation (www.pymol.org).
Results and Discussions
Asymmetric Dimerization Interface During Autophosphorylation of FGFR1.
The structure of activated FGFR1 kinase in complex with a phospholipase Cγ (PLCγ) fragment (10) shows that two symmetry-related activated kinase domains form an asymmetric dimer that we hypothesize illustrates in vivo transautophosphorylation of Y583 in the kinase insert region (Fig. 1A and Fig. 1B). The asymmetric arrangement of the two kinase molecules is mediated by an interface formed between the activation segment, the tip of nucleotide-binding loop, the β3-αC loop, the β4-β5 loop, and the N-terminal region of helix αC in a kinase molecule that serves as an enzyme (E), and the kinase insert and residues between C-lobe helices αF and αG in a second kinase molecule serving as a substrate (S) (Fig. 1C and D). Importantly, R577, a residue close to the kinase insert region of the substrate molecule, contributes to this interface (Fig. 1B). The total buried surface area is 1648 Å2 (13).
The interface formed between the two active FGFR1 molecules consists of two regions. One is the proximal substrate-binding site near the P + 1 region of the activation segment. The other is a region distal from the substrate-binding site. The distal substrate-binding site is formed between a region adjacent to the nucleotide-binding loop of molecule E and the αF-αG loop and the N-terminal residues of the kinase insert region of molecule S. In the crystal structure clear electron density is seen for R577 and D519 (Fig. S1A). It is of note that the R577 side chain faces approximately 180° opposite from that of R576, an amino acid mutated in glioblastoma (Fig. S1B) (12).
The two regions of the asymmetric dimer interface are complementary with an Sc = 0.62 (Fig. 2) (14). For the proximal substrate-binding site molecule E predominantly contributes residues from the activation segment (N659–V664) that form a short antiparallel β-sheet with residues C-terminal to Y583′ from molecule S, and R570 forms a salt bridge with E582′ (Fig. 2B). For the distal binding site, R577′ binds both the backbone carbonyl and side chain of D519, and the loop between helices αF and αG in molecule S forms multiple aliphatic contacts with the β3-αC and β4-β5 loops (Fig. 2C).
Fig. 2.

Surface distributions of residues in the asymmetric FGFR1 kinase dimer interface. (A) Overall structures of the asymmetric kinase dimer are shown in ribbon format. (B) Surface presentation of molecule E (the enzyme) is in cyan. The proximal substrate-binding region is shown in red and distal substrate-binding region is shown in yellow. Activation-loop (A-loop) and nucleotide-binding loop (N-loop) are indicated. (C) Surface representation of molecule S (substrate) is in green with the tyrosine autophosphorylation site (Y583) in the kinase insert region of molecule S indicated. Substrate site of molecule S is colored in red and the distal substrate site is in yellow.
In Vitro Tyrosine Kinase Activity of the R577E FGFR1 Mutant.
To investigate the in vitro effects of R577E mutation (FGFR1-RE) we conducted autophosphorylation experiments of wt-FGFR1 and FGFR1-RE kinase domains. Purified FGFR1 kinase domains were incubated with ATP and Mg2+ at room temperature and monitored at different times by stopping the transphosphorylation reaction with EDTA and running all samples on a nonreducing native gel (Fig. 3A and B). The reaction profiles of wt-FGFR1 and FGFR1-RE in native gels clearly showed that transphosphorylation and the reverse dephosphorylation reaction of FGFR1-RE were substantially retarded when compared to those of wt-FGFR1 kinase domain. Transphosphorylation of wt-FGFR1 kinase domain took place within 10 min, reaching a fully phosphorylated state, and then underwent the reverse dephosphorylation reaction. This contrasts with FGFR1-RE, which became fully phosphorylated within 30 min and then underwent the reverse reaction. This experiment demonstrates that the intrinsic kinase activity of FGFR1-RE kinase domain is maintained, yet it is kinetically retarded.
Fig. 3.

