

Abstract
Human T cell leukemia/lymphotropic virus type I (HTLV-I) induces adult T cell leukemia/lymphoma (ATLL). The mechanism of HTLV-I oncogenesis in T cells remains partly elusive. In vitro, HTLV-I induces ligand-independent transformation of human CD4+ T cells, an event that correlates with acquisition of constitutive phosphorylation of Janus kinases (JAK) and signal transducers and activators of transcription (STAT) proteins. However, it is unclear whether the in vitro model of HTLV-I transformation has relevance to viral leukemogenesis in vivo. Here we tested the status of JAK/STAT phosphorylation and DNA-binding activity of STAT proteins in cell extracts of uncultured leukemic cells from 12 patients with ATLL by either DNA-binding assays, using DNA oligonucleotides specific for STAT-1 and STAT-3, STAT-5 and STAT-6 or, more directly, by immunoprecipitation and immunoblotting with anti-phosphotyrosine antibody for JAK and STAT proteins. Leukemic cells from 8 of 12 patients studied displayed constitutive DNA-binding activity of one or more STAT proteins, and the constitutive activation of the JAK/STAT pathway was found to persist over time in the 2 patients followed longitudinally. Furthermore, an association between JAK3 and STAT-1, STAT-3, and STAT-5 activation and cell-cycle progression was demonstrated by both propidium iodide staining and bromodeoxyuridine incorporation in cells of four patients tested. These results imply that JAK/STAT activation is associated with replication of leukemic cells and that therapeutic approaches aimed at JAK/STAT inhibition may be considered to halt neoplastic growth.
In a small percentage of infected individuals, human T cell leukemia/lymphotropic virus type-I (HTLV-I) causes adult T cell leukemia/lymphoma (ATLL), an aggressive and often fatal disease (1–3). The epidemiology of ATLL suggests that cumulative genetic defects may be responsible for the acquisition of the neoplastic phenotype in a given T cell clone (4). T cell proliferation and selection following HTLV-I infection are dynamic processes that can be followed in vivo (5, 6) and in vitro (7) and generally result in the generation of clonal populations of mature CD4+, CD8−, and CD25+/CD7− T cells (8–10). In HTLV-I infection the time-dependent emergence of infected T cell clones is well documented, and ATLL results from the uncontrolled growth of a single clone.
In vitro, T cell immortalization (ligand-dependent) by HTLV-I occurs within a few months of culture, whereas T cell transformation (ligand-independent) requires more time and typically results in T cell lines that display constitutive activation of the JAK/STAT signaling pathway (11, 12). In physiological conditions, activation of the JAK/STAT pathway is triggered by cytokines through cell surface receptors (13). In the case of interferon and type I cytokines, the JAK family tyrosine kinases transduce the signal by phosphorylating the STAT proteins, which in turn dimerize and translocate to the nucleus to activate the expression of genes necessary for cell proliferation or differentiation (14). To ascertain whether the in vitro model of HTLV-I transformation has any bearing to the in vivo leukemogenesis, we investigated the JAK/STAT activation status in uncultured ex vivo leukemic cells from 12 HTLV-I seropositive patients with ATLL.
METHODS
Electrophoretic Mobility-Shift Assay (EMSA).
In the case of EMSA with the FcγR1 probe (5′-TGTATTTCCCAGAAAAGGAATCG-3′), cellular extracts were prepared and EMSAs were performed, as previously described (11).
In the case of the sis-inducible element (SIE), β-casein, and Iɛ probes, the binding reaction was performed by preincubating 5 μg of cell extracts with 1 μg of poly(dI-dC) in the same buffer on ice for 20 min. 32P-labeled probe (20,000 cpm) corresponding to the mammary gland factor binding site in the β-casein gene promoter (5′-TAGATTTCTAGGAATTCG-3′), 100,000 cpm of 32P-labeled HA-SIE probe (5′-GATCGTCGACATTTCCCGTAAATC-3′), and Iɛ probe (GATCTAACTTCCCAAGAACAG) (15) were added to the reaction mixture and incubated on ice for 30 min. In the supershift assay, antibodies were incubated with nuclear extracts on ice for 20 min after the addition of radiolabeled probe. Complexes were resolved on 4.5% polyacrylamide gels. The antibody anti-STAT-1 (N terminus) was purchased from Transduction Laboratory (Lexington, KY); antibodies C-20 and N-20, which recognize the C terminus of STAT-3 and N terminus of STAT-5, were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); and anti-STAT-6 was a generous gift from William J. LaRochelle (National Cancer Institute, Bethesda, MD).
