

Abstract
The molecular basis of allosteric effects, known to be caused by an effector docking to an enzyme at a site distal from the binding pocket, has been studied recently by applying directed evolution. Here, we utilize laboratory evolution in a different way, namely to induce allostery by introducing appropriate distal mutations that cause domain movements with concomitant reshaping of the binding pocket in the absence of an effector. To test this concept, the thermostable Baeyer–Villiger monooxygenase, phenylacetone monooxygenase (PAMO), was chosen as the enzyme to be employed in asymmetric Baeyer–Villiger reactions of substrates that are not accepted by the wild type. By using the known X-ray structure of PAMO, a decision was made regarding an appropriate site at which saturation mutagenesis is most likely to generate mutants capable of inducing allostery without any effector compound being present. After screening only 400 transformants, a double mutant was discovered that catalyzes the asymmetric oxidative kinetic resolution of a set of structurally different 2-substituted cyclohexanone derivatives as well as the desymmetrization of three different 4-substituted cyclohexanones, all with high enantioselectivity. Molecular dynamics (MD) simulations and covariance maps unveiled the origin of increased substrate scope as being due to allostery. Large domain movements occur that expose and reshape the binding pocket. This type of focused library production, aimed at inducing significant allosteric effects, is a viable alternative to traditional approaches to “designed” directed evolution that address the binding site directly.
Keywords: allosteric effects, enzymes, molecular dynamics simulations, protein engineering
Protein allostery has been recognized as a positive or negative cooperative event, leading to a structural change at the binding site as a result of distal docking of a molecule acting as an effector (1–5). Allosteric effects can be influenced by such factors as variation in pH, temperature, ionic strength, and covalent modification as well as mutational changes. A variety of experimental and computational techniques have been utilized to unravel the intricacies of this phenomenon (1–5), but also to achieve useful applications such as the creation of protein switches (6). Among the techniques that have been applied to address these challenges is directed evolution, a method that is generally used to engineer the catalytic profiles of enzymes or the binding properties of proteins (7–11). In the endeavor to make directed evolution of enantioselective enzymes more efficient than in the past, we recently introduced the concept of iterative saturation mutagenesis according to which sites around the binding pocket are randomized iteratively, a given site being composed of one or more amino acid positions in the protein (12–14.) When applying this technique to enzymes displaying strong allosteric effects or coupled motions along the reaction coordinate (15), it is necessary to consider such phenomena to make reasonable decisions regarding the appropriate randomization sites, that is, to identify those residues that do align the binding pocket when the actual catalytic transformation occurs. A very different approach would be the idea of “inducing” allosteric communication at a remote site that results in altered structure and dynamics at the active site, thereby influencing enzyme activity and enantioselectivity. Remote effects in the directed evolution of enantioselective enzymes have been noted earlier but these do not involve allostery (7–11, 16). Here, we describe the experimental implementation of induced allostery using a Baeyer–Villiger monooxygenase (BVMO) as the model enzyme (17–21).
Phenylacetone monooxygenase (PAMO), discovered by Fraaije and Janssen by using genome mining, is the only thermostable BVMO (22) and the only monooxygenase of its kind to be analyzed by X-ray crystallography as reported by Maleti, Mattevi and coworkers (23). Due to its robustness, it would appear to be an ideal candidate for synthetic organic applications, C-C-activating asymmetric transformations in oxidative kinetic resolution of racemic ketones, and/or desymmetrization of prochiral substrates being potentially possible. Unfortunately, the substrate scope of PAMO is limited to phenylacetone and related linear phenyl-substituted derivatives (22, 24). To address this problem, we have previously applied rational design using site-specific mutagenesis (25) as well as directed evolution based on saturation mutagenesis (26), in both cases the targeted sites being a loop next to the binding pocket. PAMO is a monomeric 62-kDa flavin-dependent enzyme that, like other BVMOs, requires the cofactor NADPH for FAD reduction (22, 23). Reduced FAD reacts with O2 forming an alkyl-hydroperoxide that, in the deprotonated form, adds nucleophilically to the carbonyl function of the substrate with formation of the short-lived Criegee intermediate in the rate determining step. Subsequent fragmentation/elimination generates an ester (or lactone) and water, like other BVMOs (17–21). In the case of PAMO, it was suggested on the basis of the X-ray structure that Arg337 participates in the stabilization of the Criegee intermediate (23). This provides a structural basis for a deeper understanding of the enzyme’s mechanism. The crystal structure was obtained without the cofactor NADP+/NADPH and in the absence of a substrate/inhibitor. Further information was recently provided by Lau, Berghuis and coworkers, who reported the X-ray structure of another BVMO, the cyclohexanone monooxygenase (CHMO) from a Rhodococcus strain, with bound FAD and NADP+ in the absence of a substrate/inhibitor (27). Importantly, two different structures were identified, a closed and an open form both, presumably, participating at various points of the catalytic cycle. These structural data illustrate the mechanistic and structural complexity of BVMOs and, consequently, the challenge in performing knowledge-driven directed evolution.
