

SUMMARY
The killer lymphocyte protease granzyme A (GzmA) triggers caspase-independent target cell death with morphological features of apoptosis. We previously showed that GzmA acts directly on mitochondria to generate reactive oxygen species (ROS) and disrupt the transmembrane potential (ΔΨm) but does not permeabilize the mitochondrial outer membrane. Mitochondrial damage is critical to GzmA-induced cell death since cells treated with superoxide scavengers are resistant to GzmA. Here we find that GzmA accesses the mitochondrial matrix to cleave the complex I protein NDUFS3, an iron-sulfur subunit of the NADH:ubiquinone oxidoreductase complex I, after Lys56 to interfere with NADH oxidation and generate superoxide anions. Target cells expressing a cleavage site mutant of NDUFS3 are resistant to GzmA-mediated cell death but remain sensitive to GzmB.
INTRODUCTION
Mitochondrial disruption is a hallmark of caspase-dependent apoptosis (Kroemer et al., 2007). The mitochondrial outer membrane (MOM) is disrupted, causing the release of proapoptotic molecules such as cytochrome c, AIF, HtrA2/Omi, Smac/Diablo, and endoG. Released cytochrome c activates caspase-9. At the same time, damaged mitochondria generate excessive reactive oxygen species (ROS) and the mitochondrial transmembrane potential (ΔΨm) dissipates. Most mitochondrial apoptosis research has emphasized the importance of MOM permeabilization (MOMP), leaving it unclear whether ROS generation is incidental or essential to apoptosis.
Killer lymphocytes (cytotoxic T lymphocytes [CTLs] and NK cells) trigger both caspase-dependent and -independent apoptosis by releasing the contents of specialized secretory lysosomes, called cytotoxic granules, into the immune synapse formed with a cell targeted for destruction (Chowdhury and Lieberman, 2008). The cell death inducers are the granzyme (Gzm) serine proteases, of which there are five isoforms in humans and ten in mice. Gzms are delivered into the target cell cytosol by perforin (PFN). Granzyme A (GzmA) and B (GzmB) are the most abundant Gzms. GzmA induces cell death morphologically indistinguishable from apoptosis, but caspase inhibition or bcl-2 family expression does not affect it (Beresford et al., 1999; Chowdhury and Lieberman, 2008; Shresta et al., 1999). GzmA causes single-stranded DNA damage rather than oligonucleosomal double-stranded DNA breaks by activating two nucleases working sequentially, an endonuclease (NM23-H1) and an exonuclease (TREX1) (Chowdhury et al., 2006; Fan et al., 2003). NM23-H1 and TREX1 are in the ER-associated SET complex, together with an inhibitor (SET). Upon GzmA exposure, the SET complex translocates to the nucleus, where GzmA cleaves SET, liberating NM23-H1 to initiate DNA damage (Fan et al., 2003). GzmB cleaves and activates effector caspases and many critical caspase substrates. Superoxide scavengers block cell death by CTLs expressing both GzmA and GzmB, suggesting that generating ROS may be critical for both caspase-dependent and -independent programmed cell death (Aronis et al., 2003; Hildeman et al., 1999; Malassagne et al., 2001; Martinvalet et al., 2005; Sandstrom et al., 1994).
Within minutes of being introduced into cells, GzmA causes increased ROS and ΔΨm dissipation. GzmA perturbation of isolated mitochondria is insensitive to bcl-2, caspase inhibition, or cytosolic factors (Martinvalet et al., 2005; Pardo et al., 2004). Bid is not cleaved and apoptogenic factors are not released (Martinvalet et al., 2005). Moreover, mitochondrial damage by GzmA does not require bak or bax, the bcl-2 family members involved in bid-mediated mitochondrial damage (Zhu et al., 2006). Therefore GzmA activates a caspase-independent mitochondrial cell death pathway without MOMP. The goal of this study is to define its molecular basis.
GzmA is a highly selective tryptase. Only a handful of intracellular substrates, including SET, Ape1, and HMGB2 in the SET complex, as well as lamins, histones, and Ku70, have been identified as physiologically relevant (Chowdhury and Lieberman, 2008). GzmA binds to its substrates by an extended exosite, which may explain both its substrate specificity and the fact that its substrate cleavage sites do not share a common peptide sequence (Bell et al., 2003; Hink-Schauer et al., 2003). Since GzmA acts directly on mitochondria, mitochondrial damage by GzmA is likely activated by GzmA proteolysis of mitochondrial substrate(s). To begin to define the GzmA mitochondrial pathway, we used two-dimensional (2D) differential gel electrophoresis to identify potential GzmA substrates in isolated intact mouse liver mitochondria. This approach was first adapted by Bredemeyer et al. (2004) to identify Gzm substrates. NDUFS3 in complex I, one of two complexes in the electron transport chain (ETC) capable of generating ROS (Camello-Almaraz et al., 2006; Grivennikova and Vinogradov, 2006), is a GzmA target. NDUFS3 cleavage disrupts complex I function to initiate superoxide-dependent, but MOMP-independent, cell death.
RESULTS
GzmA Induces ROS by Directly Targeting Mitochondria
A sublytic dose of PFN, which causes death in ~10% of cells on its own, was used to deliver recombinant human GzmA into human K562 cells. PFN loading of GzmA caused dose-dependent increased ROS, measured by conversion of superoxide anion-reactive hydroethidine (HE) to ethidium (Figure S1A available online). Cells treated with either GzmA or PFN alone showed little change in ROS. GzmA- and PFN-treated cells also stained with the vital ROS indicator 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCFDA), which reacts with cytosolic ROS (Figure S1B). Pretreatment with pan-caspase inhibitors at caspase-inhibiting concentrations had little effect on ROS production (Figure S1C). Therefore, GzmA induces caspase-independent increased ROS. GzmA acts directly on mitochondria since GzmA treatment of isolated mitochondria rapidly increased ROS production, which was unaltered by adding S100 cytosol (Figure S1D).
To determine whether mitochondria are the source of ROS in intact cells treated with GzmA and PFN, we imaged live HeLa cells, prelabeled with Mitotracker Deep Red and MitoSOX Red, a vital mitochondrial dye that fluoresces selectively in response to superoxide anion (Mukhopadhyay et al., 2007). PFN or GzmA alone did not increase MitoSOX staining (Figure 1A and data not shown). Within a few minutes of adding GzmA with PFN, MitoSOX fluorescence increased and mitochondria appeared to round up (Figure 1A). This morphological change was confirmed by electron microscopy (Figure 1B). Within 10 min of adding GzmA and PFN, mitochondria swelled, rounded, and lost many of their cristae.
Figure 1. GzmA Targets Mitochondria to Produce ROS and Alters Mitochondrial Morphology.

(A) HeLa cells were loaded with MitoTracker Deep Red and MitoSOX, a mitochondrial superoxide dye, and then treated with PFN alone (top) or PFN plus recombinant human GzmA (bottom) and analyzed by live-fluorescence microscopy. Superoxide anion is detected within 2 min in GzmA-exposed mitochondria, and mitochondria lose their characteristic elongated shape (insets).
(B) Electron micrographs of HeLa cells treated with GzmA alone (left) or PFN + GzmA (right). Higher magnification images are below. Mitochondria round up and lose their cristae when GzmA is delivered by PFN. Plasma membrane (P), nucleus (N), and mitochondrion (m). Results are representative of at least three experiments.
Although mitochondria are the primary site of superoxide anion generation, other organelles, such as the ER, may be involved in free radical-induced cell death (Haynes et al., 2004). Live-fluorescence microscopy was performed on cells colabeled with MitoSox and H2DCFDA, which reacts with cytosolic ROS. Cells treated with PFN and GzmA showed only MitoSOX, but not H2DCFDA, staining (not shown). Since treated cells stained with H2DCFDA in the absence of MitoSox costaining (Figure S1B), the lack of cytosolic ROS is likely due to MitoSOX scavenging superoxide anions before they are released into the cytosol. Therefore mitochondria are the major source of GzmA-induced ROS.