Immunoprecipitation and Immunoblotting.
Cells were lysed at 80 × 106/ml in 10 mM Tris (pH 7.4), 150 mM NaCl, 0.5% Nonidet P-40, 1% Triton X-100, 1 mM Na3VO4, 1 mM DTT, 1 mM AEBSF, 20 μg/ml aprotinin, and 20 μg/ml leupeptin. Immunoprecipitations of each lysate were performed at 4°C overnight with antibodies directed against phosphotyrosine (4G10, Upstate Biotechnology, Lake Placid, NY) or JAK3 (C-21, Santa Cruz Biotechnology). Immunoprecipitated proteins were separated by SDS/PAGE and transferred to Protran membranes (Schleicher & Schuell, Keene, NH). Immunoblotting was performed with antibodies against the STAT and JAK proteins as follows: anti-phosphotyrosine (4G10), anti-STAT-3 (K-15, Santa Cruz Biotechnology, or N terminus, Transduction Laboratory), anti-STAT-5 (C-17, N-20, and N-49, Santa Cruz Biotechnology), anti-JAK3 (C-21), anti-JAK1 (kinase 1, Transduction Laboratory).
Blots were developed using the ECL detection kit (Amersham), and stripping and probing were performed following the manufacturer’s directions.
Flow Cytometry [Fluorescence-Activated Cell Sorter (FACS) Analysis] and Bromodeoxyuridine (BrdU) Staining.
Cells (5 × 106) were washed twice in PBS. Cells were fixed in 1 ml of 75% ethanol and incubated on ice for 30 min. The cells were then washed twice in PBS and treated with 3.5 μg of RNase, DNase free (Boehringer Mannheim) for 30 min at 37°C. Finally, cells were pelleted and resuspended in 0.5 ml of 50 μg/ml of propidium iodide (Sigma). DNA profiles were analyzed with a Becton Dickinson (Mountain View, CA) FACScan using cell fit software.
For BrdU staining, cells were resuspended in complete media containing 10% FBS at a concentration of 106 cells/ml with 10 μM BrdU (Boehringer Mannheim) for 30 min, washed two times in PBS, and mounted on slides by using cytospin funnels. Cells were fixed for 10 min in 2% paraformaldehyde and washed with PBS, and DNA was denatured by the addition of 4 M HCl for 10 min at room temperature. The acid was neutralized by the addition of 0.1 M borate buffer, pH 8.5, added twice for 10 min each. Cells were washed three times in PBS, and anti-BrdU-FITC antibody (Boehringer Mannheim) was added at a final concentration of 25 μg/ml. Slides were placed in a humidified chamber at room temperature for 1 hr, washed four times with PBS containing 0.02% Tween 20, and visualized with a Nikon fluorescent microscope.
RESULTS
Activated STAT-1-, STAT-3-, and STAT-5-Related Proteins in ATLL.
Twelve patients, nine with acute ATLL and three with chronic ATLL (16), were included in the study. All had a previous history of HTLV-I infection and scored positive for HTLV-I provirus (data not shown). Each patient had more than 60% leukemic cells in the blood at the time of analysis, as inferred by the phenotypic markers characteristic of ATLL cells (Table 1). Blood samples were obtained after patient consent according to the National Institutes of Health Office of Human Subject Research. Each patient’s peripheral blood mononuclear cells (PBMC) were either lysed without any stimulation or, in some cases, were exposed first to a 15-min pulse with recombinant interleukin 2 (IL-2).
Table 1.
Clinical diagnosis and phenotypic features of ATLL cells
| Patient designation | Diagnosis | WBC/mm3 | CD3+, % | CD4+, % | CD8+, % | CD25+ | CD7−/CD25+ | CD7− | STAT binding activity |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Acute ATL | 77,000 | 100 | 97 | 0 | 98 | 97 | None detected | |
| 2 | Acute ATL | 62,000 | 93 | 97 | 2 | 86 | 84 | None detected | |
| 3 | Acute ATL/TSP-HAM | 35,000 | 94 | 93 | 2 | 96 | 88 | STAT1/STAT3 | |
| 4 | Acute ATL | 34,700 | 95 | 93 | 5 | 61 | 7 | STAT5 | |
| 5 | Acute ATL | 10,000 | 92 | 89 | 4 | 68 | 87 | STAT5 | |
| 6 | Acute ATL | 7,600 | 5 | 97 | 3 | 90 | 97 | None detected | |
| 7 | Acute ATL | 13,800 | 92 | 95 | 3 | 83 | 92 | STAT1/STAT3/STAT5 | |
| 8 | Chronic ATL | 36,900 | 90 | 70 | 8 | 69 | 79 | STAT3 | |
| 9 | Chronic ATL | 17,900 | 94 | 90 | 5 | 85 | 84 | None detected | |
| 10 | Chronic ATL | 6,000 | 83 | 70 | 14 | 63 | 73 | STAT1/STAT3 | |
| 11 | ATLL | 18,300 | 96 | 76 | 12 | 77 | 72 | STAT3/STAT5 | |
| 12 | Acute ATL | 19,000 | 94 | 92 | 2 | 93 | 89 | STAT1/STAT3 |
WBC, white blood cell; TSP-HAM, tropical spastic paraparesis–HTLV-I-associated myelopathy.
