

Abstract
The olefin cross-metathesis (CM) reaction is used extensively in organic chemistry and represents a powerful method for the selective synthesis of differentially substituted alkene products. Surprisingly, efforts to integrate this remarkable process into strategies for aromatic and heteroaromatic construction have not been reported. Such structures represent key elements of the majority of small molecule drug compounds; methods for the controlled preparation of highly substituted derivatives are essential to medicinal chemistry. Here we show that the olefin CM reaction, in combination with an acid cocatalyst or subsequent Heck arylation, provides a concise and flexible entry to 2,5-di- or 2,3,5-tri-substituted furans. These cascade processes portend further opportunities for the regiocontrolled preparation of other highly substituted aromatic and heteroaromatic classes.
Keywords: catalysis, cembranolide, Heck reaction
Ruthenium catalyzed olefin metathesis has become a remarkably powerful tool for organic chemistry (1–5). In recent years, the potential of ring-closing olefin metathesis (RCM) as a key step for the preparation of aromatic and heteroaromatic compounds has begun to be realized (6–8). Studies from our own laboratory have delineated versatile RCM-based protocols for the regiodefined synthesis of pyridines, pyridazines, furans, and pyrroles (9–15). Despite the evident potential of these methods it is notable that any RCM-based approach is reliant upon the a priori construction of a suitable precursor, which necessarily detracts from synthetic convergency and flexibility. A related approach involves the use of olefin cross-metathesis (CM) as a key step. Here both fragment coupling and strategic C=C bond formation are unified and, as such, potentially greater efficiencies are afforded. Despite this potential, and presumably due to perceived problems associated with double bond geometry, olefin CM-based methodologies for aromatic and heteroaromatic construction have not, to the best of our knowledge, been disclosed. Herein we report the successful implementation of this concept in terms of a versatile and direct entry to disubstituted and trisubstituted furans. It is likely that these studies will set the stage for the development of general olefin CM-based entries to other aromatic and heteroaromatic classes of varying ring sizes (Scheme 1, top).
Scheme 1.

Cross-metathesis as an approach to di- and trisubstituted furans.
Substituted furans are prevalent in natural products and pharmaceuticals and also represent useful intermediates in synthesis. Convergent de novo approaches to furans that embody high levels of flexibility and predictable regiocontrol are of particular value yet are relatively uncommon (16–22). Specifically, methods that employ simple starting materials, enable facile substituent introduction, and are concise are likely to be of significance to both medicinal chemistry and total synthesis. Our approach to furans relies upon the intermediacy of trans-γ-hydroxyenones 3 that are accessible via the established olefin CM of readily available allylic alcohols 1 and enones 2 (Scheme 1, Bottom) (23). Here we show that a tandem olefin CM-acid catalysed isomerization cascade provides a direct entry to differentially 2,5-disubstituted furans 4. Additionally, Heck reaction of the olefin CM products enables a tandem arylation-furan formation cascade to afford 2,3,5-trisubstituted furans 5 directly. As such, the trans-γ-hydroxyenone intermediates serve as highly versatile surrogates for 1,4-dicarbonyl compounds, especially as the final furan substitution pattern is programmed by the oxidation level of the allylic alcohol and enone components. The net result is an exceptionally concise and controlled approach to this class of heteroaromatic compounds.
Results and Discussion
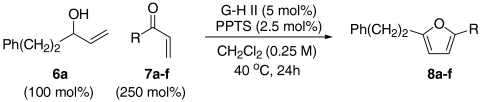
In our initial studies, we focused upon the formation of furan 8a by the olefin CM of allylic alcohol 6a with methyl vinyl ketone 7a. Here we found that, using the Grubbs–Hoveyda second generation catalyst (G-H II), the synthesis of the trans-hydroxyenone intermediate was straightforward. Subsequent addition of a variety of Brønsted or Lewis acids to the reaction medium was then evaluated to promote isomerization and condensation. From these initial assays we established that catalytic quantities of PPTS effected efficient furan formation. Furan formation presumably occurs via initial isomerization of the trans-hydroxyenone metathesis product to either the corresponding 1,4-dicarbonyl compound or the cis-hydroxyenone. We have not, as of yet, been able to unambiguously distinguish these pathways. However, our preliminary investigations suggest that furan formation from trans-hydroxyenones is faster than from analogous 1,4-dicarbonyl compounds. This would implicate a trans- to cis-isomerization pathway (24).