Ndufs3 in Mitochondrial Complex I Is a Putative GzmA Substrate
A proteomics approach was used to identify potential mitochondrial targets. Purified mouse liver mitochondria were mock treated or treated with 2.5 μM recombinant human GzmA for 10 min and then lysed in the presence of protease inhibitors. Mitochondrial proteins were resolved by 2D gel electrophoresis (Figures 2A and 2B) and analyzed using Protetix 2D software (Nonlinear Dynamics). Approximately 307 protein spots were detected per gel. The mitochondria project 2 (MitoP2) has annotated 612 mouse mitochondrial proteins and estimated that it covers approximately half of the mitochondrial proteome (Prokisch et al., 2006). A systematic mass spectrometry survey of mouse mitochondrial proteins isolated from various tissues estimated the size of the mitochondrial proteome at 591 (Mootha et al., 2003). Therefore, our 2D gel analysis displays a large proportion of the predicted mouse mitochondrial proteome.
Figure 2. GzmA Cleaves Ndufs3, the 30 kDa Subunit of Mitochondrial Complex I.
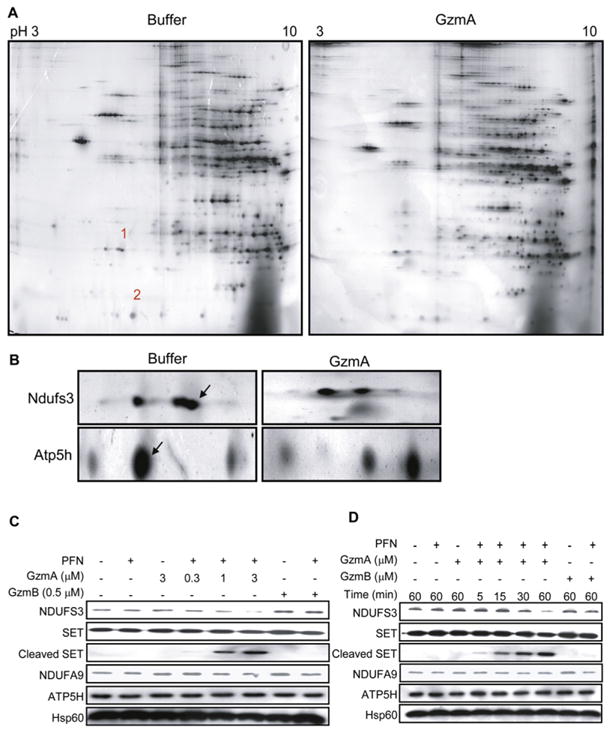
(A) Proteomic analysis. Freshly purified mouse liver mitochondria were treated with buffer or human GzmA for 10 min and resolved by 2D gel electrophoresis and silver staining. Most spots were unchanged. Spots 1 and 2 on the control gel, corresponding to missing proteins in the GzmA-treated sample, were excised and characterized by mass spectrometry.
(B) Magnification of the region corresponding to spots 1 and 2. Excised spots are indicated with arrows. Mass spectroscopy identified spot 1 as Ndufs3 and spot 2 as Atp5h. Spot 1 partially overlaps with another unchanged spot of higher apparent molecular mass and more acidic isoelectric point.
(C and D) Human K562 cells treated with human GzmA and PFN confirmed that GzmA specifically cleaves NDUFS3, but not ATP5H, in a dose- (C) and time-dependent (D) manner. SET is a previously described GzmA target. NDUFS3 was cleaved by GzmA but not GzmB. However, ATP5H was not a substrate for either Gzm. NDUFA9 is another complex I protein and Hsp60 is a mitochondrial loading control. Results are representative of at least four experiments.
Only a few spots disappeared in the GzmA-treated gels. These spots were picked from the control gel, digested with trypsin, and characterized by mass spectrometry. Two candidate mitochondrial substrates, Atp5H and Ndufs3, were selected for further investigation. Three peptides covering 14% of the amino acid sequence of Ndufs3 and nine peptides covering 60% of Atp5h were identified. Atp5h is the 15 kDa δ subunit in the connecting stalk between the ATP synthase F1 catalytic domain and the F0 integral membrane portion of the ATP synthase complex in the inner membrane (Papa et al., 2000). Ndufs3 is a 30 kDa subunit of the NADH:ubiquinone oxidoreductase ETC complex I, which catalyzes the first step of electron transfer from NADH to a noncovalently bound flavin mono-nucleotide and then, via a series of iron-sulfur clusters, to the terminal acceptor ubiquinone (Hirst et al., 2003). Ndufs3 is one of the iron-sulfur cluster proteins. Both Atp5h and Ndufs3 are good candidates for ROS generation since they are directly involved in electron transfer and coupling. Since either complex I or complex III can generate superoxide anion, Ndufs3 in complex I is especially attractive (Grivennikova and Vinogradov, 2006).
To determine whether Ndufs3 and Atp5h are physiologically relevant targets, we evaluated their cleavage in human K562 cells treated with PFN and recombinant human GzmA. Although the proteomic analysis was performed using mouse mitochondria, we chose to validate our results in human cells since proteins targeted in both species would be more likely to be significant. NDUFS3 was cleaved in cells treated with GzmA and PFN in a dose-dependent manner that paralleled cleavage of SET, a well-characterized GzmA substrate (Figure 2C) (Chowdhury and Lieberman, 2008). No immunoreactive NDUFS3 cleavage product was detected, most likely because the cleavage fragments are labile within cells. This is a feature of most other GzmA substrates (only SET and Ku70 fragments are detected in vivo) (Zhu et al., 2006). No difference in NDUFS3 cleavage was observed in the presence of o-phenanthroline, an inhibitor of AAA ATP-dependent proteases (data not shown), eliminating a role for these mitochondrial proteases in NDUFS3 cleavage. Processing of NDUFS3 was specific since NDUFA9, another complex I subunit, was not cleaved. Although Atp5h was a putative GzmA target, ATP5H was unaltered in K562 cells treated with both PFN and GzmA, suggesting that it was not a physiologically relevant substrate in human cells (Figures 2C and 2D). Since no antibody to mouse Atp5h is available, we were unable to test Atp5h cleavage in mouse cells. However, since human and mouse Atp5h are highly homologous (81% identity) and GzmA substrates are recognized by an extended interaction domain, mouse Atp5h is not a likely relevant substrate. NDUFS3 cleavage in K562 cells was evident within 30 min of treating cells with GzmA and PFN (Figure 2D). In both experiments the highly homologous GzmB (recombinant mouse protein used throughout) had no effect on NDUFS3. Blotting for Hsp60 verified comparable mitochondrial protein loading across samples. The specificity and cross-species importance of NDUFS3 as a substrate were also tested by treating mouse complex I, purified by immunocapture as described (Keeney et al., 2006), with cytotoxic granules isolated from a rat NK cell line (Figure S2). Both recombinant human GzmA and rat NK cell granules, but not recombinant mouse GzmB, cleaved mouse Ndufs3, but not Ndufb6, another complex I protein. Therefore NDUFS3, but not ATP5H, is a good candidate for a physiologically relevant GzmA substrate in both human and rodent cells.
GzmA Directly Cleaves NDUFS3 after Lys56
To determine whether NDUFS3 is a direct substrate, recombinant human NDUFS3 (rhNDUFS3), expressed with a C-terminal His6 tag, was incubated with human recombinant GzmA, enzymatically inactive GzmA produced by mutating the active site Ser176 to Ala (S-AGzmA), or mouse GzmB. GzmA efficiently cleaved rhNDUFS3 at GzmA concentrations as low as 250 nM (Figure 3A). A 23.6 kDa C-terminal fragment was readily detected with the tag antibody. Neither S-AGzmA nor GzmB had any effect on rhNDUFS3. To determine the cleavage site, both the full-length rhNDUFS3 and the C-terminal fragment were analyzed by mass spectrometry. Tryptic peptides covering the entire sequence were identified for the full-length protein, but the fragment generated 10 peptides with residue 57 being the most N-terminal amino acid of peptide QLSAFGEYVAEILPK, indicating cleavage after Lys56. Cleavage after a basic residue is consistent with GzmA being a tryptase. Moreover, the ~6.4 kDa difference between NDUFS3 and the fragment is consistent with removal of a 56 amino acid peptide. Therefore, GzmA likely cleaves NDUFS3 after Lys56.
Figure 3. GzmA Directly Cleaves Human NDUFS3 and Mouse Ndufs3.