The activation status of the STAT proteins in the cell extracts of the 10 patients was assessed by using oligonucleotide probes corresponding to the interferon γ-activation site (GAS) motifs from the FcγR1 promoter. Protein lysates were analyzed by EMSA. In 10 of 12 patients, no constitutive activation of STAT proteins was observed. Thus, by the use of the FcγR1 DNA sequence it appeared that only 2 out of 10 patients displayed constitutive STAT protein activation in the DNA-binding assay.
Identification of Activated STAT Proteins Bound to the SIE, β-Casein, and Iɛ Oligonucleotides.
To increase the sensitivity of the DNA-binding assay and to test the binding specificity of the STAT proteins present in the ATLL cells, the DNA motifs SIE, β-casein, and Iɛ (15, 18, 19) were used in conjunction with antibodies that recognize each of the STAT proteins. These probes bind with high affinity to STAT-1 and STAT-3, STAT-5, and STAT-6, respectively. In both patients 7 and 10, the SIE probe generated three bands in EMSA (Fig. 1A, lanes 7 and 10), and two of the bands were supershifted by anti-STAT-1 (Fig. 1A, lanes 5, 8, and 11). The lower band likely was composed of STAT-1 homodimers because it was completely shifted by the anti-STAT-1 antibody, whereas the middle band may have contained STAT-1 heterodimers. The addition of STAT-3 antibodies shifted the upper band in both patients’ extracts, but the supershifted band migrated faster than the STAT-3 containing complexes identified by the same antibody (even at higher concentration) in the extract from phytohemagglutinin (PHA)-PBMC (compare lanes 6, 9, and 12 of Fig. 1A). Because the anti-STAT-3 antibody used is directed against the C terminus of STAT-3, it is unlikely that the aberrant DNA shift observed is a result of the presence in the extracts of the β STAT-3 isoform (20), which lacks the epitope recognized by this antibody.
Figure 1.
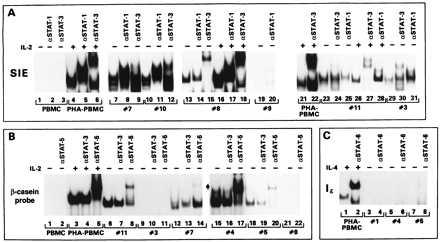
EMSA with β-casein, SIE, and Iɛ DNA oligonucleotides. EMSA obtained with SIE (A), β-casein (B), and Iɛ (C) probes. The specificity of the antibodies added to the DNA–protein complexes is indicated on the top of each panel. In some patients’ extracts the addition of the antibodies increased the signal, and in others a decrease binding was observed: in the case of STAT-5, compare lanes 6–8 and 12–14 of B, and in the case of STAT-1, compare lanes 25 and 28 of A. The reasons for this variability are unclear at present and do not appear to be related to the loading of different amounts of proteins or labeled DNA.
STAT-5 constitutive activation was detected in the extract from patient 7 but not patient 10 (data not shown), as demonstrated by the ability of the antibody directed against the amino terminus of STAT5a and STAT5b to supershift part of the DNA complex (Fig. 1B, lanes 12 and 14) in extracts of untreated cells.
The cellular extract from patients 3, 8, 11, and 12 bound to the SIE probe in absence of IL-2 stimulation (Fig. 1A, lanes 13–18, 23–25, 29–31 and Fig. 2B), but of these extracts, only those from patient 11 reacted with the β-casein probe (compare lanes 6–11, 21, and 22 of Fig. 1B and Fig. 2B). Finally, patients 4 and 5 displayed constitutive STAT-5 DNA-binding activity (Fig. 1B, lanes 15–20) but did not appear to have activated STAT-1 and STAT-3 (data not shown). None of the patients’ extracts displayed binding activity to the Iɛ oligonucleotide (a representative example for patients 1, 4, and 5 is given in Fig. 1C, lanes 3–8), suggesting the absence of STAT-6 (21).
