

Abstract
A hypoxic/anoxic microenvironment has been proposed to exist within a vascular lesion due to intimal or medial cell proliferation in vascular diseases. Here, we examined whether hypoxia alters macrophage function by exposing murine macrophage-like RAW 264.7 (RAW) cells to hypoxia (2% O2). When cells were exposed to hypoxia, a significant number of RAW cells underwent apoptosis. Additionally, small subpopulations of RAW cells were resistant to hypoxia-induced apoptosis. Through repeated cycles of hypoxia exposure, hypoxia-induced apoptosis-resistant macrophages (HARMs) were selected; HARM cells demonstrate >70% resistance to hypoxia-induced apoptosis, as compared with the parental RAW cells. When heat shock protein (HSP) expression was examined after hypoxia, we observed a significant decrease in constitutive heat shock protein 70 (HSC 70) in RAW cells, but not in HARMs, as compared with the control normoxic condition (21% O2). In contrast, the expression level of glucose-regulated protein 78 (GRP 78) in RAW and HARM cells after hypoxia treatment was not altered, suggesting that HSC 70 and not GRP 78 may play a role in protection against hypoxia-induced apoptosis. When tumor necrosis factor α (TNF-α) production was examined after hypoxic treatment, a significant increase in TNF-α production in HARM but decrease in RAW was observed, as compared with cells cultured in normoxic conditions. HARM cells also exhibit a much lower level of modified-LDL uptake than do RAW cells, suggesting that HARMs may not transform into foam cells. These results suggest that a selective population of macrophages may adapt to potentially pathological hypoxic conditions by overcoming the apoptotic signal.
Oxygen (O2) is a fundamental substance critical to life by virtue of its role as the final electron acceptor in oxidative phosphorylation; stress brought on by lack of O2 may affect cell survival. Many studies have demonstrated hypoxia- (low oxygen tension) induced apoptotic cell death in several cell types (1–4). For example, renal tubular cells have been observed to undergo apoptosis under hypoxic conditions (1). Muschel et al. (2) also demonstrated hypoxia-induced apoptosis in WEHI 7.1 cells. Using PC12 cells, Shimizu et al. (3) demonstrated that hypoxia-induced apoptosis was prevented by Bcl-2 and Bcl-xL. Recently, Graeber et al. (4) showed that apoptosis in response to hypoxia is mediated by p53. Conversely, hypoxia has also been shown to induce resistance to apoptosis. As demonstrated in a study by Hannah et al. (5), a significant decrease in neutrophil apoptosis occurs under hypoxic conditions. This paradoxical dual effect of hypoxia (induction vs. inhibition of apoptosis) on cellular function may play a role in the pathophysiology of diseases that exhibit a hypoxic microenvironment, such as atherosclerosis or restenosis.
Macrophages have been shown to respond to hypoxia by induction of specific gene expression (6, 7). Scannell et al. (6) demonstrated that hypoxia can induce the expression of tumor necrosis factor α (TNF-α) and its soluble receptors by a human macrophage cell line. Others have reported that hypoxia increases the production of interleukin (IL)-1 and TNF-α by human mononuclear cells (7). Additionally, Albina et al. (8) demonstrated that macrophages grown in an anoxic environment increased total l-arginine metabolism, arginase activity, and release of TNF-α and IL-6, suggesting that anoxia may also act as an inducer of macrophage activation.
The presence of macrophage apoptosis in human atherosclerotic lesions has been demonstrated by several investigators (9–11). Additionally, the production of TNF-α, IL-1, and IL-8 by macrophages from human atheromatous plaques has also been reported (12, 13). Most importantly, Nagornev and Maltseva (14) recently demonstrated that macrophages within atherosclerotic lesions have at least two distinct phenotypes, one type that is transformed into foam cells and another type that does not incorporate oxidized low density lipoprotein (LDL) and may be involved in inflammation. Further, they showed evidence that nontransformed activated macrophages are involved in expression of IL-1β and TNF-α, whereas no evidence of cytokine expression was found in foam cells located in the atherosclerotic plaques. These studies strongly suggest that different populations of macrophages recruited to lesion sites may play different roles in the course of atherogenesis. Furthermore, these subpopulations of macrophages may differentially respond to the hypoxic microenvironment within the lesion. In this study, we examine these possibilities and demonstrate that hypoxia induces apoptosis in murine macrophage-like RAW-264.7 (RAW) cells. We demonstrate that subpopulations of RAW cells are resistant to hypoxia-induced apoptosis, suggesting an adaptive response to the apoptotic signal. These hypoxia-induced apoptosis-resistant macrophages (HARMs) were selected from parental RAW cells and characterized with regard to heat shock protein expression, cytokine production, and modified-LDL uptake.
MATERIALS AND METHODS
Materials.