At this stage, and given the known compatibility of Grubbs’ type catalyst systems and acids (25), we considered whether both the ruthenium and acid catalysts could be employed from the outset. Gratifyingly, this proved to be the case and, under optimized conditions, allylic alcohol 6a was converted to furan 8a in 82% yield. Notably, increased overall efficiencies were observed using this tandem approach compared to the stepwise alternative. These conditions are generally applicable to a range of enone components and enable the introduction of aryl, heteroaryl, cyclopropyl, and diverse alkyl substituents in good to excellent yield (Table 1).
Table 1.
Tandem CM-furan formation: Scope of the enone component
 | |||
| Entry | Enone | Product | Yield, % |
| 1 | 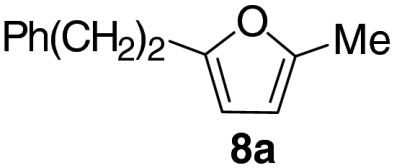 |
82 | |
| 2 | 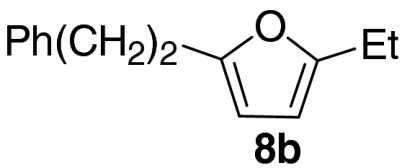 |
71 | |
| 3 | 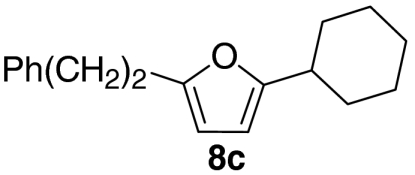 |
54 | |
| 4 | 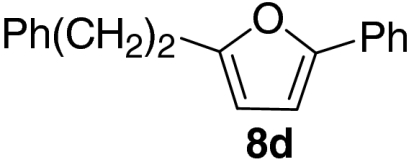 |
76 | |
| 5 | 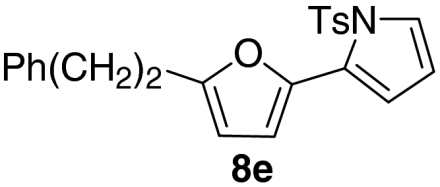 |
56 | |
| 6 | 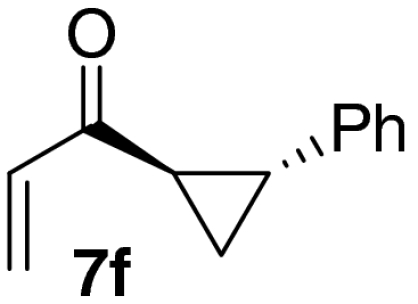 |
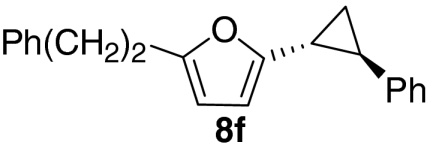 |
63 |
Examination of the scope of the allylic alcohol component revealed a greater degree of substrate sensitivity, presumably as this is the site of cross-metathesis initiation (Table 2). Under optimized conditions, alkyl, aryl, and heteroaryl substituents could all be introduced onto the furan core via the corresponding allylic alcohol (6b-g) in moderate to excellent yield. The catalyst system is, however, sensitive to the steric bulk of this component, and the introduction of a t-butyl substituent was achieved in only 36% yield (Entry 6) with the remainder of the mass balance consisting largely of starting material. This defines a current limitation but also portends further increases in scope as more efficient metathesis catalysts become available.
Table 2.