(A) rhNDUFS3 was specifically and directly cleaved by human GzmA, but not mouse GzmB, to a 26.3kDa C-terminal fragment detected by immunoblot with His6 antibody. Mass spectroscopy of this fragment identified Lys56 as the NDUFS3 cleavage site.
(B–D) To confirm the cleavage site and its relevance in intact cells, WT or K56A-NDUFS3 or control vector were expressed with a HA tag in K562 cells (B, immunoblot with HA antibody) and verified to be incorporated into complex I by HA antibody immunoprecipitation (C). NDUFA9, MTND6, and NDUFB6 are other complex I proteins (D). When transduced K562 cells were treated with GzmA and PFN for 1 hr, WT NDUFS3 was degraded, verifying Lys56 as the in vivo GzmA cleavage site. K56A-NDUFS3 is cleaved somewhat by GzmA but is more resistant than WT NDUFS3. Cleavage of the GzmA target SET serves as a positive control and Hsp60 is a negative loading control.
(E) Similarly K56A-NDUFS3 overexpressed in mouse EL4 cells (immunoblot, left) was relatively resistant to cleavage by GzmA. EL4 cells transduced with WT or K56A-NDUFS3 were lysed and treated for 20 min with the indicated GzmA concentration (right) or treated for indicated times with 250 nM GzmA (bottom). In (B)–(E) transduced NDUFS3 was detected by HA antibody.
(F–H) GzmA also cleaves mouse Ndufs3 in purified mouse complex I and inhibits complex I activity. Complex I was purified either by immunoprecipitation using a cocktail of anticomplex I subunit antibodies (F) or by anion exchange and size exclusion chromatography (G). Ndufs3 in complex I was cleaved to a 24 kDa C-terminal fragment. Treatment with GzmB or inactive S-AGzmA had no effect on Ndufs3. Another complex I protein, Ndufb6, was unchanged. (H) GzmA disrupts complex I activity. Complex I activity was assayed 15 min after mitochondria were treated for 15 min with S-AGzmA or GzmA by measuring NADH oxidation by absorbance at 340 nm. Inhibition of NADH oxidation required active GzmA and increased in both a dose- and time-dependent manner (data not shown). Means±standard deviation (SD) are indicated. *p ≤ 0.05 by 2-sided t test.
The assignment of Lys56 as the cleavage site was confirmed by comparing GzmA cleavage of HA-tagged wild-type (WT) NDUFS3 and a mutant variant in which Lys56 was replaced by Ala (K56A-NDUFS3) in stably transfected K562 (Figures 3B–3D) or EL-4 cells (Figure 3E). Overexpressed NDUFS3 and K56A-NDUFS3 were incorporated into complex I since they coimmunoprecipitated with NDUFA9, MTND6, and NDUFB6 using an HA antibody (Figures 3B and 3C). Overexpression of WT or K56A-NDUFS3 did not interfere with ETC function since cells overexpressing WT or K56A-NDUFS3 had comparable proliferation rates, cellular ATP levels, mitochondrial ΔΨm, and background ROS as cells transfected with vector (Figure S3; data not shown). When transduced K562 or EL-4 cells were treated with PFN and increasing amounts of GzmA, K56A-NDUFS3 was relatively resistant to GzmA-mediated degradation, confirming Lys56 as the GzmA cleavage site (Figures 3D and 3E). However, K56A-NDUFS3 was cleaved to some extent. We previously found that GzmA inefficiently cleaves two other GzmA substrates, SET and Ku70, at alternate sites when the primary cleavage site is mutated (Zhu et al., 2006). These findings might be explained by GzmA binding to substrates by an extended exosite; if the preferred cleavage site is unavailable, the strong interaction with residues away from the active site may allow an alternative basic residue to be targeted.
Recombinant human GzmA also produced a 24 kDa Ndufs3 fragment when added to purified mouse complex I, isolated by immunocapture (Figures 3F and S2A). Cleavage was specific since GzmB had no effect on Ndufs3. Since the method used to purify complex I involved trichloroacetic acid (TCA) precipitation, which might have altered the native conformation of Ndufs3, we also examined GzmA cleavage of Ndufs3 in mouse liver complex I, purified by sequential anion exchange, size exclusion chromatography, detergent removal by dilution, and concentration by hollow membrane cross flow filtration as described (Sazanov et al., 2000) (Figure 3G). Only active GzmA, but not S-AGzmA or GzmB, cleaved Ndufs3. Two other complex I proteins, Ndufb6 and Mtnd6, were also not cleaved, again supporting specificity (data not shown). Therefore, human GzmA specifically cleaves both human and mouse NDUFS3. This is not too surprising given the high degree of identity between the proteins (87%). In fact, in residues 50–70 surrounding the cleavage site, there is only a single Ala52 to Thr difference. The similar apparent molecular masses of the mouse and human C-terminal cleavage fragments also suggest a shared cleavage site.
GzmA Cleavage of Ndufs3 Interferes with Complex I Activity
Complex I catalyzes the first reaction of the respiratory chain, the oxidation of NADH to NAD+, and provides two electrons to reduce ubiquinone to ubiquinol (Hirst et al., 2003). To determine whether Ndufs3 cleavage alters complex I activity, we measured NADH oxidation by GzmA-treated purified mouse mitochondria. GzmA, but not S-AGzmA or diisocoumarin-inactivated GzmA, significantly inhibited NADH oxidation (Figure 3H; data not shown). Therefore, GzmA cleavage of native Ndufs3 disrupts complex I electron transport.
GzmA Translocates to the Mitochondrial Matrix
When PFN delivers Gzms into target cells, Gzms are first endocytosed into early endosomes and then released within minutes into the target cell cytoplasm (Keefe et al., 2005). Some Gzm translocates to the target cell nucleus, but neither GzmA nor GzmB is known to enter mitochondria (Chowdhury and Lieberman, 2008). Because NDUFS3 is a mitochondrial matrix protein, our finding that GzmA cleaves NDUFS3 suggests that GzmA traffics into the matrix. Immunoelectron microscopy was used to determine whether GzmA might be internalized into mitochondria. Hsp60 staining labeled the mitochondrial matrix (Figure 4A). GzmA colocalized with Hsp60 only in cells treated with both GzmA and PFN. These results were confirmed by quantitative morphometry of the number of each type of gold particle per mitochondrial surface area (Figure 4B). Moreover, GzmA and PFN treatment severely altered mitochondrial ultrastructure as previously noted (Figure 1B). GzmA therefore penetrates the mitochondrial matrix to cleave NDUFS3.
Figure 4. GzmA Enters Mitochondria.

(A) HeLa cells, treated with recombinant human GzmA with (right) or without (left) PFN, were analyzed by immunoelectron microscopy for the mitochondrial matrix protein Hsp60 (10 nm particles, black arrows) and GzmA (15 nm, blue arrows). Without PFN, there is no staining for GzmA. GzmA colocalizes with Hsp60 in the matrix in cells treated with both GzmA and PFN.
(B) Morphometry quantification of the number of matrix Hsp60 (black bars) or GzmA (white bars) gold particles per mitochondrial surface area. Means ± SD are indicated.
GzmA Is Internalized into Isolated Mitochondria
As another test of mitochondrial import, isolated mitochondria were incubated with Alexa 488-labeled GzmA (GzmA 488) or with control SET 488 and then treated with trypsin to remove any protein that had not been internalized. Mitochondria incubated with GzmA 488, but not with buffer or SET 488, stained for Alexa 488 by flow cytometry, providing additional evidence of GzmA internalization (Figure 5A).
Figure 5. GzmA Interacts with the Cytosolic Chaperones Hsp70 and Hsp90 and Requires ΔΨm to Cleave Ndufs3 and Generate ROS.

(A) Fluorescently labeled GzmA (GzmA 488) is internalized by purified mouse liver mitochondria, but the control fluorescent recombinant SET protein (SET 488) is not. Purified mouse liver mitochondria were incubated with SET 488 or GzmA 488, gently trypsinized to remove surface proteins, and then analyzed by flow cytometry. Valinomycin, which disrupts ΔΨm, inhibits GzmA entry into mitochondria. Results are representative of at least three independent experiments.