Figure 2.

Persistence of STAT activation in cell extracts of patients 3 and 12. (A) White blood cell (WBC) counts over time in patient 12. The arrows indicate the times of sample collection. (B) EMSA on cell extracts from patient 12 at different sampling times (first 3 panels). The last panel represents the EMSA on patient 3 at the second sampling time.
In summary, 6 of 12 patients’ extracts had STAT-3 binding activity, and among these 6, 4 displayed STAT-1 binding activity as well. Activated STAT-5 was demonstrated in 4 of 12 patients, and all patients lacked activated STAT-6 (Table 1).
Persistence of STAT Activation in ATLL.
To ascertain whether the constitutive activation of STAT proteins found in the blood cells represented a temporary status of activation rather than being an intrinsic characteristic of the leukemic cells, two ATLL patients were followed longitudinally. One patient (patient 12) was followed in the last 26 days of his/her life and another patient (patient 3) at a 6-month interval.
PBMC from patient 12 were obtained at three separate intervals: the first sample was collected before the last therapeutic intervention; the second sample was obtained during relapse; and the third sample was obtained 2 days before the death of the patient. As indicated in Fig. 2A and Table 1, at the first sampling time 19,000 white blood cells (WBC)/mm3 were present. The latter two samples had twice the amount of leukemic cells, and more than 90% of them displayed the characteristic phenotype of CD4+, CD8−, and CD25+ leukemic cells.
Cell extracts from patient 12, at all three sampling points, formed DNA complexes with the SIE DNA sequence, and the mobility of the DNA complexes was altered by the addition of anti-STAT-1 and -STAT-3 antibodies (Fig. 2B, lanes 5, 6, 17, 18, 29, and 30) consistent with the presence of activated STAT-1 and STAT-3 proteins at all sampling points. As in most of the other patients studied, the extracts of patient 12 did not form DNA complexes with either the β-casein probe, before IL-2 triggering (Fig. 2B, lanes 9, 10, 21, 22, 33, and 34), or Iɛ oligonucleotides (data not shown), suggesting the absence in all the extracts of activated STAT-5 and STAT-6 proteins.
In the case of patient 3, STAT-1 and STAT-3 binding activity was demonstrated at the time of the first sampling (Fig. 1A, lanes 29–31) as well as in the cells obtained 6 months later (Fig. 2B, lanes 40–42). Thus, STAT activation does not appear to be a transient status in ATLL patients but rather an intrinsic feature of ATLL cells.
JAK3 Activation in ATLL.
Phosphorylated STAT proteins are found to be associated with phosphorylated JAK proteins in cells triggered with cytokines (13, 21–23). To identify which JAKs were phosphorylated and associated with STAT proteins in ATLL samples, cellular extracts, available in sufficient amounts from patients 3, 8, and 12, were first immunoprecipitated with anti-phosphotyrosine antibody (4G10) and immunoblotted with 4G10 antibody or with antibodies against JAK1, JAK2, JAK3, Tyk2, STAT-1, STAT-3, and STAT-5 (13, 22).
Immunoblotting with 4G10 revealed several phosphorylated protein bands in IL-2-treated PHA-PBMC as well as in patients 3, 8, and 12 (Fig. 3a). Immunoblotting with antibodies against JAK3 demonstrated the presence of phosphorylated JAK3 in the extracts of all three patients (Fig. 3 a and d), whereas JAK1, STAT-3, and STAT-5 were clearly detected only in IL-2-treated PHA-PBMC and in patient 8 (Fig. 3 a–e). None of the patients scored positive for JAK2 or Tyk2 (data not shown).
Figure 3.
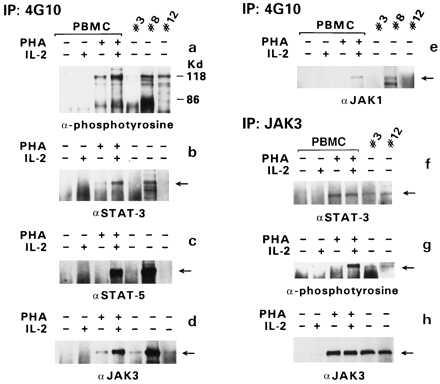
Association of JAK/STAT proteins. Immunoprecipitation (IP) was performed with the anti-phosphotyrosine antibody 4G10, and immunoblotting was performed with antibodies specific for various STAT or JAK proteins (as indicated in a–e). In the case of patient 3, protein extracts from the first sampling were used for the 4G10 IP, and those from the second sampling were used for the JAK3 IP. The amount of protein in sample 1 of patient 3 was half of that in sample 2. IP with anti-JAK3 antibodies was followed by immunoblotting with anti-STAT-3 antibody (f), anti-phosphotyrosine antibody (g), or anti-JAK3 antibody (h).