Results and Discussion
Experimental Platform.
It is not a trivial task to identify remote sites in an enzyme at which saturation mutagenesis can be expected to generate mutations that would trigger allosterically-induced domain movements with the creation of a structurally new binding pocket in the absence of an effector. Suggestions about movements of the NADP-binding domain believed to be coupled to the catalytic mechanism of PAMO (23) led us to consider mutations strategically located in regions distal from the active site that might induce such structural reorganizations (28–30). Close inspection of the PAMO X-ray structure shows that the number of likely possibilities is in fact limited. One option would be to focus on hinge regions but, in the case of PAMO and other BVMOs, these are characterized by highly conserved sequences. Therefore, we devised a different strategy by searching for potential randomization sites at or near two domain interfaces. For example, residues at the interface between the NADP-binding domain and the so-called helical domain of PAMO could be chosen in the expectation that appropriate mutations would lead to strong attractive interactions with concomitant structural changes. A different possibility appeared more promising, namely, to identify hot spots in PAMO that would bring the NADP-binding domain spatially closer to the FAD-binding domain (Fig. 1).
Fig. 1.

Cartoon representations of the different domains in the PAMO structure. FAD domain is shown in Green, Dark Blue corresponds to the NADP-binding domain, and Light Blue to the helical domain. The NADP- and FAD-binding domains are linked by two antiparallel β-strands in a hinge-like structure configuration (Black). NADP-binding and helical domains can be considered as separate entities connected by two segments (Red). The randomization site Gln93/Pro94 in the N-terminal region of the α-helix is also labeled.
We spotted in the PAMO X-ray structure several already strongly attractive contacts between the two domains, structurally not far from the hinge region. They occur between the loop segment Trp177-Glu180 (NADP-binding domain) and the loop segment Tyr56–Tyr60 (FAD-binding domain), the π-cation interaction between His179 and Arg59 being an example (Figs. 1 and 4). Located directly behind, and in loose contact with the loop segment Tyr56-Tyr60, is the N-terminal region (Ala91-Glu95) of an α-helix that, by nature, is a rigid secondary element. We speculated that appropriate mutations in this part of the helix could lead to strong attractive interactions with loop segment Tyr56-Tyr60 that, in turn, would result in the movement of loop segment Trp177-Glu180. Therefore, Gln93/Pro94 was chosen as the randomization site, a decision that was also guided by the realization that position 92 is the last member of a long loop extending parallel to loop segment Asp66-Tyr64 that is in direct contact with the flavin ring. Exploratory docking experiments in which phenylacetone was placed in the binding pocket of WT PAMO, indicated that the substrate is about 18 Å away from position 93 (Fig. S1).
Fig. 4.

Comparison of the backbone and side-chain shift between WT PAMO (Green) and mutant Gln93Asn/Pro94Asp (Blue). New H-bonds and a salt bridge between Arg59 and Asp94 in the mutant variant are illustrated in this figure. It is also possible to observe a shorter distance between the loop Tyr56-Tyr60 and the NADP-binding domain loop Trp177- Glu180.
In the present study, we opted for positions 93/94 as a “combined site” for simultaneous saturation mutagenesis (31) using NDT codon degeneracy (14) encoding 12 amino acids (Phe, Leu, Ile, Val, Tyr, His, Asn, Asp, Cys, Arg, Ser, and Gly). About 400 transformants were screened for activity and enantioselectivity using gas chromatography (GC), the model reaction being the oxidative kinetic resolution of 2-ethylcyclohexanone (rac-1d) (Scheme 1) in conjunction with the known thermostable secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus as the NADPH regeneration enzyme and isopropanol as the reductant (32). The aim was to perform laboratory evolution using a single substrate, subsequently, to isolate the enzyme variants, and to test the hit(s) as catalysts in the analogous transformation of a set of other compounds without performing any additional mutagenesis/screening experiments. The model compound 1d as well as most of the other substrates (1b, 1c, and 1e–k) are not accepted by the WT PAMO (22–24), meaning that under the standard reaction conditions (Materials and Methods), compounds rac-1b–k do not react to provide lactones to any detectable extent, not even when increasing the reaction time to 48 h or longer. 2-Phenylcyclohexanone (1a) is the only compound in the list that shows at least some activity (7 U/mg) using WT PAMO, but conversion after a prolonged reaction time is less than 10% under standard conditions and enantioselectivity as measured by the selectivity factor is poor (E = 1.5).
Scheme 1.