(B) GzmA interacts with Hsp70 and Hsp90, two chaperones implicated in mitochondrial import. K562 cells treated with buffer or freeze-thawed human CTL granules were lysed and immunoprecipitated (IP) using GzmA antibody in the presence or absence of 10 mM of ATP and blotted for Hsp70, Hsp90, GzmA, and GAPDH. GzmA interacts with Hsp70 and Hsp90, but not with GAPDH, but only in the absence of 10 mM ATP, which inhibits Hsp chaperone function. GzmA was not detected in the input, which was loaded with 10% of the reaction mixture.
(C) Valinomycin also inhibits GzmA cleavage of Ndufs3 in isolated mouse liver mitochondria. (D and E) Valinomycin at nanomolar concentrations that disrupt ΔΨm (D) but are not toxic interferes with ROS production in K562 cells treated with GzmA and PFN (E). Cells treated with buffer or GzmA or PFN alone are background controls.
GzmA Binds to the Cytosolic Chaperones Hsp70 and Hsp90
GzmA does not contain a mitochondrial import sequence, raising the question of how GzmA enters the mitochondrial matrix to access NDUFS3. Moreover, GzmA does not disrupt the MOM since apoptogenic factors in the intermembrane space are not released (Martinvalet et al., 2005). Therefore, GzmA access to the matrix is not due to indiscriminate mitochondrial disruption. Protein trafficking into the mitochondrial matrix is highly regulated. One pathway involves binding to cytosolic chaperones Hsp70 and Hsp90 that dock onto the cytosolic portion of Tom70, the 70 kDa subunit of the translocase of the outer mitochondrial membrane, as a first step in mitochondrial import (Geissler et al., 2001; Young et al., 2003). Tom70 and Tom20 then hand off their cargo to the translocase core channel Tom40 (Rehling et al., 2004). The 50 kDa subunit of the translocase of the inner mitochondrial membrane Tim50 is the first component of the Tim23 complex that interacts with cargo in the intermembrane space. Although defining the pathway by which GzmA traffics into the matrix is beyond the scope of this study, we hypothesized that GzmA takes advantage of this pathway for entry. In support of this idea, we previously found that recombinant GzmA and S-AGzmA bind with exceedingly high affinity to Hsp70 and Hsp27 (Beresford et al., 1998). However, Hsp chaperones can also bind to improperly folded proteins that might be in recombinant protein preparations. Therefore, as a first step to validating the interaction of GzmA with putative mitochondrial chaperones, we evaluated whether native GzmA binds to Hsp70 or Hsp90. Human CTL granules were a source of native GzmA. K562 cells were treated with freeze-thawed granules, and lysates were immunoprecipitated using GzmA antibody and then blotted for Hsp70 and Hsp90. GzmA coprecipitated with both Hsp70 and Hsp90, but not with GAPDH (Figure 5B). Furthermore, GzmA binding to Hsp70 and Hsp90 was disrupted by a molar excess of ATP, suggesting that this interaction was specific since ATP-bound Hsp70 and Hsp90 have reduced affinity for their client proteins. In addition, recombinant human GzmA, but not recombinant mouse GzmB, was specifically coimmunoprecipitated with both Hsp70 and Hsp90 (data not shown). The lack of an interaction with GzmB may not be surprising since GzmB has been reported to cleave the Hsp cofactor proteins, Hop and Hip, as well as Hsp70 and Hsp90 (Bredemeyer et al., 2006; Hostetter et al., 2007; Loeb et al., 2006). Nonetheless, GzmB in the human granules did not abolish the interaction of GzmA with Hsps. Further study will be required to determine whether GzmB processing of Hsps and their activating cofactors affects the GzmA mitochondrial pathway. These data suggest that although GzmA does not have a mitochondrial import signal, it may access the mitochondrial matrix by interacting with an Hsp.
Since GzmA disrupted complex I activity (Figure 3H) and was internalized into isolated mitochondria (Figure 5A) in the absence of added cytosol, we wanted to know whether Hsps copurified with the mitochondrial preparation. Purified mouse mitochondria were either washed with bicarbonate to remove loosely adherent proteins or left untreated and the mitochondrial pellet was analyzed by immunoblot for Hsp proteins (Figure S4). Hsp70 and Hsp60, but not Hsp90, were present in our mitochondrial preparation. Washing with bicarbonate partially released Hsp70, but not the mitochondrial matrix protein Hsp60. Therefore, some Hsp70 protein was associated with the outer membrane of the isolated mitochondria and potentially available to chaperone GzmA to the mitochondrial translocases.
Treating cells with the Hsp90 inhibitor 17-allylaminogeldanamycin (17-AAG) did not inhibit GzmA-induced ROS, cell death, or SET cleavage and in fact enhanced each of these downstream events (Figure S5). This suggests that Hsp90 is unlikely to be the GzmA mitochondrial chaperone. Hsp70 may be a better candidate. We were unable to test an Hsp70 inhibitor because it was too toxic to our cells. Hsp90 performs many functions in cells and has been shown to protect mitochondria from death stimuli in tumor cells (Kang et al., 2007), so our finding that inhibiting Hsp90 enhances GzmA-induced cell death may not be surprising. Sorting out the roles of Hsp70 and Hsp90 in this pathway requires further study.
GzmA Cleavage of NDUFS3 and ROS Generation Requires ΔΨm
Protein translocation across the mitochondrial double membranes by the Tom/Tim twin translocases requires an intact ΔΨm (Rehling et al., 2004). To begin to test the hypothesis that GzmA mitochondrial translocation is mediated by the Tom/Tim system, we examined whether GzmA is able to enter mitochondria and cleave Ndufs3 in purified mitochondria treated with valinomycin, which disrupts ΔΨm (Daum et al., 1982). GzmA 488 entry into isolated mitochondria and cleavage of Ndufs3 were inhibited by preincubation with valinomycin (Figures 5A and 5C). To assess whether valinomycin also inhibited ROS generation in intact cells, we first verified that treatment of K562 cells with low nanomolar concentrations of valinomycin dissipated ΔΨm without excessive cytotoxicity (Figure 5D; data not shown). Pre-treatment of K562 cells with 16 nM valinomycin inhibited GzmA-and PFN-induced ROS generation (Figure 5E). Therefore, GzmA requires ΔΨm to cleave Ndufs3 and induce mitochondrial damage in cells. These data support a model in which GzmA is carried to mitochondria by an Hsp and then internalized via the ΔΨm-dependent mitochondrial import machinery. However, additional experiments are needed to test this hypothesis.
GzmA-Mediated ROS Production and Cell Death Are Impaired in Pseudo ρ0 Cells Depleted of Mitochondrial DNA
We next looked at the effect of transient mitochondrial DNA (mtDNA) depletion on GzmA-induced ROS and cell death. Pseudo ρ0 cells are partially depleted of mtDNA by culture in the presence of ethidium bromide (Kaminski et al., 2007). Because mtDNA encodes for genes in complex I and other key ETC proteins, these cells rely on glycolysis to produce ATP. K562 cells or K562 pseudo ρ0 cells, which contain about 1/5 of normal cellular mtDNA (Figure 6A), were treated with PFN and GzmA to assess ROS production and cell death. Pseudo ρ0 cells did not generate ROS and were resistant to GzmA-induced cell death (Figures 6B and 6C). These results indicate that an intact and functional ETC is needed for GzmA-generated ROS and cell death.
Figure 6. GzmA-Induced ROS Production and Cell Death Require Complex I Activity.

(A–C) Pseudo ρ0 K562 cells with reduced mtDNA, assessed by quantitative PCR (A), are resistant to GzmA-induced ROS (B) and cell death (C), assayed by annexin V and PI staining. In (C), black bars indicate K562 cells; gray bars, pseudo ρ0 K562 cells.
(D and E) The complex I inhibitor rotenone (Rot) and the superoxide anion scavenger Tiron (80 mM) inhibit GzmA-induced ROS (D) and cell death (E). K562 cells were pretreated with rote-none or Tiron and then incubated with PFN and/or GzmA. Light and dark gray bars indicate cells pretreated for 2 hr with 0.25 μM and 1 μM of rote-none, respectively.
(F) Scavenging superoxide anion does not affect upstream cleavage of NDUFS3 but inhibits downstream cleavage of SET when K562 cells are exposed to GzmA and PFN.
For all panels, results are means ± SD for representative experiments performed at least three times.