Tandem CM-furan formation: scope of the allylic alcohol component
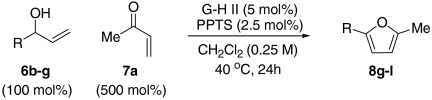
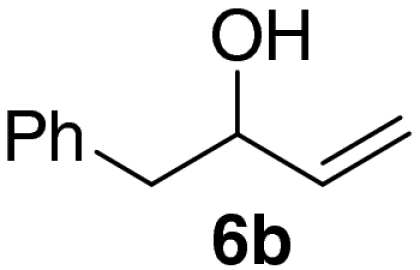
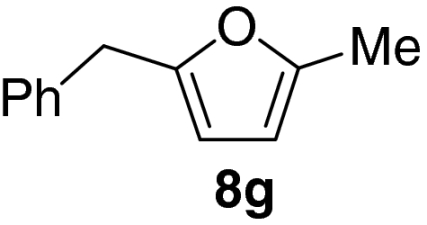
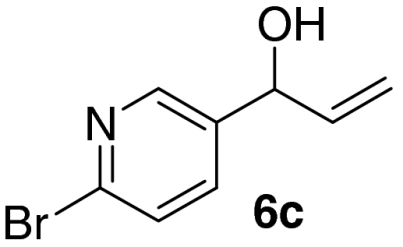
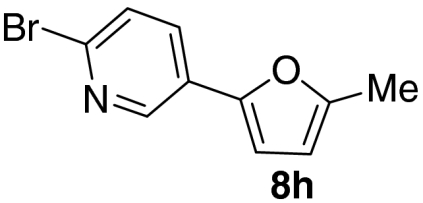
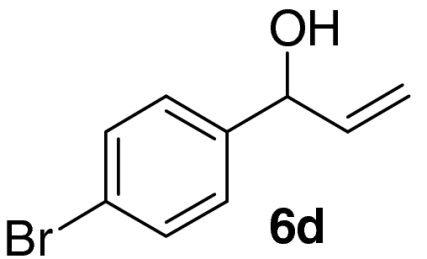
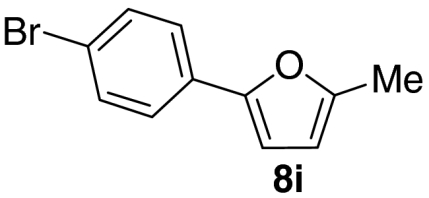
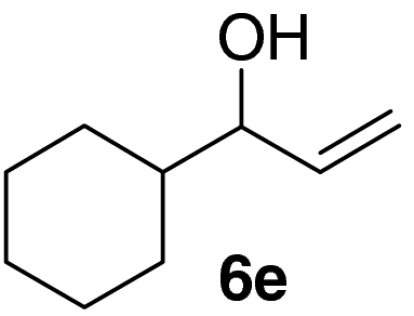
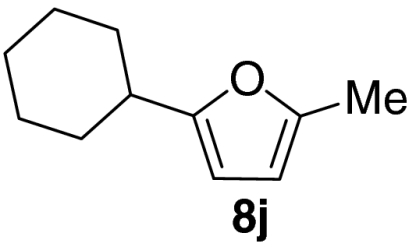
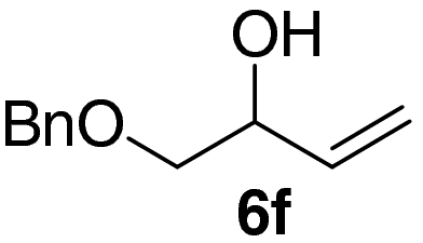
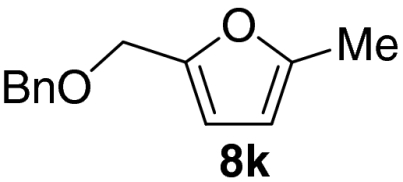
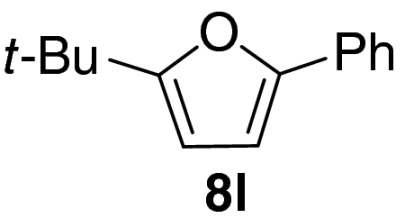
*The reaction was run at 70 °C.
†The reaction was run at 0.17 M.
‡Phenyl vinyl ketone was employed as the enone component and the reaction was run at 60 °C.
It is pertinent to consider the role of the allylic alcohol hydroxyl group in promoting the efficient CM reactions described here. Recent work has highlighted the enhanced metathesis efficiency of allylic alcohols compared to their corresponding ethers (26, 27), and our own preliminary observations are consistent with these findings. As evidenced by Hoveyda, it is possible that hydrogen bonding between the allylic alcohol derived carbene and a ruthenium chloride ligand is crucial in expediting subsequent CM to the electron deficient enone (27).