Autophosphorylation of FGFR1 in vitro and in vivo. Profiles of in vitro phosphorylation reactions of isolated kinase domains of (A) wt-FGFR1 and (B) FGFR1-RE at room temperature as a function of time. (C) Kinase activity of FGFR1-RE in vitro is maintained. Lysates of L6 cells expressing wt-FGFR1 or the FGFR1-RE mutant were subjected to immunoprecipitation with anti-FGFR1 antibodies. The immunoprecipitates were then incubated in the presence or absence of an FGFR1 substrate (PLCγ fragment, described in the results) for 30 min at room temperature followed by SDS-PAGE and immunoblotting with anti-pTyr or anti-FGFR1 antibodies. (D) Autophosphorylation of FGFR1-RE in vivo, is strongly compromised. L6 cells expressing either wt-FGFR1 or its RE mutant were stimulated with increasing concentrations of FGF (as indicated) for 10 min at 37 °C. Lysates of unstimulated or FGF-stimulated cells were subjected to immunoprecipitation using anti-FGFR1 antibodies followed by SDS-PAGE and immunoblotting with antipTyr or anti FGFR1 antibodies. (E) L6 cells expressing wt-FGFR1 or FGFR1-RE were stimulated with 100 ng/ml FGF for different times (as indicated). Lysates of unstimulated or FGF stimulated cells were subjected to SDS-PAGE followed by immunoblotting with anti-pTyr or anti-FGFR1 antibodies.
To study how the R577E mutation affects the activity and transphosphorylation of full-length FGFR1, we stably expressed wt-FGFR1 and FGFR1-RE in L6 myoblasts (Fig. 3C). Lysates from cells expressing wt-FGFR1 or FGFR1-RE were immunoprecipitated and subjected to an in vitro autophosphorylation reaction at room temperature (7). The experiment presented in Fig. 3C shows that both full-length wt-FGFR1 and FGFR1-RE become tyrosine autophosphorylated to a similar extent and are capable of phosphorylating an exogenous substrate molecule composed of the two SH2 domains and a phosphorylation site of PLCγ. These results show that the tyrosine kinase activity of full-length R577E FGFR1 mutant is maintained in vitro.
Tyrosine Autophosphorylation of the R577E Mutant Is Strongly Compromised in Living Cells.
We next compared autophosphorylation of WT or the R577E FGFR1 mutant in FGF-stimulated live cells. Stable L6 cell lines matched for expression level of wt-FGFR1 or FGFR1-RE were stimulated with different FGF concentrations for 10 min at 37 °C (Fig. 3D) or with 100 ng/ml FGF at different time points (Fig. 3E). The level of receptor tyrosine phosphorylation was determined by subjecting lysates from unstimulated or FGF-stimulated cells to immunoprecipitation with anti-FGFR1 antibodies followed by immunoblotting with antipTyr antibodies. FGF stimulation of cells expressing wt-FGFR1 resulted in ligand-dependent receptor tyrosine phosphorylation. By contrast, FGF stimulation of cells expressing FGFR1-RE resulted in a very weak phosphorylation even at the highest dose of the ligand. The drastic reduction in tyrosine autophosphorylation of FGFR1-RE in vivo, is not caused by the loss of its intrinsic kinase activity because both isolated full-length R577E mutant and the purified kinase domain of the R577E mutant maintained kinase activity in vitro.
Crystal Structure of R577E Mutant.
We next examined the effect of the R577E mutation on the integrity of the kinase domain by determining the crystal structure of the kinase domain of an FGFR1 mutant protein. The R577E mutant protein was expressed in E. coli and purified by affinity, size exclusion, and anion exchange chromatography. Rod-shaped crystals of mutant protein grew in 2 wk at room temperature and diffract to 3.2 Å resolution. These crystals belong to space group C2 and include four copies of FGFR1-RE in the asymmetric unit. All four molecules of FGFR1-RE are in very similar conformations and superpose with RMSDs < 0.4 Å over residues 461–762 without the kinase insert region (aa. 576–594) (Fig. S2) (15); the kinase insert is flexible and modeled in only two of the four molecules. All four FGFR1-RE molecules are in the active state and exhibit few changes in overall conformation when compared to 3GQI. We have also determined the structure of unphosphorylated FGFR1 in the inactive conformation to 2.70 Å resolution (Table S1).