The Complex I Inhibitor Rotenone Hinders GzmA-Mediated ROS Production and Cell Death
To probe further the importance of complex I in GzmA-induced cell death, we treated K562 cells, preincubated with rotenone or the superoxide scavenger Tiron as a positive control, with GzmA and PFN and analyzed ROS production and cell death. As previously reported (Martinvalet et al., 2005), Tiron completely inhibits ROS production and cell death (Figures 6D and 6E). Rotenone also inhibits both ROS generation and cell death, although not as completely as Tiron. Pretreatment of cells with Piericidin A, another complex I inhibitor, also blocked GzmA-mediated cell death (data not shown).
We also investigated whether scavenging superoxide anion with Tiron interfered with GzmA NDUFS3 cleavage. Tiron had no effect on NDUFS3 cleavage (Figure 6F), as would be expected if GzmA cleavage of NDUFS3 is upstream of ROS production. However, when K562 cells were preincubated with Tiron, SET cleavage was completely inhibited at concentrations that scavenge all the ROS (40–80 mM) (Figure 6F). This lends additional support to the importance of mitochondrial superoxide production in completing the GzmA cell death program since SET cleavage is required to activate the GzmA nucleases (Fan et al., 2003).
Expression of an NDUFS3 Cleavage Site Mutant Inhibits GzmA-Mediated ROS Production and Cell Death
To test the importance of NDUFS3 cleavage in GzmA-mediated cell death, we treated EL4 cells stably overexpressing WT or K56A-NDUFS3 with GzmA and PFN. ROS production, assayed by HE staining and flow cytometry, was reduced almost to background in cells expressing K56A-, but not WT, NDUFS3 (Figure 7A). EL4 cells overexpressing K56A-NDUFS3 were also more resistant to PFN- and GzmA-induced death measured by annexin V + propidium iodide (PI) staining compared to cells expressing vector or WT NDUFS3 (Figure 7B). Overexpression of K56A-NDUFS3 did not reduce caspase-dependent ROS generation following UV irradiation, demonstrating that this protection was GzmA specific (Figure 7C). Cells expressing K56A-NDUFS3 were also protected from ΔΨm dissipation (data not shown). Expression of the NDUFS3 cleavage site mutant in 293T and K562 cells also inhibited cell death by GzmA and PFN (data not shown).
Figure 7. Overexpression of a Cleavage Site Mutant of NDUFS3 Inhibits GzmA-Induced Mitochondrial Damage and Cell Death.

Mouse EL4 cells were stably transfected with vector, HA-tagged wild-type (WT) NDUFS3, or K56A-NDUFS3 expression vectors (Figure 3E).
(A) EL4 cells overexpressing K56A-NDUFS3 generate reduced ROS in response to GzmA and PFN, compared to cells transduced with WT NDUFS3 or vector. (Adding GzmA without PFN tended to reduce background ROS detection in cells for all conditions for unclear reasons, suggesting that GzmA may scavenge ROS.)
(B and C) Overexpression of K56A-NDUFS3 also protects EL4 cells from GzmA- and PFN-induced cell death measured by annexin V-PI staining (B) but does not substantially affect UV-induced ROS generation (C) or death (data not shown). Cells were treated with buffer (−), 0.5 μM GzmA (A), PFN (P), PFN plus 0.25 μM GzmA (gray bars), or PFN plus 0.5 μM GzmA (black bars).
(D) Mutant NDUFS3 also inhibits cleavage of the SET homolog I2PP2A in overexpressing EL4 cells. (E and F) These stably transfected EL4 cell lines were coated with gp33 peptide and used as targets for CTL effector cells derived from GzmB−/− (E) or GzmA−/− (F) mice in a 6 hr 51Cr-release assay. E:T ratio, effector:target cell ratio. Black bars represent EL4 cells transduced with vector; white bars, WT NDUFS3; gray bars, K56A-NDUFS3. Overexpression of K56A-NDUFS3 protects cells from GzmA-expressing GzmB−/− CTLs but not from GzmA−/− CTLs.
For all panels, results shown are means ± SD of at least 4–6 independent experiments. *p ≤ 0.02 compared with vector-transduced target cells and p < 0.03 compared with WT NDUFS3-transduced target cells.
We previously showed that mitochondrial superoxide anion induced by GzmA triggers the nuclear translocation of the SET complex, which contains SET and the GzmA-activated DNases, NM2321-H1 and TREX1 (Martinvalet et al., 2005). Since cells overexpressing K56A-NDUFS3 generate much less ROS when exposed to GzmA, we investigated the effect of K56A-NDUFS3 overexpression on GzmA-induced SET cleavage. In both control and WT NDUFS3-overexpressing EL4 cells, GzmA and PFN triggered nuclear translocation of full-length I2PP2A (the mouse SET homolog, data not shown) and I2PP2A cleavage (Figure 7D). However, overexpression of K56A-NDUFS3 inhibited both I2PP2A translocation and cleavage.
NDUFS3 Cleavage Is Critical for Cell Death by GzmA-Expressing CTLs
We next wanted to determine whether GzmA-mediated NDUFS3 cleavage is critical for cell death in the most relevant model, CTL-mediated cell death. CTLs from P14 TCR transgenic mice kill EL4 cells coated with a lymphochoriomeningitis virus gp33 peptide (Pircher et al., 1989). We backcrossed the P14 Tg mice with mice genetically deficient in GzmA or GzmB to generate gp33-specific CTL expressing only GzmB or GzmA, respectively. We then compared the susceptibility of gp33-coated EL4 target cells overexpressing WT or K56A-NDUFS3 or control vector to lysis by GzmB−/− P14 or GzmA−/−P14 CTLs by 51Cr-release assay (Figures 7E and 7F). As expected, killing by GzmA−/− CTLs was not affected by overexpressing either WT or mutant NDUFS3 (Figure 7F). However, target cells overexpressing K56A-NDUFS3 were about 2-fold more resistant to cell death triggered by GzmB−/− CTLs (Figure 7E). Overexpression of WT NDUFS3 did not significantly protect cells from GzmB−/− CTLs at any effector: target (E:T) cell ratio (Figure 7E). However, cytolysis of K56A-NDUFS3-transduced EL4 cells was significantly reduced at each E:T ratio when compared to vector-transduced (p < 0.02) or WT NDUFS3-transduced cells (p < 0.03). Similar results were observed when transduced 293T cells were substituted for EL4 target cells (data not shown). Therefore, NDUFS3 cleavage plays an important role in GzmA, but not GzmB and/or caspase, cell death.
DISCUSSION
GzmA activates a mitochondrial MOMP-and caspase-independent death pathway by cleaving NDUFS3 in complex I. NDUFS3 cleavage is likely the first step of GzmA programmed cell death. In addition to generating ROS, disrupting ΔΨm, and interfering with ATP generation, NDUFS3 cleavage is required for DNA damage. The SET complex, which contains the two GzmA-activated DNases NM23-H1 and TREX1 and their inhibitor SET, is normally extra-nuclear (Chowdhury et al., 2006; Martinvalet et al., 2005). It translocates to the nucleus in response to superoxide anion, generated by NDUFS3 cleavage. Both SET complex translocation and SET cleavage, required to activate DNA damage, as well as cell death, are blocked by superoxide scavengers or overexpression of GzmA-resistant K56A-NDUFS3.
NDUFS3 cleavage is specific since other complex I proteins are not substrates and GzmB does not cleave NDUFS3. Moreover, NDUFS3 is a GzmA substrate in both rodents and humans. Both mouse Ndufs3 and human NDUFS3 are cleaved by human GzmA and both human and rat GzmA cleave mouse Ndufs3. Furthermore, GzmA disrupts complex I function. Functional complex I is needed to generate ROS to initiate cell death since pseudo ρ0 cells with a deficit in ETC function are GzmA resistant. Electron flow along the ETC is probably critical to initiate death since electrons are needed to generate reactive free radicals.
Although ROS generation and SET complex translocation are detected within minutes of introducing GzmA into cells, NDUFS3 cleavage is only obvious after 30 min. If NDUFS3 cleavage triggers both ROS and SET complex translocation, why this apparent discrepancy in kinetics? Immunoblot may be relatively insensitive to protein cleavage since clear cut changes are detected only when a significant fraction of a protein has been degraded. However, ROS may be continuously generated every time an NDUFS3 molecule is degraded. Native ROS scavengers may get saturated before a large fraction of NDUFS3 molecules have been proteolyzed.