Given our ongoing interest in cembranolide natural products such as deoxypukalide 15 (28), we were interested in assessing whether this furan synthesis could be employed in an intramolecular setting to provide an entry to the macrocyclic furan core present in many compounds of this class (Scheme 2). Such an approach would unite the direct nature of the methodology presented herein with the established utility of olefin metathesis for macrocycle formation (29). Wadsworth–Emmons olefination of undecenal using the modified phosphonate reagent 9 afforded acrylamide 10 in 90% yield. This species underwent efficient conjugate addition with the Gilman cuprate derived from n-BuLi to generate 11. This route demonstrates the feasibility of installing the C-1 substitution present in the cembranolide natural products. Note that organocuprate 1,4-additions onto Weinreb acrylamides have not previously been reported; we have found that careful control of reaction temperature and time are essential in preventing the known ene-type degradation of the intermediate silyl ketene aminal (30). Oxidative cleavage of the terminal olefin yielded 12, and this was subjected to double addition of vinyl Grignard to set-up the tethered allylic alcohol-enone metathesis precursor 13. Exposure of this species to our previously described conditions but under higher dilution, afforded the 13-ring macrocyclic furan system 14 in excellent yield. Here prolonged reaction times were required to effect (a) complete metathesis and (b) complete conversion of the intermediate hydroxyenone to the corresponding furan.
Scheme 2.

RCM-furanization approach to the cembranolide macrocycle.
The formation of trisubstituted furans is potentially enabled by this methodology. However, the use of allylic alcohols or enones possessing 1,1-disubstitution affords only traces of the corresponding trisubstituted furan under the conditions reported in Tables 1 and 2. This is due to inefficiencies associated with the metathesis step. Undeterred, we decided to examine Heck arylation of the trans-hydroxyenone CM products, and after some experimentation we found that exposure of hydroxyenone 16a and bromotoluene to the mild Heck protocol reported by Fu (31) resulted in the formation of furan 17a in 80% yield (Scheme 3). Under these conditions, only traces of furan 8a (< 10%) resulting from cyclisation of hydroxyenone 16a in advance of Heck arylation were observed. This sequence is amenable to the introduction of diverse aryl substituents at C-3 and generally excellent overall yields of the furan products 17a-f are achieved. Variation of the hydroxyenone component adds further flexibility and facilitates regiodefined switching of the substituents at the 2- or 5-positions of the furan ring (c.f. 16b→18 and 16c→20). In the case of furan 18, traces of 1,4-dicarbonyl 19 were also isolated, presumably formed via either base or Pd-H mediated isomerization of the starting enone 16b; notably this component did not cyclize to the corresponding furan under the reaction conditions. This sequence can also be used in an intramolecular setting. Accordingly, enone 16d underwent efficient Heck cyclization to afford tricyclic furan 21 along with significant quantities of 1,4-dicarbonyl derivative 22. Again, this component did not cyclize under the reaction conditions, even upon extended reaction times, and presumably arises via isomerization after the Heck reaction.
Scheme 3.

CM-Heck approach to trisubstituted furans. Asterisks indicate contamination with ca. 5% of non-arylated furan.
The use of a Heck reaction to trigger aromatization is of interest. Our current understanding of this process finds precedence in the known stereochemical outcome of Heck reactions onto trans-acrylates under the conditions employed here (Scheme 4) (31). Syn-carbopalladation of the enone I mandates σ-bond rotation of II in advance of β-hydride elimination to deliver intermediate III; this scenario assumes that β-hydride elimination occurs faster than epimerization of the σ-Pd bond via an η-3 oxy-π-allyl palladium species (32). Cis-hydroxyenones such as III are known to exist in equilibrium with their corresponding hemiketals IV (33) and have been observed to undergo spontaneous furan formation (to V) by loss of water (34). To corroborate this mechanism, trans-enone 23 was subjected to the Heck conditions and found to deliver arylated product 24 where the substituents present in the starting material now reside in a cis-arrangement, as evidenced by diagnostic NOE enhancements (see SI Text). At higher conversions, the ratio of 24 to isomeric products decreases. Although this result establishes the feasibility of trans- to cis-isomerization during the Heck process, the concomitant formation of isomeric products (as an inseparable mixture and characterized by mass spectrometry only) prevents definitive confirmation that β-hydride elimination generates the cis-product directly. Note that general Heck protocols involving β-alkylated enones have not been reported, presumably due to facile secondary isomerization of the initial adducts (35, 36). Furan formation via the intermediacy of the corresponding 1,4-dicarbonyl (which could potentially arise after Heck arylation) is discounted as 25 did not afford furan 18 when exposed to the Heck conditions. Additional evidence discounting this pathway lies in our inability so far to convert 1,4-dicarbonyl 22 to furan 21, even under forcing conditions (see Scheme 3).