Because the 4G10 antibody could also precipitate nonphosphorylated protein bound to phosphorylated proteins, the extracts from patients 3 and 12 were first immunoprecipitated with anti-JAK3 antibody, and immunoblotting was performed with 4G10 antibody, anti-STAT-3 antibody, and anti-JAK3. STAT-3 coprecipitated with JAK3 in both cases (Fig. 3f). In addition, JAK3 was detected by the 4G10 antibody in both immunoprecipitates as well as by the anti-JAK3 antibody (Fig. 3 g and h), demonstrating its activation in both patients’ extracts. Altogether, these results demonstrate that JAK3 is phosphorylated and associated with STAT-3, at least in the leukemic cells of patients 3 and 12. Sufficient cell extracts were not available from the remaining patients to perform similar analyses.
Constitutive JAK/STAT Activation Is Associated with Leukemic Cell Proliferation.
To determine whether the constitutive activation of STAT proteins was coupled with cell proliferation, the PBMC from patients 2, 3, 11, and 12 were stained with propidium iodide (PI) and analyzed for DNA content by FACS cytofluorometry. In addition, the cells from patients 3 and 12 were also incubated with BrdU and stained with a monoclonal antibody against BrdU to identify morphologically the cells in S phase.
The leukemic cells from patient 12 were cycling at all three time points (data not shown for sample 1) as demonstrated by PI staining and BrdU incorporation (Fig. 4A). Consistent with the ongoing DNA replication, which occurs during the S phase of the cell cycle, the multinucleated flower-like leukemic cells from patient 12 were also stained with anti-BrdU antibody (Fig. 4A). Both analyses performed on the cells of patient 3, at the second sampling time, revealed that DNA synthesis was occurring in the large, multinucleated cycling leukemic cells, as well (Fig. 4B). Also, in the case of patient 11, whose cell extracts, similar to patients 3 and 12, contained activated STATs, the PI staining was consistent with a cycling cell population. In contrast, 95% of the cells from patient 2, whose extracts lacked DNA-binding activity for all the STAT proteins tested (data not shown), were arrested at the G0/G1 phase of the cell cycle (Fig. 4B Upper). Altogether, these results demonstrated that the cells of three patients, whose extracts revealed constitutive activation of STAT proteins, actively synthesized DNA, whereas in the case of patient 2, the absence of STAT activation was consistent with a much lower percentage of cells in S phase and 94% of cells arrested in G0/G1.
Figure 4.

Analysis of DNA synthesis and DNA content in leukemic cells. (A) PI staining and FACS analysis (Upper) and BrdU staining of cells from patient 12 obtained at the second and third sampling times. (B) PI staining and FACS analysis of the cells from patients 2, 11, and 3 (second sampling time). BrdU staining of the cells from patient 3 at the second sampling time.
DISCUSSION
In this study, we analyzed the functional status of the JAK/STAT pathway in uncultured leukemic cells from several cases of ATLL. In 8 of the 12 patients, STAT protein’s DNA-binding activity was detected by EMSA in the leukemic cell extracts. Six of the eight samples that were positive showed activated STAT-3, and two of these six also had active STAT-5. The remaining two positive samples showed activation of STAT-5 alone. None of the patients’ samples scored positive for STAT-6, as measured by the GAS element of the Iɛ promoter. Importantly, the identity of the shifted bands was confirmed by supershifting by using specific anti-STAT antibodies. In most of the patients’ extracts, the DNA–protein complex supershifted by the STAT-3 antibody migrated faster than the complex observed in IL-2-stimulated PBMC, suggesting that the STAT involved could either be a STAT-3 isoform, different from β STAT-3, or a STAT-3-related protein. DNA affinity purification of the activated STAT proteins present in the leukemic cells’ extracts followed by Western blotting will presumably help the precise identification of these proteins.