Oxidative kinetic resolution of rac-1 catalyzed by PAMO mutants.
Identification and Characterization of Hits.
The library generated by saturation mutagenesis at positions 93/94 by using NDT codon degeneracy was found to contain two mutants that catalyze measurable conversion of ketone rac-1d within a reaction time of 24 h as specified in the GC screening process, namely Gln93Val/Pro94Phe and Gln93Asn/Pro94Asp. Due to amino acid bias inherent in the nature of the saturation mutagenesis protocol (14, 31), the possibility that hits were missed cannot be excluded inspite of the high percentage of coverage. Because the double mutant Gln93Asn/Pro94Asp proved to be the much more active of the two identified hits, we subsequently focused all further efforts on this variant. It leads to a selectivity factor of E = 50 in the model reaction, favoring (S)-2d. Remarkably, all other substrates are likewise accepted by this mutant, and with the exception of ketone 1b, good to excellent enantioselectivity was uniformly achieved (Table 1). Although substrate acceptance has been made possible by our method, activities are modest. Combining the present approach with such techniques as DNA shuffling (7–11) or iterative saturation mutagenesis at sites aligning the reshaped binding pocket (11, 12) may allow for further improvements.
Table 1.
Oxidative kinetic resolution of ketones rac-1a–k using PAMO variant Gln93Asn/Pro94Asp under standard reaction conditions (Materials and Methods). Details of the procedure for expression, purification, and enzymatic activity assays are included in SI Material and Methods. Note that the R/S assignment may change in accordance with the Cahn-Ingold-Prelog convention; the absolute configuration is as shown in Scheme 1.
| Entry | Substrate | Specificactivity (U/mg) | Conv. (%) | Residual ketone 1 |
Lactone 2 |
E-value | ||
| ee (%) | Abs. conf. | ee (%) | Abs. conf. | |||||
| 1 | rac-1a | 64 | 45 | 38 | S | 95 | R | 92 |
| 2 | rac-1b | 40 | 17 | 2.7 | S | 18 | R | 1.5 |
| 3 | rac-1c | 35 | 15 | 46 | R | 91 | S | 25 |
| 4 | rac-1d | 49 | 21 | 25 | R | 95 | S | 50 |
| 5 | rac-1e | 42 | 37 | 37 | R | 95 | S | 68 |
| 6 | rac-1f | 51 | 36 | 14 | R | 92 | S | 40 |
| 7 | rac-1g | 24 | 23 | 19 | S | 94 | R | 42 |
| 8 | rac-1h | 39 | 15 | 17 | S | > 99 | R | > 200 |
| 9 | rac-1i | 48 | 42 | 43 | S | 86 | R | 25 |
| 10 | rac-1j | 58 | 43 | 58 | S | 98 | R | > 200 |
| 11 | rac-1k | 52 | 11 | 17 | S | 96 | R | 55 |
We also tested the catalytic potential of the best PAMO variant Gln93Asn/Pro94Asp as a catalyst in the desymmetrization of 4-substituted cyclohexanone derivatives 3a–c. Scheme 2 shows that, in all cases, excellent enantioselectivity resulted. Interestingly, the absolute configuration of the lactone products are uniformly opposite to what is observed when using CHMO as the catalyst (17–21). The latter has been reported to deliver an ee-value of only 52% in the desymmetrization of substrate 4c, whereas the evolved PAMO mutant Gln93Asn/Pro94Asp is highly enantioselective in the opposite absolute sense (Scheme 2).
Scheme 2.

Desymmetrization of ketones 3a–c using PAMO variant Gln93/Pro94Asp.
Another point of interest concerns the question of whether the mutational change in going from WT PAMO to the double mutant Gln93Asn/Pro94Asp shifts the substrate acceptance from phenylacetone to the above compounds, or whether substrate scope has been widened to include all of the compounds. The latter pertains as shown by kinetic experiments using phenylacetone as the substrate: WT PAMO (Km = 0.071 mM, kcat = 2.3 s-1, and kcat/Km = 32,000 M-1 s-1), mutant Gln93Asn/Pro94Asp (Km = 0.054 mM, kcat = 1.5 s-1, and kcat/Km = 27,000 M-1 s-1).
Thermostability Experiments.
To check whether substrate acceptance and enantioselectivity were evolved at the expense of the robustness of PAMO, 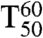 values were measured, the temperature at which 50% of the activity has been lost following a heat treatment for 60 min (33, 34). The results show that thermostability is essentially maintained: WT PAMO (
values were measured, the temperature at which 50% of the activity has been lost following a heat treatment for 60 min (33, 34). The results show that thermostability is essentially maintained: WT PAMO (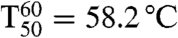 ); variant Gln93Asn/Pro94Asp (
); variant Gln93Asn/Pro94Asp (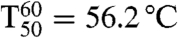 ).