ETC I and III are the major sources of spontaneous superoxide anion production (Camello-Almaraz et al., 2006; Grivennikova and Vinogradov, 2006). Complex I not only generates superoxide radicals but also maintains the proton gradient across the inner membrane (ΔΨm) and is needed for ATP synthesis. Since complex I is the entry point of electrons, disabling complex I disrupts energy metabolism at its very root. Of note, caspase-3 also cripples complex I by cleaving NDUFS1 (Ricci et al., 2004). Moreover, treatment with rotenone, a specific complex I inhibitor, can by itself induce apoptosis (Li et al., 2003). Silencing NDUFS3 or another complex I subunit (GRIM-19) sensitizes some cancer cells to cell death by IFN-β and retinoic acid (Huang et al., 2007). A recent study also implicates complex I in activation-induced T cell death (Kaminski et al., 2007). Both HIV-1 and cytomegalovirus have devised strategies to interfere with complex I expression or mitochondrial localization (Ladha et al., 2005; Reeves et al., 2007). Since complex I is the first step in energy metabolism and redox pathways, it is not surprising that multiple cell death pathways have disabled it. Our results suggest that oxygen radicals produced by disrupting complex I in these various pathways are not mere byproducts of cell death but are central to its execution.
Superoxide scavengers also block cell death by GzmB-expressing CTLs (Malassagne et al., 2001; Martinvalet et al., 2005). GzmB induces MOMP by cleaving bid and activating caspase-3 but can also activate caspase-independent mitochondrial damage (Heibein et al., 1999). In fact, GzmB cell death is unimpaired in cells lacking both bax and bad, mediators of bid-induced MOMP, or by caspase inhibition (Heibein et al., 1999; MacDonald et al., 1999; Thomas et al., 2001; Wang et al., 2001). Preliminary data suggest that GzmB might also directly target complex I. Another Gzm, mouse GzmC (most homologous to human GzmH), dramatically alters mitochondrial morphology without cleaving bid or activating the caspases (Johnson et al., 2003). Therefore, mitochondrial disruption may be central to all Gzm death pathways.
A proteomics approach would be a good starting point to identify mitochondrial substrates of other Gzm. The manageable number of mitochondrial proteins, compared to the whole cell proteome, and using intact mitochondria, rather than mitochondrial lysates, likely enhanced our ability to identify a relevant substrate. Reducing the incubation time and granzyme concentration also favored identifying relevant substrates. Our 2D gels demonstrate the lack of indiscriminate proteolysis by GzmA. NDUFS3 now joins a small list of validated GzmA substrates.
We were surprised to find that GzmA cleaves a mitochondrial matrix protein. Complex I is embedded in the mitochondrial inner membrane with a stalk that sticks into the matrix. The exact structure of mammalian complex I is unknown, although its structure has been inferred from subcomplexes that remain intact after exposure to detergents, by subassemblies formed in cells carrying mutations of different subunits, from low-resolution structures of the bovine complex, and from the crystal structure of the bacterial complex I (NDH-1) resolved at 3.3 Å (Sazanov et al., 2000). NDH-1 has 14 subunits, which all have mammalian orthologs and constitute the core of complex I. NDUFS3 is a core component, the ortholog of bacterial Nqo5. Nqo5 is well exposed in the connecting stalk that links the peripheral arm to the membrane arm of the complex. Although the mammalian enzyme has additional subunits, NDUFS3 likely occupies a similar position in the stalk. Furthermore, the connecting stalk is the narrowest segment of bovine complex I and therefore is unlikely to house additional peptides. GzmA cleavage of NDUFS3 supports the leading current model of complex I topology in which both NDUFS3 and the caspase substrate NDUFS1 are accessible in the matrix-protruding stalk (Hirst et al., 2003; Ricci et al., 2004). However, both GzmA and caspase-3 need to access the matrix to cleave their complex I targets. Even if MOMP allows caspase-3 into the intermembrane space, caspase-3 would still need to transit across the inner mitochondrial membrane to cleave NDUFS1.
GzmA lacks a mitochondrial import signal but nonetheless traffics to the mitochondrial matrix without causing MOMP. Disrupting ΔΨm before adding GzmA inhibits NDUFS3 cleavage and ROS generation. Therefore, ΔΨm is required for GzmA to enter the matrix, and thus ΔΨm dissipation occurs after GzmA has gotten into mitochondria. The ΔΨm-dependent twin-pore translocase might be involved in GzmA mitochondrial import. Imported cargo is brought to the outer membrane translocase by Hsps (Geissler et al., 2001; Young et al., 2003). Hsp27 and Hsp70 bind to immobilized recombinant S-AGzmA so tightly that 6 M urea is needed to disrupt binding (Beresford et al., 1998). Now we have verified this Hsp-GzmA interaction by showing that Hsp70 and Hsp90 coprecipitate in an ATP-dependent manner with native GzmA.
EXPERIMENTAL PROCEDURES
Gzm and PFN Treatment
Cells were washed in HBSS and 5 × 104 cells were resuspended in 60 μl loading buffer (HBSS with 10 mM HEPES, pH 7.2, 0.4% BSA, 3 mM CaCl2). A sublytic concentration of PFN (the concentration that lyses 5%–15% of cells) and/or GzmA or GzmB (1 μM or as indicated) was added and the cells were cultured at 37°C for the indicated time. When inhibitors were used, cells were preincubated for up to 2 hr with the inhibitor before adding GzmA and PFN for 1 hr. NP40 cell lysates obtained after 1 hr incubation were analyzed by immunoblot. For cell death assays, cells were stained with annexin V and PI 1 hr after adding PFN and/or Gzm and analyzed by flow cytometry (Martinvalet et al., 2005). Dead cells were defined as the sum of all annexin V+ and/or PI+ cells. For electron microscopy, 106 HeLa cells in aliquots of 2 × 104/60 μl were treated with GzmA and/or PFN for 10 min and washed in HBSS before fixation in 4% paraformaldehyde for 2 hr.
ROS Production and ΔΨm
ROS production was monitored by adding 2 μM HE just before flow cytometry analysis. Changes in ΔΨm were monitored with the potentiometric dye JC-1 using the Mitochondrial Membrane Potential Detection Kit (BioCarta, San Diego, CA, USA). Control cells were cultured overnight after UV irradiation (200 μJ/cm2 × 5 min). Flow cytometry was performed using a Becton Dickinson FACSCalibur flow cytometer and Cellquest Pro software.
Isolated Mitochondria Assays
Mitochondria and S100 supernatants were freshly prepared from mouse liver as described (Susin et al., 2000). Mitochondria (0.5 mg/ml protein) were incubated with or without S100 supernatant (1 mg/ml) plus Gzms at indicated concentrations in 60 μl of mitochondrial buffer (220 mM sucrose, 68 mM mannitol, 10 mM KCl, 5 mM KH2PO4, 2 mM MgCl2, 0.5 mM EGTA, 5 mM succinate, 2 mM rotenone, 10 mM HEPES, pH 7.2) for 30 min at 37°C. Mitochondria were stained with 150 μM HE for flow cytometry as above. To determine whether Hsps were contained in mitochondrial preparations, mitochondria were washed at 4°C with 0.2 M HCO3− (pH 10) or mitochondrial buffer.
Mitochondrial import was assessed as reported (Young et al., 2003) with slight modification. Purified mouse mitochondria were preincubated at room temperature for 20 min with or without valinomycin before adding 1 μM SET-Alexa 488 or 2 μM GzmA-Alexa 488 for 20 min at room temperature (RT). Treated mitochondria were washed in the same buffer and then incubated with 2.5 μg/ml trypsin for 10 min at 4°C. The reaction was stopped with 1 mM PMSF. After further washing, fluorescence was evaluated.
Live-Cell Imaging for ROS Production
HeLa cells (5 × 104 cells/well), plated overnight on collagen-coated coverslips (VWR) in 6-well plates, were stained for 1 hr at 37°C with 25 nM MitoTracker Deep Red plus 2.5 μM MitoSOX Red in DMEM complete medium. After three washes, cells were incubated in complete medium for 30 min at 37°C. Just before imaging, cover slides were washed three times in warm HBSS and incubated in a heated imaging chamber containing 1 ml loading buffer with PFN and/or 0.4 μM GzmA. Fluorescence was observed immediately with a Zeiss laser-scanning fluorescent microscope and SlideBook 4.0 OS × digital imaging software.