Constitutive activation of STAT protein correlated with phosphorylation of JAK3 (but not with that of JAK1, JAK2, and Tyk-2) and at least in three cases with proliferation of leukemic cells. The finding of JAK3 activation in ATLL suggests an involvement of the γc receptor, which is known to mediate the signal for IL-2, IL-4, IL-7, IL-9, and IL-15 (24). STAT-3 and, more peripherally, STAT-1 are phosphorylated in response to all of these cytokines, and, with the exception of IL-4, which activates STAT-6 (19), all of the other cytokines activate STAT-5 as well (22, 23).
STAT protein activation in ATLL may be a response to an autocrine or paracrine cytokine stimulation or may result from activation of one or more proteins involved in the signaling cascade. In uncultured ATLL cells, the expression of mRNA as assessed by Northern blotting for cytokines that signal through the γc receptor, including IL-15, IL-2, and IL-4 (25–29), appear to be absent, and the expression of the remaining known γc users, IL-7 and IL-9, has not been studied. HTLV-I-infected cultures produce a large number of cytokines including granulocyte/macrophage colony-stimulating factor, TNFα, IL-6, IL-9, and oncostatin-M. In the case of the HTLV-I-infected T cell line HuT-102, IL-15 has been demonstrated to be expressed at high levels (30).
HTLV-I-infected cultures become, with time, IL-2-independent and acquire a constitutive activation of JAK/STAT proteins (11, 12). Antibodies against IL-2 and IL-15 (unpublished observation) do not abolish the JAK/STAT pathway activation, suggesting that the activation may not be dependent on cytokine stimulation. In fact, in the Hut102 T cell line, which expresses high levels of IL-15, antibodies against IL-15 do not substantially decrease the activation level of JAK/STAT proteins (unpublished observation). However, as in the case of ATLL cells, the expression of other users of the γc receptor, namely IL-7, IL-9, and IL-4, has not been thoroughly investigated. Thus, at present, an autocrine mechanism in vivo and in vitro cannot be rigorously excluded.
Constitutive activation of JAKs and/or STATs has been correlated with cell transformation in other models of viral transformation. In murine pre-B lymphocytes transformed by the Abelson murine leukemia virus, constitutive JAK1, JAK3, and STAT activation (31) has been shown and transformation of human B cells by Epstein–Barr virus has been correlated with STAT-1 and STAT-3 phosphorylation (32). In mice, the erythroleukemia, induced by the spleen focus-forming virus, is also associated with constitutive STAT activation (33).
In human hematopoietic malignancies, JAK2 has been found activated in a few cases of childhood acute B cell lymphocytic leukemia (ALL) (34, 35). STAT-1 and STAT-5, and STAT-3 and STAT-5 activation have also been demonstrated in adult ALL patients and in acute myeloblastic leukemia (AML) patients, respectively (32, 35). In Sézary syndrome, a disease of CD4+ T cells similar to ATLL, activation of JAK3 (36) was found in 10 of 14 patients and was associated with activated STAT-5 in 9 patients and activated STAT-3 in 4 patients. Thus, the participation of the JAK/STAT pathway in tumorigenesis is a definite possibility, and the identification of the JAK kinase involved in different hematopoietic malignancies may be instrumental in targeting specific kinases for therapy.
JAK3 is activated by the cytokines that use the γc receptor chain, including IL-2, IL-4, IL-7, IL-9, and IL-15 (22), but it is nonessential for signaling by other cytokines. JAK3 is defective in an autosomal form of severe combined immunodeficiency disease (SCID) in humans (37–39). Of the JAK family members, JAK-3 is restricted in its pattern of expression, which is mainly confined to hematopoietic cells (22). In fact, JAK3 knock-out mice, although deficient in natural killer, T cell, and B cell function, do not have disorders in nonimmunological systems (40–42).
Thus, drugs that inhibit JAK3 activation may be of value as immunosuppressive and antileukemic agents, and the activation of JAK3, in HTLV-I infection in vivo as well as in vitro, suggests that HTLV-I-transformed T cell lines may be an appropriate tool to assess the efficacy of anti-JAK3 agents for use in ATLL.
Acknowledgments
We are grateful to Drs. Jacalyn Pierce, William Farrar, and John O’Shea for helpful discussion, to Dr. Tatiana Kislyakova for help with some of the experiments, to Cathryn Lee for her assistance with patient samples, and to Kelli Carrington for editorial assistance.