).
Deconvolution Experiments.
The immediate question arose as to whether the double mutant is really necessary for achieving activity and enantioselectivity, or whether the corresponding single variants, characterized by point mutations, would likewise function well. For this purpose, PAMO single mutants Gln93Asn and Pro94Asp were generated by site-specific mutagenesis and tested for activity as catalysts in the model reaction involving rac-1d. Neither of the single mutants showed any activity whatsoever. The same was found for all other substrates, except for 1a in which case very low activity and stereorandom behavior (E ≈ 1) was registered, similar to WT PAMO. This proves that a strong cooperative effect is operating in the double mutant. The results also indicate, but do rigorously prove, that our initial choice regarding simultaneous randomization at two positions was the better of the two possible strategies for targeting positions 93 and 94. Finally, we tested the activity of the two single mutants in the reaction of phenylacetone and discovered that they are similar to WT PAMO: Mutant Gln93Asn (Km = 0.143 mM, kcat = 3.1 s-1, and kcat/Km = 21,000 M-1 s-1), mutant Pro94Asp (Km = 0.48 mM, kcat = 1.3 s-1, and kcat/Km = 27,000 M-1 s-1).
Single Site Saturation Mutagenesis Libraries.
Although the deconvolution experiments show that the two single point mutants Gln93Asn and Pro94Asp are inactive, it was not absolutely clear whether other point mutations at these two positions could lead to active single mutants. Therefore, two separate single residue saturation mutagenesis libraries were generated by randomization at positions 93 and 94, respectively, using, in this case, NNK codon degeneracy. In each case 200 transformants were screened, but no active variants were found. It is, thus, clear once more that simultaneous randomization at a site comprising more than one amino acid position opens the door for potentially achieving pronounced cooperativity between the respective residues, that is, more than additivity. We do not suggest, however, that saturation mutagenesis at sites comprising single amino acid positions is always a futile exercise.
MD Simulations.
To gain insight into the source of enhanced activity and thereby to check our original postulate regarding possible allosteric communication, we turned to MD simulations of WT PAMO and of the superior double mutant Gln93Asn/Pro94Asp. Being guided by the X-ray structure of PAMO (23), the Schrödinger software package was applied to the two proteins harboring FAD, initially in the absence of NADP+ and, subsequently, with bound cofactor. The actual MD simulations were performed with the Desmond program by using an algorithm for high-speed parallel execution (35). Two different strategies for applying MD simulations were tested to avoid any possible misinterpretations due to the uncertainty regarding the catalytic mechanism of PAMO, that is, the question whether the presence of bound NADP+ influences the shape of the binding pocket during the different steps in the catalytic cycle (23). The first MD simulations were performed with the protein structures having bound FAD, but excluding the coenzyme NADP+. The productive MD for WT PAMO, the evolved mutant Gln93Asn/Pro94Asp, and the point mutant Pro94Asp were run during 2 ns. Subsequently, average structures of the WT and mutants were calculated from the conformers obtained in the second ns of simulation following equilibration (Fig. 2).
Fig. 2.

Binding pocket surfaces of WT PAMO (Green), mutant Pro94Asp (Orange), and mutant Gln93Asn/Pro94Asp (Blue). The binding pocket surfaces were obtained from the average structure of 1 ns of MD trajectory after 1 ns of equilibration. Productive MD simulations were run during 2 ns including coenzyme FAD.
It is logical to expect that substrate accessibility of the active site is determined primarily by the residues in close proximity to the flavin on its re-side. The flavin ring binds at the bottom of the cleft between the FAD- and NADP-binding domains. This active pocket in the WT PAMO appears as an enclosed cavity lacking good solvent accessibility and capability to bind large substrates. In sharp contrast, the MD simulations show that the space above the re-side of the bound flavin ring in the double mutant is much more solvent accessible, offering sufficient room for substrates to bind. (Fig. 2). In agreement with the observed lack of activity of the single mutant Pro94Asp and WT PAMO, the respective average structures in both simulations do not show significant conformational differences. Inspection of the active site of WT PAMO, of the single mutant Pro94Asp, and of the active double mutant Gln93Asn/Pro94Asp reveals distinct conformational changes resulting in different spatial relationships of residues near FAD and, thus, a change in the shape of the binding pocket. Fig. S2 and Table S1 pinpoint the major geometric differences.
Second round MD calculations were performed with WT PAMO and the double mutant Gln93Asn/Pro94Asp, each with bound FAD and NADP+ during 6 ns of productive simulation. The same basic tendency regarding conformational differences between WT PAMO and mutant Gln93Asn/Pro94Asp was observed (Figs. S3 and S4 and Table S2).