Briefly, the FGFR1-RE mutant crystal structure is an active-state kinase domain with an extended activation loop. The N-lobe is rotated toward the C-lobe of the kinase structure as has previously been seen in activated phosphorylated FGFR structures (9, 10). In the structure of FGFR1-RE, no density for ATP analog, ACP-PCP, was found in the catalytic cleft between the N-lobe and C-lobe. Helix αC in the N-lobe of FGFR1-RE is rotated slightly closer to the activation loop than in previously determined structures of FGFR1 (Fig. 4A–C). The two unphosphorylated activation loop tyrosines (Y653, Y654) in FGFR1-RE are located in the same positions as phosphotyrosines (pY653, pY654) in the active FGFR1 structure probably because of incompatibility of this crystal form with the inactive conformation of the kinase (Fig. S3).
Fig. 4.

The structures of kinase domains of (A) wt-FGFR1 (PDB ID: 3KY2), (B) FGFR1-RE mutant (PDB ID: 3KXX), and (C) activated FGFR1 (FGFR1-3P) (PDB ID: 3GQI) in a simplified cartoon (Upper) and in a ribbon diagram (Below). The catalytic loop is shown in yellow, and the activation loop in green, helix αC is depicted as a cylinder in the cartoon. Phosphotyrosines are colored in red in the cartoon and in stick representation in the ribbon diagram. (D) Ribbon diagrams of kinase insert loops of FGFR1, FGFR1-RE, and FGFR1-3P are in green, cyan, and blue, respectively. Side chains of R576, R577 and R577E are shown in stick representation. (E) Superposition of kinase insert regions of FGFR1 (Green), FGFR1-RE (Cyan), and FGFR1-3P (Blue) revealing multiple conformations of the kinase insert regions in the three structures.
Comparison of FGFR1 Crystal Structures.
The conformation of the FGFR1 kinase insert region shows significant conformational flexibility between all three FGFR1 structures (FGFR1 and FGFR1-RE determined in this study and 3GQI that we term FGFR1-3P), with rmsd between 4 and 5.4 Å over residues 576–594) (15) (Fig. 4). For FGFR1-RE the asymmetric dimer discussed above, where one kinase domain presents itself as a substrate for a partner kinase domain, is not seen; in FGFR1-RE we find that in none of the four molecules in the asymmetric unit does the kinase insert region present itself to the catalytic cleft of another FGFR1 molecule. In all three structures R576 maintains a similar orientation, however, in the FGFR1-RE structure the orientation of side-chain R577E is flipped approximately 180° compared to wild-type FGFR1 (Fig. 4D). We hypothesize that this crystallographically seen alteration in the conformation of residue 577 illustrates a change in structural space that this loop samples over time.
Upon receptor activation and initiation of transphosphorylation there is a specific sequence of tyrosine phosphorylation that ensues. This means that dimerization surfaces between two kinase domains are sequentially utilized to allow phosphorylation in the correct order and implies that specific interactions between the two kinase molecules will play important roles in phosphorylation of each specific tyrosine. This is surprising, as the same enzymatic reaction occurs at each phosphorylation site within the protein, suggesting similar surface properties of the intermolecular interaction.