Scheme 4.

Proposed mechanism for the tandem Heck-furan formation sequence and some preliminary mechanistic experiments.
Conclusions
In summary, unique examples of the use of olefin CM as a means of accessing heteroaromatic compounds have been described. The present work illustrates the power of this approach in terms of highly direct and controlled entries to di- and trisubstituted furans. The synthesis of the latter relies upon the strategic employment of a Heck arylation to effect C-C bond formation with concomitant aromatisation. The chemistry described here, which capitalizes upon readily available components, is both direct and regiocontrolled. As such, widespread application is anticipated.
Methods
Full experimental details and compound characterization data are included in SI Text.
General Procedure for the Synthesis of 2,5-Disubstituted Furans.
A resealable reaction tube, fitted with a magnetic follower, was charged with Grubbs–Hoveyda second generation catalyst (5 mol%) and PPTS (2.5 mol%). The tube was then sealed with a rubber septum and purged with argon. Argon sparged CH2Cl2 (0.25 M with respect to the allylic alcohol employed) was then added via syringe, and the tube was cooled to -78 °C prior to sequential addition of the requisite enone (250 mol% or 500 mol%) and allylic alcohol (100 mol%). The rubber septum was then replaced with a screw cap and the tube was heated at 40 °C (oil bath temperature) for 24 h. After cooling to room temperature, the reaction mixture was concentrated in vacuo and the residue was filtered through a short cartridge of SiO2 (eluting with 10∶1 petrol-EtOAc). The fractions containing the target furan were combined and concentrated in vacuo. Purification of the residue by flash column chromatography afforded the corresponding furan.
General Procedure for the Synthesis of 2,3,5-Trisubstituted Furans.
(A) Olefin CM step: An argon purged reaction flask, fitted with a magnetic follower, was charged with Grubbs–Hoveyda second generation catalyst (2.5 mol%) and argon sparged CH2Cl2 (0.25 M with respect to the allylic alcohol employed). The requisite enone (500 mol%) and allylic alcohol (100 mol%) were then added sequentially via syringe. The mixture was stirred at room temperature for 24 h and then concentrated in vacuo. Purification of the residue by flash column chromatography afforded the corresponding trans-γ-hydroxyenone. (B) Heck step: A reaction tube, fitted with magnetic follower, was charged with Pd2(dba)3 (5 mol%), Pt-Bu3HBF4 (20 mol%), and (if solid) the corresponding aryl bromide (250 mol%). The tube was sealed with a septum and purged with argon. The requisite trans-γ-hydroxyenone (100 mol%), the corresponding aryl bromide (if liquid) (250 mol%), anhydrous p-dioxane (0.1 M with respect to the enone component) and then Cy2NMe (250 mol%) were added sequentially via syringe. The mixture was then heated at either 70 °C or 85 °C for 16 h. The mixture was cooled to room temperature and filtered through a short cartridge of SiO2 (eluting with 10∶1 petrol-EtOAc). The fractions containing the target furan were combined and concentrated in vacuo. Purification of the residue by flash column chromatography afforded the corresponding furan.
Supplementary Material
Acknowledgments.
We thank the EPSRC for supporting this project. Merck is thanked for unrestricted funding.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 3279.
This article contains supporting information online at www.pnas.org/cgi/content/full/0913466107/DCSupplemental.