Superposition of the average structures of WT PAMO and the double mutant using FAD and the FAD-binding residues as anchors points to a structurally crucial backbone rotation of the NADP-binding domain in a movement that is reminiscent of a scissors-opening, reaching displacements between 3–5 Å of the backbone of distal residues, and conformational changes of the same order of magnitude occurring at some residues aligning the active site (Fig. 3). Table S3 summarizes the distance values of displacement between equivalent residues forming the active site in the superposed MD simulations of WT PAMO and mutant Gln93Asn/Pro94Asp. Surprisingly, the NADP+ cofactor adopts a somewhat different position in its binding domain (Fig. 3B). Some NADP+ binding residues in WT PAMO no longer participate in the double mutant, Ser196, Glu200, and Arg337 being prime examples, whereas previous non-involved residues form new side-chain and backbone interactions with the cofactor as, for example, in the case of Phe387. In summary, the MD simulation results show that the combined action of the two point mutations not only orchestrates distinct structural changes globally, but also locally at the binding pocket.
Fig. 3.

Cartoon representation of the superimposed WT PAMO (Green) and mutant Gln93Asn/Pro94Asp (Blue) structures. (A) Average structures were obtained from the conformers of 1 ns of MD trajectory after 5 ns of equilibration. Productive MD simulations were run during 6 ns including coenzyme FAD and NADP+. Structural superposition was performed using FAD molecule and FAD-binding residues (residues within 3 Å) as anchor. It is possible to observe important backbone rotation especially in the NADP domain. (B) Details of the NADP+ binding, comparing WT PAMO (Green) and mutant Gln93Asn/Pro94Asp (Blue). Arg337 in the mutant shifts away from the flavin ring and does not show the H-bond present in the WT.
It is informative to view our findings in the light of the two recently reported crystal structures of the enzyme cyclohexanone monooxygenase (CHMO) from an environmental Rhodococcus strain with bound FAD and NADP+, fortuitously obtained under different crystallization conditions (27). This BVMO has a sequence identity of 43% and high structure similarity with WT PAMO. Interestingly, the conformational differences between WT PAMO and the evolved double mutant Gln93Asn/Pro94Asp are similar to the observed structural differences between the two reported crystalline forms of the CHMO. The two conformers correspond to a closed and a more open conformation, interconversion resulting in domain shifts with concomitant sliding of the cofactor NADP+. In the open form of CHMO, the increase in surface accessibility of the binding pocket relative to the situation in the closed form was associated with the movement of the NADP-binding domain, much like the mutation-induced allosteric effect in our system (27). Backbone movements around the active site in addition to repositioning of NADP+ were also described in the two forms of the CHMO (27), very similar to the changes that we observe when going from WT PAMO to the double mutant. For example, the two crystal structures reveal the reorientation of two residues (Arg132/Lys328) much like that of Arg337 and Lys 336 in our system, as well as similar modifications of the H-bonding around NADP+. Moreover, reasonable hypotheses regarding the nature of the domain movement required for catalysis in the Baeyer–Villiger monooxygenase family were suggested (27) that have direct bearing on our results. The allosterically induced domain movements in our system, similar in nature, were brought about by strategically located mutations. This resulted in a highly altered catalytic profile with respect to substrate acceptance and enantioselectivity.
In an attempt to unveil the reason(s) for the pronounced domain movements caused by the evolved double mutant, a deeper analysis of the region nearest to the saturation mutagenesis site 93/94 was necessary, specifically by analyzing the MD simulations trajectories of WT PAMO, of the double mutant Gln93Asn/Pro94Asp, and also of the inactive single mutant variant Pro94Asp. Fig. 4 shows the average backbone structure and side-chain position of WT PAMO and of the double mutant Gln93Asn/Pro94Asp. New H-bond interactions such as Asp94/Arg59 and Trp57/Trp177 are visible. The interaction Asp94/Arg59 implies a strong salt bridge, anchoring the rigid helix structure to the loops Tyr56-Tyr60 and Trp177-Glu180. In sharp contrast, the strong Asp94/Arg59 salt bridge is not found in the single mutant Pro94Asp, due to the steric clash between Arg59 and Glu93 that would arise if this interaction were to occur. The situation is reversed in the double mutant in that Gln is mutated to Asn at position 93.
A few more explanatory comments are in order. Our MD simulations of WT PAMO point to a strong interaction between Arg59 and His179 that is also revealed by its crystal structure. It involves a cation-π interaction with histidine serving as the aromatic partner. Such interactions have been found to be stronger than salt bridges in other systems (36, 37). In the double mutant, this interaction is maintained but now Arg59 also interacts with mutated Asp94 via a salt bridge. This strengthens the connection between the rigid α-helix, where Asp94 is located, and the NADP-binding domain, an effect that causes exposure of the binding pocket together with repositioning of the backbone and side-chain structural components of residues surrounding it. Such reorganization has a direct bearing on activity and enantioselectivity.