Recently, an asymmetric dimer was described for the kinase domain of FGFR2 (9). In the FGFR2 crystal structure Y769 (equivalent to Y766 in FGFR1) is trapped in a position that seems poised to be a substrate for the other kinase domain. Y769 is located at the extreme C-terminus of the kinase domain. Comparison of these two structures (PDB IDs: 3CLY and 3GQI) vividly illustrates the relationship between two FGFR family kinase domains that act as either the enzyme (molecule E) or the substrate (molecule S) (Fig. 5A and B). In both structures the buried surface area of the interface is in the range of 800–900 Å2 (13) and is comprised of a proximal and a distal binding site. The two structures show that at the proximal substrate-binding region there is high structural similarity in the C-lobe of molecule E. However, the distal substrate-binding region is significantly different between the two structures. The N-lobe residues of molecule E that comprise the distal substrate-binding surface are conformationally divergent (Figs. 2 and 5A and B). In the structure of FGFR1 the distal binding site is formed by residues in the β3-αC loop and by amino acids from the nucleotide-binding loop that contributes only A488 and F489 to the interaction. However, although the conformation of the β3-αC loop is largely unchanged from FGFR1, the nucleotide-binding loop of FGFR2 is significantly altered in conformation and all residues from G488 to G493 contribute to binding. We therefore hypothesize that structural differences in the kinase N-lobe alter the distal substrate-binding site and could be important for the sequential nature of transautophosphorylation.
Fig. 5.

Overall structures of asymmetric FGFR1 and FGFR2 kinase dimers, and distances between sequentially ordered FGFR1 tyrosine autophosphorylation sites. (A, B) Overall structures of asymmetric FGFR1 and FGFR2 kinase dimers are shown in ribbon diagrams (Upper) or as cartoons (Bottom). The proximal and the distal substrate interfaces are marked by a yellow or a red sphere, respectively. The phosphorylated regions and activation loops of both structures are shown. Helix αC is shown as a cylinder. The proximal substrate interface of both structures is marked by a yellow circle, and the distal substrate interface is marked by a red circle. (C) A model of FGFR1 (including residue Y766 not yet observed in an inactive FGFR1 structure) is shown in ribbon diagram and six phosphotyrosine sites in stick representation and colored in red. The sequence of autophosphorylation of the six autophosphorylation sites of FGFR1 is marked with numbers and approximate distances between inter autophosphorylation sites shown. Distances between two phosphotyrosine sites are the average of distance between unphosphorylated and phosphorylated FGFR1 structures, and summarized in the table.
Structure-based sequence alignment of FGFRs shows conservation of residues involved in the formation of interfaces found in structures of both active FGFR1 and FGFR2 (Fig. S4A) (16). Interestingly, the N-terminal tip of the helix αG and the adjacent region in the N-terminal loop of the helix αG in FGFR1 is involved in interface formation as a part of the substrate (molecule S) whereas the same region in FGFR2 structure is part of the enzyme (molecule E). In addition, the loop C-terminal to the helix αG is involved in interface formation as a part of the substrate in the FGFR2 structure. On both loops connecting helix αG to the main body of the kinase several loss-of-function mutations have been clinically discovered (Fig. S4B) (17). The loss of autophosphorylation activity of the receptors in vivo may come from the disruption of interface formation for transphosphorylation.