References
- 1.Adv Synth Catal. 2007;349:1–265. doi: 10.1002/adsc.200600264. Special issue dedicated to olefin metathesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoveyda AH, Zhugralin AR. The remarkable metal-catalysed olefin metathesis reaction. Nature. 2007;450:243–251. doi: 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]
- 3.Nicolaou KC, Bulger PG, Sarlah D. Metathesis reactions in total synthesis. Angew Chem Int Edit. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]
- 4.Fürstner A. Olefin metathesis and beyond. Angew Chem Int Ed. 2000;39:3012–3043. [PubMed] [Google Scholar]
- 5.Armstrong SK. Ring closing diene metathesis in organic synthesis. J Chem Soc Perk T. 1998;1:371–388. [Google Scholar]
- 6.van Otterlo WAL, de Koning CB. Metathesis in the synthesis of aromatic compounds. Chem Rev. 2009;109:3743–3782. doi: 10.1021/cr900178p. [DOI] [PubMed] [Google Scholar]
- 7.Donohoe TJ, Fishlock LP, Procopiou PA. Ring-closing metathesis: Novel routes to aromatic heterocycles. Chem Eur J. 2008;14:5716–5726. doi: 10.1002/chem.200800130. [DOI] [PubMed] [Google Scholar]
- 8.Donohoe TJ, Orr AJ, Bingham M. Ring-closing metathesis as a basis for the construction of aromatic compounds. Angew Chem Int Ed. 2006;45:2664–2670. doi: 10.1002/anie.200503512. [DOI] [PubMed] [Google Scholar]
- 9.Donohoe TJ, et al. Ring-closing metathesis for the synthesis of heteroaromatics: Evaluating routes to pyridines and pyridazines. Tetrahedron. 2009;65:8969–8980. [Google Scholar]
- 10.Donohoe TJ, et al. Synthesis of substituted pyridines and pyridazines via ring closing metathesis. Chem Commun. 2009:3008–3010. doi: 10.1039/b904363b. [DOI] [PubMed] [Google Scholar]
- 11.Donohoe TJ, et al. Flexible metathesis-based approaches to highly functionalised furans and pyrroles. Tetrahedron. 2008;64:809–820. [Google Scholar]
- 12.Donohoe TJ, Fishlock LP, Procopiou PA. A metathesis-based approach to the synthesis of 2-pyridones and pyridines. Org Lett. 2008;10:285–288. doi: 10.1021/ol702684d. [DOI] [PubMed] [Google Scholar]
- 13.Donohoe TJ, Fishlock LP, Procopiou PA. Ring-closing metathesis as a key step in the synthesis of 2-pyridones and pyridine triflates. Synthesis. 2008:2665–2667. [Google Scholar]
- 14.Donohoe TJ, Fishlock LP, Lacy AR, Procopiou PA. A metathesis-based approach to the synthesis of furans. Org Lett. 2007;9:953–956. doi: 10.1021/ol062933r. [DOI] [PubMed] [Google Scholar]
- 15.Donohoe TJ, Orr AJ, Gosby K, Bingham M. A metathesis approach to aromatic heterocycles. Eur J Org Chem. 2005:1969–1971. [Google Scholar]
- 16.Kirsch SF. Syntheses of polysubstituted furans: recent developments. Org Biomol Chem. 2006;4:2076–2080. doi: 10.1039/b602596j. [DOI] [PubMed] [Google Scholar]
- 17.Hou XL, et al. Regioselective syntheses of substituted furans. Tetrahedron. 1998;54:1955–2020. [Google Scholar]
- 18.Jung CK, Wang JC, Krische MJ. Phosphine-mediated reductive condensation of γ-acyloxy butynoates: A diversity oriented strategy for the construction of substituted furans. J Am Chem Soc. 2004;126:4118–4119. doi: 10.1021/ja049377l. [DOI] [PubMed] [Google Scholar]
- 19.Dudnik AS, Gevorgyan V. Metal-catalyzed [1,2]-alkyl shift in allenyl ketones: synthesis of multisubstituted furans. Angew Chem Int Ed. 2007;46:5195–5197. doi: 10.1002/anie.200701128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barluenga J, Riesgo L, Vicente R, López LA, Tomás M. Cu(I)-catalyzed regioselective synthesis of polysubstituted furans from propargylic esters via postulated (2-furyl)carbene complexes. J Am Chem Soc. 2008;130:13528–13529. doi: 10.1021/ja8058342. [DOI] [PubMed] [Google Scholar]