Finally, to check our conclusions derived from the MD simulations, we considered appropriate covariance maps that are known to constitute useful indicators of correlated and anti-correlated motions in enzymes (38–40). Cross-correlation coefficients of the Cα atoms were first constructed by using a 6 ns simulation of WT PAMO, including FAD and NADP+. Analysis confirms that strong correlating motions exist between the regions Trp177-Glu180 (NADP-binding domain), Tyr56-Tyr60 (FAD-binding domain), Ala91-Glu95, and the residues forming the active site, particularly Asp66, Leu153, Ser196, Arg337, and Phe389 (Fig. S5). The analogous procedure was then applied to the superior double mutant Gln93Asn/Pro94Asp. The respective covariance map shows increased correlation between the mutation site and the FAD segment in the interface FAD-NADP domains that supports our previous conclusion regarding increased contact as a result of mutagenesis (Fig. S6). Upon comparing the two covariance maps, important differences are observed, particularly around the active site residues, hinges, and the NADP-binding domain (Fig. S7), likewise, in line with our mechanistic conclusions.
Conclusions
Baeyer–Villiger monooxygenases are not considered to be allosterically controlled enzymes (5, 17–21), yet their structures consist of semirigid domains connected via flexible regions or hinges that are typical features present in allosteric systems. It has been stated that the involvement of both flexibility and semirigidity in allosteric enzymes is likely to be a result of evolution, ensuring a balance between sensitivity or specificity and robustness relating to functional importance (1). In the case of BVMOs, the movement of domains harboring cofactors is essential for catalysis (23, 27). In the present study, we have successfully applied knowledge-based directed evolution to a thermostable Baeyer–Villiger monooxygenase, PAMO, specifically in the quest to induce domain movements via allostery that lead to a different shape of the binding pocket, very much like traditional allosteric communication brought about by effectors docking at remote sites. Random mutagenesis was focused on a two-residue site distal from the binding pocket, the choice being based on the analysis of the PAMO X-ray structure. This structure-based approach led to the identification of an active double mutant that accepts a set of otherwise inert 2-substituted cyclohexanone derivatives with high enantioselectivity in the respective oxidative kinetic resolutions. MD simulations and covariance maps proved to be useful both in interpreting the data and in choosing appropriate mutations sites that can be expected to induce allosteric effects (39).
Our work bears some relationship to certain diseases caused by mutations that induce allosteric effects (2, 39). These can shut down the functionality of allosteric enzymes, or they can cause conformational changes at the active site that lead to constitutive activation regardless of whether an effector is bound, as in the case of the G-protein (2). We believe that knowledge-guided directed evolution, with the aim to induce allostery of the kind described herein, may be useful in future directed evolution studies of enzymes, especially when induced domain movements can be predicted with a sufficient degree of certainty. This also applies to laboratory evolution of binding properties, as in the case of therapeutic proteins.
Materials and Methods
Library Screening.
Individual colonies were placed into 2.2-mL 96-deep-well plates containing 800 μL of LB media with 100 μg mL-1 carbenicilline by a colony picker QPIX (Genetix). After cell growth at 37 °C overnight with shaking at 800 rpm, 10 μL of each preculture was transferred into a new plate containing 800 μL of TB media supplemented with 0.1% L-arabinose as inducer and 100 μg mL-1 carbenicilline. The duplicate plates were grown for an additional 24 h to induce PAMO expression. The cultures were centrifuged at 3200 × g and 4 °C for 6 min and the supernatants were discarded. The original plates were stored. Each cell pellet was resuspended in 600 μL of 50 mM Tris-HCl (pH 8.0) containing 1 mg mL-1 lysozyme and four units of DNase I. Lysis were performed at 37 °C and shaken at 800 rpm for 3 h. Cell debris was precipitated by centrifugation at 3200 × g and 4 °C for 30 min and 50 μL of each cleared supernatant transferred to a 1.1 mL 96-deep-well plate. Then in each well, 50 μL of the secondary alcohol dehydrogenase (20ADH) crude extract (about 10 U) (37), 10 μL of 1 mM NADP+, and 20 μL of 100 mM rac-1d in acetonitrile and 370 μL of 50 mM Tris-HCl (pH 8.0) containing 5 mM isopropanol were added. The reaction plates were incubated at 37 °C and shaken at 800 rpm for 24 h. Four Hundred μL of ethyl acetate was then added to extract the substrate and product from the solution. After centrifugation, 200 μL of organic layer in each well was transferred into a new glass-made 96-deep-well plate, and subjected to GC analysis for screening. Active clones were collected and the results reproduced. The entire gene of the indentified hits was sequenced to confirm there were not any other mutations.