Conclusions
Receptor tyrosine kinases transphosphorylate in response to ligand activation in a specific sequence, however, the molecular mechanism responsible for this sequential order of transphosphorylation events is not understood. In this study we show that for FGFR1 there are two regions that mediate asymmetric dimer formation when Y583 is transphosphorylated. Furthermore, we show that the single point mutation of a residue intrinsic to this interface, R577E, drastically reduces autophosphorylation of FGFR1-RE in live cells. To confirm that this mutation did not alter the kinase fold and did not introduce significant conformational changes we crystallized FGFR1-RE and found no significant differences to wt-FGFR1 (Fig. 4B and C). This confirms that the loss of ligand-induced FGFR1 autophosphorylation in living cells is not caused by a conformational change in the kinase domain. The drastic reduction of autophosphorylation of FGFR1-RE mutant in live cells can be addressed by the steric constraints driven by the ligand-induced dimerization in vivo. In living cells autophosphorylation is mediated by FGF and heparan sulfate proteoglycan induced FGFR1 dimerization. Under these conditions interactions among kinase domains in two dimensions increase the steric constraints and decrease the probability of positions between kinase domains within a dimeric complex. That is, only a limited number of modes of interaction between kinase domains of receptor molecules in the cytoplasmic face of the cell membrane are allowed. In an in vitro environment on the other hand, the kinase domains is not subjected to steric constraints generated by receptor dimerization in the cell membrane allowing for freedom to move in three dimension enabling transphosphorylation of Y583 and other tyrosine residues. However, ligand-induced dimeric FGFR1-RE in vivo cannot bypass the disrupted interface due to the steric constraints generated by dimerization resulting in the failure of transphosphorylation. We have previously demonstrated that autophosphorylation and kinase activation of FGF-stimulated FGFR1 in cells is prevented by mutating the two tyrosine autophosphorylation sites in the activation loop. However, autophosphorylation and kinase activation of ligand stimulated FGFR1 in cells is not prevented by mutating a single or the four other tyrosine phosphorylation sites in the kinase domain (18). The experiments presented in our current report demonstrate that a point mutation that prevents asymmetric kinase dimer formation has a stronger impact on autophosphorylation and kinase activity of ligand-stimulated FGFR1 in living cells.
We previously showed that transphosphorylation of FGFR1 occurs in precisely ordered sequence (7, 8). The order of transphosphorylation sites of FGFR1 is as follows: Y653, Y583, Y463, Y766, Y585, and Y654. Strikingly, the distance between each of these sequential tyrosine phosphorylation sites is between 35–50 Å (Fig. 5C). The phosphorylation of Y583 comes second in the order after the phosphorylation of Y653 in the activation loop. Furthermore, the full activation of FGFR1 is achieved by the phosphorylation of Y654 in the activation loop, which is the last residue to be phosphorylated in sequence. The failure of the phosphorylation of Y583 may result in attenuation or termination of transphosphorylation, perhaps by failure to form an asymmetric dimer necessary for Y463 phosphorylation resulting in strong inhibition of receptor autophosphorylation in living cells. Moreover, it is of note that the autophosphorylation experiments of ligand stimulated living cells were performed with cells expressing full-length FGFR1 containing a cytoplasmic region with intact juxtamemberane (JM) and C-terminal tail, enabling potential formation of additional asymmetric contacts between the cytoplasmic regions of ligand stimulated FGFR1 that might be supported by asymmetric contacts by the JM or the C-terminal tail of FGFR1.
Materials and Methods
Protein Expression and Purification.
Site-directed mutagenesis was performed to introduce the mutant (R577E), and transformed into E. coli strain BL21 (codon+). Cultures were grown in terrific broth media at 37 °C to an OD600 of 0.8 and induced with 1 mM isopropyl-thiogalactopyranoside at 18 °C for 10 h. Cells were harvested and resuspended in lysis buffer (20 mM Tris-HCl pH 8.0, 20 mM NaCl, and 2 mM PMSF) then lysed by French press followed by centrifugation to remove cellular debris. Expression and purification of WT and FGFR1 R577E mutant (aa 458–765) were performed as previously described (7). Proteins were first isolated by affinity chromatography on Ni-NTA beads (GE Healthcare) and eluted with an imidazole gradient up to 250 mM. The eluted sample was subsequently subjected to size-exclusion chromatography using Superdex-200 (S200, GE Healthcare), and further purified by MonoQ (GE Healthcare) ion-exchange chromatography. The purity and mass of the purified protein was verified by electrospray mass spectroscopy.
Crystallization and Structure Determination.