ABBREVIATIONS
- JAK
Janus kinases
- STAT
signal transducer/activator of transcription
- SIE
sis-inducible element
- ATLL
adult T cell leukemia/lymphoma
- HTLV-I
human T cell leukemia/lymphotropic virus type-I
- EMSA
electrophoretic mobility-shift assay
- IL
interleukin
- PBMC
peripheral blood mononuclear cells
- FACS
fluorescence-activated cell sorter
References
- 1.Poiesz B J, Ruscetti F W, Gazdar A F, Bunn P A, Minna J D, Gallo R C. Proc Natl Acad Sci USA. 1980;77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallo R C. Sci Am. 1986;255:88–98. doi: 10.1038/scientificamerican1286-88. [DOI] [PubMed] [Google Scholar]
- 3.Hinuma Y, Nagata K, Hanaoka M, Nakai M, Matsumoto T, Kinoshita K I, Shirakawa S, Miyoshi I. Proc Natl Acad Sci USA. 1981;78:6476–6480. doi: 10.1073/pnas.78.10.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy E L, Hanchard B, Figueroa J P, Gibbs W N, Lofters W S, Campbell M, Goedert J J, Blattner W A. Int J Cancer. 1989;43:250–253. doi: 10.1002/ijc.2910430214. [DOI] [PubMed] [Google Scholar]
- 5.Cavrois M, Wain-Hobson S, Gessain A, Plumelle Y, Wattel E. Blood. 1996;88:4646–4650. [PubMed] [Google Scholar]
- 6.Wattel E, Vartanian J P, Pannetier C, Wain-Hobson S. J Virol. 1995;69:2863–2868. doi: 10.1128/jvi.69.5.2863-2868.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn B, Gallo R C, Franchini G, Popovic M, Aoki T, Salahuddin S Z, Markham P D, Wong-Staal F. Mol Biol Med. 1984;2:29–36. [PubMed] [Google Scholar]
- 8.Markham P D, Salahuddin S Z, Kalyanaraman V S, Popovic M, Sarin P, Gallo R C. Int J Cancer. 1983;31:413–420. doi: 10.1002/ijc.2910310404. [DOI] [PubMed] [Google Scholar]
- 9.Popovic M, Lange-Wantzin G, Sarin P S, Mann D, Gallo R C. Proc Natl Acad Sci USA. 1983;80:5402–5406. doi: 10.1073/pnas.80.17.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyoshi I, Kubonishi I, Yoshimoto S, Akagi T, Ohtsuki Y, Shiraishi Y, Nagata K, Hinuma Y. Nature (London) 1981;294:770–771. doi: 10.1038/294770a0. [DOI] [PubMed] [Google Scholar]
- 11.Migone T S, Lin J X, Cereseto A, Mulloy J C, O’Shea J J, Franchini G, Leonard W J. Science. 1995;269:79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- 12.Xu X, Kang S H, Heidenreich O, Okerholm M, O’Shea J J, Nerenberg M I. J Clin Invest. 1995;96:1548–1555. doi: 10.1172/JCI118193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schindler C, Darnell J E. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 14.Darnell J E, Jr, Kerr I M, Stark G R. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 15.Patel B K R, Wang L M, Lee C C, Taylor W G, Pierce J H, LaRochelle W J. J Biol Chem. 1996;271:22175–22182. doi: 10.1074/jbc.271.36.22175. [DOI] [PubMed] [Google Scholar]
- 16.Shimoyama M members of The Lymphoma Study Group. Br J Haematol. 1991;79:428–437. doi: 10.1111/j.1365-2141.1991.tb08051.x. [DOI] [PubMed] [Google Scholar]
- 17.Salahuddin S Z, Markham P D, Wong-Staal F, Franchini G, Kalyanaraman V S, Gallo R C. Virology. 1983;129:51–64. doi: 10.1016/0042-6822(83)90395-1. [DOI] [PubMed] [Google Scholar]
- 18.Zhong Z, Wen Z, Darnell J E., Jr Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 19.Paul W E. Blood. 1991;77:1859–1870. [PubMed] [Google Scholar]
- 20.Caldenhoven E, van Dijk T B, Solari R, Armstrong J, Raaijmakers J A M, Lammers J W J, Koenderman L, de Groot R P. J Biol Chem. 1996;271:13221–13227. doi: 10.1074/jbc.271.22.13221. [DOI] [PubMed] [Google Scholar]