Procedure for the Enzymatic Oxidation of Substrates.
In a typical protocol, 1 mL reaction system consists of 2.5 mM substrate (added as a concentrated solution in acetonitrile), 5 mM isopropanol, 100 μM NADP+, 1.0 μM purified PAMO mutant enzyme, and 20ADH (2 U) and 50 mM Tris-HCl buffer (pH 8.0). The mixture in 8 ml glass tube with a sealed cap (to avoid evaporation of 2-alkyl substituted substrates) was shaken at 200 rpm and 30 °C for times established (between 5–16 h) to control the conversion rate less than 50% for kinetic resolution. For desymmetrization of rac-3a–c, the reaction time is 24 h. The reaction was stopped, worked up by extraction with ethyl acetate (containing 200 mg L-1 internal standard as nonane, dodecane, hexadecane, and octadecane). The sample was analyzed by achiral and chiral GC to determine the conversion of and the enantiomeric excess of the residual ketones and produced lactones. Control experiments were performed for all the tested substrates with purified WT PAMO enzymes (about 5 μM). Further details can be found in SI Text.
MD Simulations.
Using the published X-ray structural data of 1W4X, the in silico structure determinations of the wild type and mutants were performed with the Schrödinger software package. MD simulations were performed with the Desmond program by using an algorithm for high-speed parallel execution (35). Details can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments.
We thank the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (Schwerpunkt 1170) for generous support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.O. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911656107/DCSupplemental.
References
- 1.Cui Q, Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai C-J, del Sol A, Nussinov R. Protein allostery, signal transmission and dynamics: A classification scheme of allosteric mechanisms. Mol Biosyst. 2009;5:207–216. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 4.Ozkan E, Yu H, Deisenhofer J. Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. P Natl Acad Sci USA. 2005;102:18890–18895. doi: 10.1073/pnas.0509418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Dynamic coupling and allosteric behavior in a nonallosteric protein. Biochemistry. 2006;45:7693–7699. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guntas G, Mansell TJ, Kim JR, Ostermeier M. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. P Natl Acad Sci USA. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lutz S, Bornscheuer UT. Protein Engineering Handbook. Weinheim: Wiley-VCH; 2009. [Google Scholar]
- 8.Turner NJ. Directed evolution drives the next generation of biocatalysts. Nat Chem Biol. 2009;5:567–573. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- 9.Jäckel C, Kast P, Hilvert D. Protein design by directed evolution. Annu Rev Biophys Biomol Struct. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- 10.Bershtein S, Tawfik DS. Advances in laboratory evolution of enzymes. Curr Opin Chem Biol. 2008;12:151–158. doi: 10.1016/j.cbpa.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 11.Reetz MT. Directed evolution as a means to engineer enantioselective enzymes. In: Gotor V, Alfonso I, Garcia-Urdiales E, editors. Asymmetric Organic Synthesis with Enzymes. Weinheim: Wiley-VCH; 2008. [Google Scholar]
- 12.Reetz MT, Wang L-W, Bocola M. Directed evolution of enantioselective enzymes: Iterative cycles of CASTing for probing protein-sequence space. Angew Chem. 2006;118:1258–1263. doi: 10.1002/anie.200502746. Erratum 2556; Angew Chem Int Ed 45:1236–1241; Erratum 2494. [DOI] [PubMed] [Google Scholar]
- 13.Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on the basis of B-factors as a strategy for increasing protein thermostability. Angew Chem. 2006;118:7909–7915. doi: 10.1002/anie.200602795. Angew Chem Int Ed 45:7745–7751. [DOI] [PubMed] [Google Scholar]
- 14.Reetz MT, Carballeira JD. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc. 2007;2:891–903. doi: 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- 15.Wong KF, Selzer T, Benkovic SJ, Hammes-Schiffer S. Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. P Natl Acad Sci USA. 2005;102:6807–6812. doi: 10.1073/pnas.0408343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reetz MT, et al. Learning from directed evolution: Further lessons from theoretical investigations into cooperative mutations in lipase enantioselectivity. ChemBioChem. 2007;8:106–112. doi: 10.1002/cbic.200600359. [DOI] [PubMed] [Google Scholar]