- 21.Xiao Y, Zhang J. Tetrasubstituted furans by a Pd(II)-catalyzed three-component Michael addition/cyclization/cross-coupling reaction. Angew Chem Int Ed. 2008;47:1903–1906. doi: 10.1002/anie.200704531. [DOI] [PubMed] [Google Scholar]
- 22.Zhang M, Jiang HF, Neumann H, Beller M, Dixneuf PH. Sequential synthesis of furans from alkynes: successive ruthenium(II)- and copper(II)-catalyzed processes. Angew Chem Int Ed. 2009;48:1681–1684. doi: 10.1002/anie.200805531. [DOI] [PubMed] [Google Scholar]
- 23.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. A general model for selectivity in olefin cross metathesis. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 24.Avery TD, Taylor DK, Tiekink ERT. A new route to diastereomerically pure cyclopropanes utilizing stabilized phosphorus ylides and γ-hydroxy enones derived from 1,2-dioxines: Mechanistic investigations and scope of reaction. J Org Chem. 2000;65:5531–5546. doi: 10.1021/jo0002240. [DOI] [PubMed] [Google Scholar]
- 25.Hong SH, Sanders DP, Lee CW, Grubbs RH. Prevention of undesirable isomerization during olefin metathesis. J Am Chem Soc. 2005;127:17160–17161. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- 26.Hoye TR, Zhao H. Some allylic substituent effects in ring-closing metathesis reactions: allylic alcohol activation. Org Lett. 1999;1:1123–1125. doi: 10.1021/ol990947+. [DOI] [PubMed] [Google Scholar]
- 27.Hoveyda AH, Lombardi PJ, O’Brien RV, Zhugralin AR. H-bonding as a control element in stereoselective Ru-catalyzed olefin metathesis. J Am Chem Soc. 2009;131:8378–8379. doi: 10.1021/ja9030903. [DOI] [PubMed] [Google Scholar]
- 28.Donohoe TJ, Ironmonger A, Kershaw NM. Synthesis of (-)-(Z)-deoxypukalide. Angew Chem Int Ed. 2008;47:7314–7316. doi: 10.1002/anie.200802703. [DOI] [PubMed] [Google Scholar]
- 29.Gradillas A, Pérez-Castells J. Macrocyclization by ring-closing metathesis in the total synthesis of natural products: Reaction conditions and limitations. Angew Chem Int Ed. 2006;45:6086–6101. doi: 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]
- 30.Keck GE, McHardy SF, Murry JA. Some unusual reactions of Weinreb amides. Tetrahedron Lett. 1993;34:6215–6218. [Google Scholar]
- 31.Littke AF, Fu GC. A versatile catalyst for Heck reactions of aryl chlorides and aryl bromides under mild conditions. J Am Chem Soc. 2001;123:6989–7000. doi: 10.1021/ja010988c. [DOI] [PubMed] [Google Scholar]
- 32.Albéniz AC, Catalina NM, Espinet P, Redón R. Bonding modes in palladium(II) enolates: consequences for dynamic behavior and reactivity. Organometallics. 1999;18:5571–5576. [Google Scholar]
- 33.Greatrex BW, Taylor DK. Ring-opening of unsymmetrical 1,2-dioxines using Cobalt(II) salen complexes. J Org Chem. 2005;70:470–476. doi: 10.1021/jo040241f. [DOI] [PubMed] [Google Scholar]
- 34.Marshall JA, Wang XJ. Synthesis of furans by Ag(I)-promoted cyclization of allenyl ketones and aldehydes. J Org Chem. 1991;56:960–969. [Google Scholar]
- 35.Stadler M, List B. Heck reactions of crotonaldehyde. Synlett. 2008:597–599. [Google Scholar]
- 36.Li X, Zeng X. Sequential transhalogenation and Heck reaction for efficient access to dioxo-tetrasubstituted 2,4E,E-dienes: Synthesis of segment C1–C6 of apoptolidin. Tetrahedron Lett. 2006;47:6839–6842. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.