Complex I Purification and Activity
Complex I was purified as reported by Carroll et al. (2003) and Sazanov et al. (2000). Complex I activity was monitored by measuring the conversion of NADH into NAD+, as reported (Yamaguchi et al., 1998).
Supplementary Material
Figure S1. GzmA induces a caspase-independent increase in ROS by directly targeting the mitochondria
(A) GzmA loading of K562 cells with PFN induces a dose-dependent increase of ROS. K562 cells were treated with buffer, a sublytic dose of PFN, GzmA alone or PFN plus increasing GzmA concentrations and stained for ROS with HE.
(B) Cells were treated as in (A) and ROS was measured by H2DCFDA staining. Grey histograms are buffer-treated cells and white histograms are cells treated as indicated.
(C) PFN loading of GzmA in K562 induces a caspase-independent increase in ROS. K562 cells, preincubated or not with 100 μM ZVAD and DEVD as pan-caspase inhibitors (not shown), were treated with PFN and/or GzmA and stained for ROS. Caspase inhibition did not inhibit ROS production.
(D) GzmA treatment of purified mitochondria induces a rapid increase in ROS. Freshly purified mouse liver mitochondria were either untreated or treated with GzmA in the presence or absence of cytosolic S100 fraction and stained for ROS with HE. On its own, GzmA induced ROS. Results shown are representative of at least 3 independent experiments.
Figure S2. Ndufs3 in purified ETC complex I is targeted by purified rat CTL granules and recombinant human GzmA
(A) Mouse liver mitochondrial complex I was purified by immunocapture and analyzed by gel electrophoresis and silver stain. Annotation on the right identifies the most prominent complex I subunits.
(B) Mouse Ndufs3 is cleaved by human and rodent GzmA. Complex I was treated with purified rat CTL granules or recombinant human GzmA or GzmB. Nudfs3, but not Ndufb6, was cleaved by both sources of GzmA, but not by GzmB.
Figure S3. Over-expression of K56A-NDUFS3 doesn’t alter cellular ATP levels
EL4 cells stably transfected with empty vector, WT NDUFS3 or K56A-NDUFS3 expression vectors were grown in normal aerobic conditions and ATP content was measured and compared to ATP levels in mock transfected cells.
Figure S4. Hsp70 adheres to isolated mitochondria
Purified mouse liver mitochondria were washed or not with bicarbonate (HCO3−) and the presence of Hsps in the mitochondrial pellet assessed by immunoblot. Hsp70 was detected in isolated mitochondria, but levels decreased after bicarbonate washing, suggesting that some Hsp70 was loosely adhered to the mitochondrial outer membrane, but some was either internalized or more firmly adherent. By contrast, no Hsp90 was detected and matrix Hsp60 was detected, but was not affected by washing.
Figure S5. 17-AAG increases GzmA-induced ROS, cell death and SET cleavage.
(A) K562 cells, preincubated or not with the Hsp90 inhibitor 17-AAG as indicated, were treated with PFN and/or GzmA and stained for ROS (top panel) and cell death by annexin-V +PI (bottom panel). (B) K562 cells were treated as in A, and SET cleavage was followed by immunoblot.
Acknowledgments
This work was supported by NIH grants AI45587 (J.L.) and T32 HL66987 (R.F.). We thank Katie Doud for proteomics advice, Maria Ericsson for electron microscopy, Sandra Lee for statistical support, and members of the Lieberman laboratory for helpful suggestions. This paper is dedicated to the memory of Olivier T. Martinvalet.
Footnotes
Supplemental Data include Experimental Procedures and five figures and can be found with this article online at http://www.cell.com/cgi/content/full/133/4/681/DC1/.
References
- Aronis A, Melendez JA, Golan O, Shilo S, Dicter N, Tirosh O. Potentiation of Fas-mediated apoptosis by attenuated production of mitochondria-derived reactive oxygen species. Cell Death Differ. 2003;10:335–344. doi: 10.1038/sj.cdd.4401150. [DOI] [PubMed] [Google Scholar]
- Bell JK, Goetz DH, Mahrus S, Harris JL, Fletterick RJ, Craik CS. The oligomeric structure of human granzyme A is a determinant of its extended substrate specificity. Nat Struct Biol. 2003;10:527–534. doi: 10.1038/nsb944. [DOI] [PubMed] [Google Scholar]
- Beresford PJ, Jaju M, Friedman RS, Yoon MJ, Lieberman J. A role for heat shock protein 27 in CTL-mediated cell death. J Immunol. 1998;161:161–167. [PubMed] [Google Scholar]
- Beresford PJ, Xia Z, Greenberg AH, Lieberman J. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity. 1999;10:585–594. doi: 10.1016/s1074-7613(00)80058-8. [DOI] [PubMed] [Google Scholar]
- Bredemeyer AJ, Lewis RM, Malone JP, Davis AE, Gross J, Townsend RR, Ley TJ. A proteomic approach for the discovery of protease substrates. Proc Natl Acad Sci USA. 2004;101:11785–11790. doi: 10.1073/pnas.0402353101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredemeyer AJ, Carrigan PE, Fehniger TA, Smith DF, Ley TJ. Hop cleavage and function in granzyme B-induced apoptosis. J Biol Chem. 2006;281:37130–37141. doi: 10.1074/jbc.M607969200. [DOI] [PubMed] [Google Scholar]
- Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol. 2006;291:C1082–C1088. doi: 10.1152/ajpcell.00217.2006. [DOI] [PubMed] [Google Scholar]
- Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol Cell Proteomics. 2003;2:117–126. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J. The exonuclease TREX1 is in the SET complex and acts in concert with NM23–H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell. 2006;23:133–142. doi: 10.1016/j.molcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Chowdhury D, Lieberman J. Death by a thousand cuts: Granzyme pathways of cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G, Gasser SM, Schatz G. Import of proteins into mitochondria. Energy-dependent, two-step processing of the intermembrane space enzyme cytochrome b2 by isolated yeast mitochondria. J Biol Chem. 1982;257:13075–13080. [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. Tumor suppressor NM23–H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112:659–672. doi: 10.1016/s0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- Geissler A, Rassow J, Pfanner N, Voos W. Mitochondrial import driving forces: enhanced trapping by matrix Hsp70 stimulates translocation and reduces the membrane potential dependence of loosely folded preproteins. Mol Cell Biol. 2001;21:7097–7104. doi: 10.1128/MCB.21.20.7097-7104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikova VG, Vinogradov AD. Generation of superoxide by the mitochondrial complex I. Biochim Biophys Acta. 2006;1757:553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15:767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Heibein JA, Barry M, Motyka B, Bleackley RC. Granzyme B-induced loss of mitochondrial inner membrane potential (Delta Psi m) and cytochrome c release are caspase independent. J Immunol. 1999;163:4683–4693. [PubMed] [Google Scholar]
- Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, Marrack PC. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- Hink-Schauer C, Estebanez-Perpina E, Kurschus FC, Bode W, Jenne DE. Crystal structure of the apoptosis-inducing human granzyme A dimer. Nat Struct Biol. 2003;10:535–540. doi: 10.1038/nsb945. [DOI] [PubMed] [Google Scholar]
- Hirst J, Carroll J, Fearnley IM, Shannon RJ, Walker JE. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim Biophys Acta. 2003;1604:135–150. doi: 10.1016/s0005-2728(03)00059-8. [DOI] [PubMed] [Google Scholar]
- Hostetter DR, Loeb CR, Chu F, Craik CS. Hip is a pro-survival substrate of granzyme B. J Biol Chem. 2007;282:27865–27874. doi: 10.1074/jbc.M704312200. [DOI] [PubMed] [Google Scholar]
- Huang G, Chen Y, Lu H, Cao X. Coupling mitochondrial respiratory chain to cell death: an essential role of mitochondrial complex I in the interferon-beta and retinoic acid-induced cancer cell death. Cell Death Differ. 2007;14:327–337. doi: 10.1038/sj.cdd.4402004. [DOI] [PubMed] [Google Scholar]