- 17.Kayser MM. ‘Designer reagents’ recombinant microorganisms: New and powerful tools for organic synthesis. Tetrahedron. 2009;65:947–974. [Google Scholar]
- 18.Stewart JD. Cyclohexanone monooxygenase: A useful reagent for asymmetric Baeyer–Villiger reactions. Curr Org Chem. 1998;2:195–216. [Google Scholar]
- 19.Mihovilovic MD. Enzyme mediated Baeyer–Villiger oxidations. Curr Org Chem. 2006;10:1265–1287. [Google Scholar]
- 20.Walsh CT, Chen Y-CJ. Enzymic Baeyer–Villiger oxidations by flavin-dependent monooxygenases. Angew Chem. 1988;100:342–352. Angew Chem, Int Ed Engl 27:333–343. [Google Scholar]
- 21.Hilker I, Wohlgemuth R, Alphand V, Furstoss R. Microbial transformations 59: First kilogram scale asymmetric microbial Baeyer–Villiger oxidation with optimized productivity using a resin-based in situ SFPR strategy. Biotechnol Bioeng. 2005;92:702–710. doi: 10.1002/bit.20636. [DOI] [PubMed] [Google Scholar]
- 22.Fraaije MW, et al. Discovery of a thermostable Baeyer–Villiger monooxygenase by genome mining. Appl Microbiol Biotechnol. 2005;66:393–400. doi: 10.1007/s00253-004-1749-5. [DOI] [PubMed] [Google Scholar]
- 23.Malito E, Alfieri A, Fraaije MW, Mattevi A. Crystal structure of a Baeyer–Villiger monooxygenase. P Natl Acad Sci USA. 2004;101:13157–13162. doi: 10.1073/pnas.0404538101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zambianchi F, et al. Titration and assignment of residues that regulate the enantioselectivity of phenylacetone monooxygenase. Adv Synth Catal. 2007;349:1327–1331. [Google Scholar]
- 25.Bocola M, et al. Converting phenylacetone monooxygenase into phenylcyclohexanone monooxygenase by rational design: Towards practical Baeyer–Villiger monooxygenases. Adv Synth Catal. 2005;347:979–986. [Google Scholar]
- 26.Reetz MT, Wu S. Chem Commun. Cambridge, UK: 2008. Greatly reduced amino acid alphabets in directed evolution: Making the right choice for saturation mutagenesis at homologous enzyme positions; pp. 5499–5501. [DOI] [PubMed] [Google Scholar]
- 27.Mirza IA, et al. Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J Am Chem Soc. 2009;131:8848–8854. doi: 10.1021/ja9010578. [DOI] [PubMed] [Google Scholar]
- 28.Qu C, Akanuma S, Moriyama H, Tanaka N, Oshima T. A mutation at the interface between domains causes rearrangement of domains in 3-isopropylmalate dehydrogenase. Protein Eng. 1997;10:45–52. doi: 10.1093/protein/10.1.45. [DOI] [PubMed] [Google Scholar]
- 29.Oue S, Okamoto A, Yano T, Kagamiyama H. Redesigning the substrate specificity of an enzyme by cumulative effects of the mutations of non-active site residues. J Biol Chem. 1999;274:2344–2349. doi: 10.1074/jbc.274.4.2344. [DOI] [PubMed] [Google Scholar]
- 30.Atkin KE, et al. The structure of monoamine oxidase from Aspergillus niger provides a molecular context for improvements in activity obtained by directed evolution. J Mol Biol. 2008;384:1218–1231. doi: 10.1016/j.jmb.2008.09.090. [DOI] [PubMed] [Google Scholar]
- 31.Hogrefe HH, Cline J, Youngblood GL, Allen RM. Creating randomized amino acid libraries with the QuikChange® multi site-directed mutagenesis kit. BioTechniques. 2002;33:1158–1165. doi: 10.2144/02335pf01. [DOI] [PubMed] [Google Scholar]
- 32.Zheng C, Pham VT, Philips RS. Asymmetric reduction of ketoesters with alcohol dehydrogenase from Thermoanaerobacter ethanolicus. Bioorg Med Chem Lett. 1992;2:619–622. [Google Scholar]
- 33.Eijsink VGH, Gåseidnes S, Borchert TV, van den Burg B. Directed evolution of enzyme stability. Biomol Eng. 2005;22:21–30. doi: 10.1016/j.bioeng.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Ó’Fágáin C. Enzyme stabilization—recent experimental progress. Enzyme Microb Tech. 2003;33:137–149. [Google Scholar]
- 35.Bowers KJ, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters; Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); Tampa, Florida. 2006. [Google Scholar]
- 36.Reddy AS, Sastry GM, Sastry GN. Cation-aromatic database. Proteins. 2007;67:1179–1184. doi: 10.1002/prot.21202. [DOI] [PubMed] [Google Scholar]
- 37.Crowley PB, Golovin A. Cation-pi interactions in protein-protein interfaces. Proteins. 2005;59:231–239. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- 38.Ichiye T, Karplus M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1994;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Nussinov R. Allosteric effects in the marginally stable von Hippel-Lindau tumor suppressor protein and allostery-based rescue mutant design. P Natl Acad Sci USA. 2008;105:901–906. doi: 10.1073/pnas.0707401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.