Proteins were concentrated to 12 mg/ml. The R577E mutant protein was transferred to the buffer containing 1 mM AMP-PCP, 6 mM MgCl2, 2 mM tris[2-carboxyethyl]phosphine hydrochloride (TCEP-HCl), 20 mM Tris pH 8.0, and 100 mM NaCl, and subjected to screening and optimization. Crystals were grown at room temperature in 14 d using the hanging drop technique containing equal volumes of protein solution and reservoir buffer (15% [w/v] polyethylene glycol 3350, 200 mM lithium citrate). Crystals belonged to the centered monoclinic space group C2 with unit cell dimensions of a = 186.8 Å, b = 74.3 Å, c = 135.8 Å, and β = 97.4° with four molecules in the asymmetric unit. The solvent content of the complex was around 61%. Crystals were transferred into the cryoprotectant containing reservoir buffer with 15% glycerol then flash frozen in liquid nitrogen. Crystals of wt-FGFR1 were obtained as described (15). Wt-FGFR1 crystals belonged to C2 space group with unit cell dimensions of a = 212.0 Å, b = 49.8 Å, c = 66.5 Å, and β = 107.5°. Data were collected on beamline X29 at the National Synchrotron Light Source for R577E FGFR1 mutant and using the home source for the wild-type protein. Data were processed using HKL2000 (19). A molecular replacement solution for FGFR1 was found with Phaser (20) using the structures of the kinase domains of FGFR1 (21) (PDB code: 1FRK) and of FGFR-3P (10) (PDB code: 3GQI). Model building and the refinement of wt- and mutant FGFR1 were carried out with Coot (21) and CNS (22) to a crystallographic R and Rfree for FGFR1-RE of 22.2% and 26.3%, and for wt-FGFR1 of 20.3% and 25.2%, respectively. Figures were prepared using PYMOL (www.pymol.org). PDBsum was used to calculate intermolecular interfaces (13). Data and refinement statistics are summarized in Table 1.
Table 1.
Data collection and structure refinement statistics for WT-FGFR1 and the R577E mutant FGFR1-RE
| Data collection | FGFR1-RE | WT-FGFR1 |
| X-ray source | NSLS X29 | Rigaku R-Axis 4 |
| Wavelength (Å) | 1.0809 | 1.5418 |
| Space group | C2 | C2 |
| Unit cell dimensions | ||
| a (Å) | 185.6 | 212.0 |
| b (Å) | 74.2 | 49.8 |
| c (Å) | 135.8 | 66.5 |
| β (°) | 97.4 | 107.5 |
| Resolution (Å) | 50-3.2 (3.31–3.20) | 50-2.7 (2.80–2.70) |
| No. of total reflections | 623,854 | 164,827 |
| No. of unique reflections | 30,901 | 18,604 |
| Completeness (%) | 98.8 (97.4) | 99.9 (100.0) |
| I/σ (I) | 15.6 (4.7) | 14.6 (6.0) |
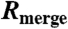 (%) (%) |
9.5 (35.2) | 8.6 (24.1) |
| Redundancy | 4.0 (3.9) | 3.6 (3.5) |
| Refinement | ||
| Resolution (Å) | 50-3.2 (3.24–3.20) | 50-2.7 (2.76–2.70) |
| Rwork (%) | 22.2 (31.0) | 20.3 (25.0) |
| Rfree (%) | 26.3 (37.3) | 25.2 (33.9) |
| No. of atoms | ||
| Protein | 9,444 | 4,673 |
| Ligand/ion | 0 | 20 |
| Water | 0 | 247 |
| rmsd | ||
| Bond lengths (Å) | 0.010 | 0.007 |
| Bond angles (°) | 0.99 | 0.97 |
| Protein Data Bank ID | 3KXX | 3KY2 |
Values in parentheses indicate statistics for the highest resolution shells.
In Vitro Transphosphorylation of wt-FGF1 and FGFR1-RE.
1 μl of purified wt-FGFR1 (aa. 458–765) or FGFR1-RE (10 mg/ml) was mixed with 1 μl of each 25 mM ATP, 125 mM MgCl2 (in 10 mM HEPES pH 7.5), and 10 mM HEPES pH 7.5, then quenched with the 1 μl of 250 mM EDTA in 10 mM HEPES pH 7.5 at every 10 min until 90 min at room temperature. Native gel electrophoresis was performed with reaction samples with 7% native gels.