- Johnson H, Scorrano L, Korsmeyer SJ, Ley TJ. Cell death induced by granzyme C. Blood. 2003;101:3093–3101. doi: 10.1182/blood-2002-08-2485. [DOI] [PubMed] [Google Scholar]
- Kaminski M, Kiessling M, Suss D, Krammer PH, Gulow K. Novel role for mitochondria: protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol. 2007;27:3625–3639. doi: 10.1128/MCB.02295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, Lieberman J. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249–262. doi: 10.1016/j.immuni.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Ladha JS, Tripathy MK, Mitra D. Mitochondrial complex I activity is impaired during HIV-1-induced T-cell apoptosis. Cell Death Differ. 2005;12:1417–1428. doi: 10.1038/sj.cdd.4401668. [DOI] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Loeb CR, Harris JL, Craik CS. Granzyme B proteolyzes receptors important to proliferation and survival, tipping the balance toward apoptosis. J Biol Chem. 2006;281:28326–28335. doi: 10.1074/jbc.M604544200. [DOI] [PubMed] [Google Scholar]
- MacDonald G, Shi L, Vande Velde C, Lieberman J, Greenberg AH. Mitochondria-dependent and -independent regulation of Granzyme B-induced apoptosis. J Exp Med. 1999;189:131–144. doi: 10.1084/jem.189.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malassagne B, Ferret PJ, Hammoud R, Tulliez M, Bedda S, Trebeden H, Jaffray P, Calmus Y, Weill B, Batteux F. The superoxide dismutase mimetic MnTBAP prevents Fas-induced acute liver failure in the mouse. Gastroenterology. 2001;121:1451–1459. doi: 10.1053/gast.2001.29590. [DOI] [PubMed] [Google Scholar]
- Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–370. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Yoshihiro K, Hasko G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun. 2007;358:203–208. doi: 10.1016/j.bbrc.2007.04.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa S, Zanotti F, Gaballo A. The structural and functional connection between the catalytic and proton translocating sectors of the mitochondrial F1F0-ATP synthase. J Bioenerg Biomembr. 2000;32:401–411. doi: 10.1023/a:1005584221456. [DOI] [PubMed] [Google Scholar]
- Pardo J, Bosque A, Brehm R, Wallich R, Naval J, Mullbacher A, Anel A, Simon MM. Apoptotic pathways are selectively activated by granzyme A and/or granzyme B in CTL-mediated target cell lysis. J Cell Biol. 2004;167:457–468. doi: 10.1083/jcb.200406115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- Prokisch H, Andreoli C, Ahting U, Heiss K, Ruepp A, Scharfe C, Meitinger T. MitoP2: the mitochondrial proteome database–now including mouse data. Nucleic Acids Res. 2006;34:D705–D711. doi: 10.1093/nar/gkj127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science. 2007;316:1345–1348. doi: 10.1126/science.1142984. [DOI] [PubMed] [Google Scholar]
- Rehling P, Brandner K, Pfanner N. Mitochondrial import and the twin-pore translocase. Nat Rev Mol Cell Biol. 2004;5:519–530. doi: 10.1038/nrm1426. [DOI] [PubMed] [Google Scholar]
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Sandstrom PA, Mannie MD, Buttke TM. Inhibition of activation-induced death in T cell hybridomas by thiol antioxidants: oxidative stress as a mediator of apoptosis. J Leukoc Biol. 1994;55:221–226. doi: 10.1002/jlb.55.2.221. [DOI] [PubMed] [Google Scholar]
- Sazanov LA, Peak-Chew SY, Fearnley IM, Walker JE. Resolution of the membrane domain of bovine complex I into subcomplexes: implications for the structural organization of the enzyme. Biochemistry. 2000;39:7229–7235. doi: 10.1021/bi000335t. [DOI] [PubMed] [Google Scholar]
- Shresta S, Graubert TA, Thomas DA, Raptis SZ, Ley TJ. Granzyme A initiates an alternative pathway for granule-mediated apoptosis. Immunity. 1999;10:595–605. doi: 10.1016/s1074-7613(00)80059-x. [DOI] [PubMed] [Google Scholar]
- Susin SA, Larochette N, Geuskens M, Kroemer G. Purification of mitochondria for apoptosis assays. Methods Enzymol. 2000;322:205–208. doi: 10.1016/s0076-6879(00)22020-x. [DOI] [PubMed] [Google Scholar]
- Thomas DA, Scorrano L, Putcha GV, Korsmeyer SJ, Ley TJ. Granzyme B can cause mitochondrial depolarization and cell death in the absence of BID, BAX, and BAK. Proc Natl Acad Sci USA. 2001;98:14985–14990. doi: 10.1073/pnas.261581498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GQ, Wieckowski E, Goldstein LA, Gastman BR, Rabinovitz A, Gambotto A, Li S, Fang B, Yin XM, Rabinowich H. Resistance to granzyme B-mediated cytochrome c release in Bak-deficient cells. J Exp Med. 2001;194:1325–1337. doi: 10.1084/jem.194.9.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Belogrudov GI, Hatefi Y. Mitochondrial NADH-ubiquinone oxidoreductase (complex I). Effect of substrates on the fragmentation of subunits by trypsin. J Biol Chem. 1998;273:8094–8098. doi: 10.1074/jbc.273.14.8094. [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Zhu P, Zhang D, Chowdhury D, Martinvalet D, Keefe D, Shi L, Lieberman J. Granzyme A, which causes single-stranded DNA damage, targets the double-strand break repair protein Ku70. EMBO Rep. 2006;7:431–437. doi: 10.1038/sj.embor.7400622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. GzmA induces a caspase-independent increase in ROS by directly targeting the mitochondria
(A) GzmA loading of K562 cells with PFN induces a dose-dependent increase of ROS. K562 cells were treated with buffer, a sublytic dose of PFN, GzmA alone or PFN plus increasing GzmA concentrations and stained for ROS with HE.
(B) Cells were treated as in (A) and ROS was measured by H2DCFDA staining. Grey histograms are buffer-treated cells and white histograms are cells treated as indicated.
(C) PFN loading of GzmA in K562 induces a caspase-independent increase in ROS. K562 cells, preincubated or not with 100 μM ZVAD and DEVD as pan-caspase inhibitors (not shown), were treated with PFN and/or GzmA and stained for ROS. Caspase inhibition did not inhibit ROS production.
(D) GzmA treatment of purified mitochondria induces a rapid increase in ROS. Freshly purified mouse liver mitochondria were either untreated or treated with GzmA in the presence or absence of cytosolic S100 fraction and stained for ROS with HE. On its own, GzmA induced ROS. Results shown are representative of at least 3 independent experiments.
Figure S2. Ndufs3 in purified ETC complex I is targeted by purified rat CTL granules and recombinant human GzmA
(A) Mouse liver mitochondrial complex I was purified by immunocapture and analyzed by gel electrophoresis and silver stain. Annotation on the right identifies the most prominent complex I subunits.
(B) Mouse Ndufs3 is cleaved by human and rodent GzmA. Complex I was treated with purified rat CTL granules or recombinant human GzmA or GzmB. Nudfs3, but not Ndufb6, was cleaved by both sources of GzmA, but not by GzmB.
Figure S3. Over-expression of K56A-NDUFS3 doesn’t alter cellular ATP levels
EL4 cells stably transfected with empty vector, WT NDUFS3 or K56A-NDUFS3 expression vectors were grown in normal aerobic conditions and ATP content was measured and compared to ATP levels in mock transfected cells.
Figure S4. Hsp70 adheres to isolated mitochondria
Purified mouse liver mitochondria were washed or not with bicarbonate (HCO3−) and the presence of Hsps in the mitochondrial pellet assessed by immunoblot. Hsp70 was detected in isolated mitochondria, but levels decreased after bicarbonate washing, suggesting that some Hsp70 was loosely adhered to the mitochondrial outer membrane, but some was either internalized or more firmly adherent. By contrast, no Hsp90 was detected and matrix Hsp60 was detected, but was not affected by washing.
Figure S5. 17-AAG increases GzmA-induced ROS, cell death and SET cleavage.
(A) K562 cells, preincubated or not with the Hsp90 inhibitor 17-AAG as indicated, were treated with PFN and/or GzmA and stained for ROS (top panel) and cell death by annexin-V +PI (bottom panel). (B) K562 cells were treated as in A, and SET cleavage was followed by immunoblot.