- 21.Darnell J E., Jr Proc Natl Acad Sci USA. 1996;93:6221–6224. doi: 10.1073/pnas.93.13.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bacon C M, Cho S S, O’Shea J J. Annu N Y Acad Sci. 1996;795:41–59. doi: 10.1111/j.1749-6632.1996.tb52654.x. [DOI] [PubMed] [Google Scholar]
- 23.Leonard W J. Nat Med. 1996;2:968–969. doi: 10.1038/nm0996-968. [DOI] [PubMed] [Google Scholar]
- 24.Leonard W J, Noguchi M, Russell S M, McBride O W. Immunol Rev. 1994;138:61–86. doi: 10.1111/j.1600-065x.1994.tb00847.x. [DOI] [PubMed] [Google Scholar]
- 25.Arima N, Daitoku Y, Ohgaki S, Fukumori J, Tanaka H, Yamamoto Y, Fujimoto K, Onoue K. Blood. 1986;68:779–782. [PubMed] [Google Scholar]
- 26.Uchiyama T, Hori T, Tsudo M, Wano Y, Umadome H, Tamori S, Yodoi J, Maeda M, Sawami H, Uchino H. J Clin Invest. 1985;76:446–453. doi: 10.1172/JCI111992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kodaka T, Uchiyama T, Umadome H, Uchino H. Jpn J Cancer Res (GANN) 1989;80:531–536. doi: 10.1111/j.1349-7006.1989.tb01672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uchiyama T, Kamio M, Kodaka T, Tamori S, Fukuhara S, Amakawa R, Uchino H, Araki K. Blood. 1988;72:1182–1186. [PubMed] [Google Scholar]
- 29.Arima N, Hidaka S, Fujiwara H, Matsushita K, Ohtsubo H, Arimura K, Kukita T, Fukumori J, Tanaka H. Blood. 1996;87:2900–2904. [PubMed] [Google Scholar]
- 30.Tagaya Y, Bamford R N, DeFilippis A P, Waldmann T A. Immunity. 1996;4:329–336. doi: 10.1016/s1074-7613(00)80246-0. [DOI] [PubMed] [Google Scholar]
- 31.Danial N N, Pernis A, Rothman P B. Science. 1995;269:1875–1877. doi: 10.1126/science.7569929. [DOI] [PubMed] [Google Scholar]
- 32.Weber-Nordt R M, Egen C, Wehinger J, Ludwig W, Gouilleux-Gruart V, Mertelsmann R, Finke J. Blood. 1996;88:809–816. [PubMed] [Google Scholar]
- 33.Ohashi T, Masuda M, Ruscetti S K. Blood. 1995;85:1454–1462. [PubMed] [Google Scholar]
- 34.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder J S, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman C M. Nature (London) 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 35.Gouilleux-Gruart V, Gouilleux F, Desaint C, Claisse J F, Capiod J C, Delobel J, Weber-Nordt R, Dusanter-Fourt I, Dreyfus F, Groner B, Prin L. Blood. 1996;87:1692–1697. [PubMed] [Google Scholar]
- 36.Zhang Q, Nowak I, Vonderheid E C, Rook A H, Kadin M E, Nowell P C, Shaw L M, Wasik M A. Proc Natl Acad Sci USA. 1996;93:9148–9153. doi: 10.1073/pnas.93.17.9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russell S M, Tayebi N, Nakajima H, Riedy M C, Roberts J L, Aman M J, Migone T S, Noguchi M, Markert M L, Buckley R H, O’Shea J J, Leonard W J. Science. 1995;270:797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- 38.Notarangelo L D. Curr Opin Immunol. 1996;8:448–453. doi: 10.1016/s0952-7915(96)80028-8. [DOI] [PubMed] [Google Scholar]
- 39.Oakes S A, Candotti F, Johnston J A, Chen Y Q, Ryan J J, Taylor N, Lui X, Hennighausen L, Notarangelo L D, Paul W E, Blaese R M, O’Shea J J. Immunity. 1996;5:605–615. doi: 10.1016/s1074-7613(00)80274-5. [DOI] [PubMed] [Google Scholar]
- 40.Nosaka T, van Deursen J M, Tripp R A, Thierfelder W E, Witthuhn B A, McMickle A P, Doherty P C, Grosveld G C, Ihle J N. Science. 1995;270:800–802. doi: 10.1126/science.270.5237.800. , and erratum (1996) 271, 17. [DOI] [PubMed] [Google Scholar]
- 41.Thomis D C, Gurniak C B, Tivol E, Sharpe A H, Berg L J. Science. 1995;270:794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 42.Park S Y, Sajjo K, Takahashi T, Osawa M, Arase H, Hirayama N, Miyake K, Nakauchi H, Shirasawa T, Saito T. Immunity. 1995;3:771–782. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]