Cell Culture, Immunoprecipitation, and Immunoblotting Experiments.
A retroviral vector, pBABE, containing a puromycin resistance gene was utilized for generation of stable cell lines expressing wt-FGFR1 or the R577E FGFR1 mutant in L6 myoblasts. Cells were grown in DMEM containing 10% FBS and penicillin/streptomycin. For experiments, cells were starved overnight in DMEM containing penicillin/streptomycin, and subsequently, stimulated for 10 min with 100 ng/ml FGF. Cell lysates were subjected to immunoprecipitation followed by immunoblotting with various antibodies. Antiphosphotyrosine (4G10) antibodies were obtained from Upstate Biotechnology, and anti-FGFR1 antibodies were previously described (7, 8).
Supplementary Material
Acknowledgments.
Authors thank the staff of X29 at the National Synchrotron Light Source. This work was supported by National Institutes of Health Grants R01-AR051448 (JS), R01-AR051886 (JS), P50-AR054086 (JS), GM088240 (TJB), and AI075133 (TJB).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0914157107/DCSupplemental.
References
- 1.Schlessinger J. Signal transduction by allosteric receptor oligomerization. Trends Biochem Sci. 1988;13:443–447. doi: 10.1016/0968-0004(88)90219-8. [DOI] [PubMed] [Google Scholar]
- 2.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;10:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger J, Lemmon MA. SH2 and PTB domains in tyrosine kinase signaling. Sci STKE. 2003;19:RE12. doi: 10.1126/stke.2003.191.re12. [DOI] [PubMed] [Google Scholar]
- 4.Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- 5.Lemmon MA, Schlessinger J. Regulation of signal transduction and signal diveristy by receptor oligomerization. Trends Biochem Sci. 1994;19:459–463. doi: 10.1016/0968-0004(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 6.Lemmon MA, Schlessinger J. Transmembrane signaling by receptor oligermerization. Methods Mol Biol. 1998;84:49–71. doi: 10.1385/0-89603-488-7:49. [DOI] [PubMed] [Google Scholar]
- 7.Furdui CM, Lew ED, Schlessinger J, Anderson KS. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol Cell. 2006;21:711–717. doi: 10.1016/j.molcel.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Lew ED, Furdui CM, Anderson KS, Schlessinger J. The precise sequence of FGF receptor atuophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci Signal. 2009;2:ra6. doi: 10.1126/scisignal.2000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, et al. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc Natl Acad Sci USA. 2008;105:19660–19665. doi: 10.1073/pnas.0807752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bae JH, et al. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohmann E, et al. Mutations in different components of FGF signaling in LADD syndrome. Nat Genet. 2006;38:414–417. doi: 10.1038/ng1757. [DOI] [PubMed] [Google Scholar]
- 12.Rand V, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci USA. 2005;102:14344–14349. doi: 10.1073/pnas.0507200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laskowski RA, et al. PDBsum: A web-based database of summaries and analyses of all PDB structures. Trends Biochem Sci. 1997;22:488–490. doi: 10.1016/s0968-0004(97)01140-7. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 15.Mohammadi M, Schlessinger J, Hubbard SR. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell. 1996;86:577–587. doi: 10.1016/s0092-8674(00)80131-2. [DOI] [PubMed] [Google Scholar]
- 16.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 17.Wilkie AO. Bad bones, absent smell, selfish testes: The pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Mohammadi M, et al. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol. 1996;16:977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minor W, Tomchick D, Otwinowski Z. Strategies for macromolecular synchotron crystallography. Structure. 2000;8:R105–110. doi: 10.1016/s0969-2126(00)00139-8. [DOI] [PubMed] [Google Scholar]
- 20.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D: Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.