The murine macrophage cell line RAW 264.7 (RAW) was obtained from the American Type Culture Collection. Human plasma LDL was purchased from Calbiochem. Mouse monoclonal antibody for constitutive heat shock protein 70 (HSC 70) and polyclonal antibody for glucose-regulated protein 78 (GRP 78) were purchased from StressGen (Victoria, BC, Canada). TNF-α ELISA kit and ECL chemiluminescent reagent were purchased from Amersham. RPMI 1640 culture medium was purchased from GIBCO/BRL Life Technologies. Fetal bovine serum (FBS) was purchased from HyClone.
Selection of HARMs.
RAW cells were cultured in normal medium [RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 1% l-glutamine, penicillin (100 units/ml) and streptomycin (100 μg/ml)]. Culture experiments involving hypoxic conditions were carried out in a Forma O2/CO2 incubator (Forma Scientific, Marietta, OH) in which the O2 level was maintained using the inert gas N2 by an O2-sensor–gas control valve servomechanism. Trypsinized RAW cells were initially grown in RPMI 1640 medium supplemented with 10% FBS in 175-cm2 flasks under 5% CO2/95% air culture at 37°C. For selection of HARMs, RAW cells were initially grown to subconfluence in normoxic (21% O2) conditions. Before hypoxia (2% O2) exposure, culture medium was replaced with fresh normal medium. After 24–48 hr of hypoxia exposure, detached (viability < 20%) cells were removed with the culture medium. Remaining attached (viability >90%) cells were then recovered, inoculated into fresh normal medium, and again exposed to hypoxia. This procedure was repeated 10 times with the attached cells that survived each round of hypoxia treatment. The percentage of apoptotic macrophages induced by hypoxia was measured by fluorescence-activated cell sorting after propidium iodide (PI) staining by using a doublet discrimination protocol (15, 16). Briefly, RAW and HARM cell cycles were synchronized by a 24-hr incubation in RPMI 1640 medium containing 5% FBS followed by a second 24-hr incubation in RPMI with 1% FBS. Cells were then given fresh medium (RPMI 1640 with 1% FBS) and incubated in either normoxic or hypoxic conditions. At specific times, RAW and HARM cells were harvested and centrifuged at 500 × g for 5 min. Cells were then washed once in PBS and fixed in cold 80% (vol/vol) ethanol for 30 min. After three more washes with PBS, cells were incubated for 30 min at 37°C in PBS and stained with PI (50 μg/ml in PBS containing 0.1% Triton X-100, 0.1 mM EDTA, and 50 μg/ml RNase) overnight at 4°C (15, 16). Analysis of PI staining of RAW and HARM cells was carried out on an ELITE fluorescence-activated cell sorter (Coulter) using a cell cycle analysis doublet discrimination protocol. Apoptotic cells are indicated by the bar (E or C in Fig. 2) to the left of the G0 peak.
Figure 2.
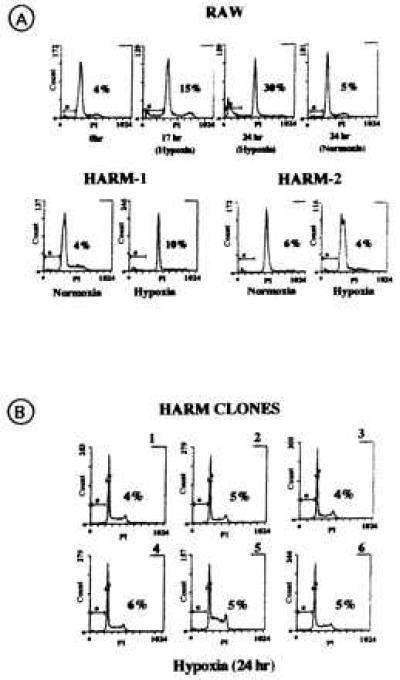
Fluorescence-activated cell sorting analysis of hypoxia-induced apoptosis in RAW and HARM cells. (A) Macrophage apoptosis induced by hypoxia in RAW (17 and 24 hr), HARM-1 and HARM-2 cells (24 hr). (B) Hypoxia (24 hr) induced-apoptosis of HARM clones 1–6. Numbers within graphs represent the percent of hypoxia-induced apoptosis.
Heat Shock Protein Western Blots.
RAW and HARM cells were exposed to either normoxia or hypoxia for 17 or 24 hr at 37°C. Cells were then lysed on ice with 500 μl of a buffer containing 25 mM Hepes at pH 7.5, 100 mM NaCl, 20 mM β-glycerophosphate, 1.5 mM MgCl2, 1 mM vanadate, 0.5 mM EGTA, 0.25 mM EDTA, 0.1% Nonidet P-40, 1 mM phenylmethanesulfonyl fluoride (PMSF), and 10 μg/ml each leupeptin. pepstatin, and aprotinin. Lysate protein concentration was determined using the BCA protein assay (Pierce). Lysates were then processed for reducing SDS/PAGE on 12% polyacrylamide gels, and then were transferred to nitrocellulose. Membranes were incubated in blocking solution [5% nonfat dry milk in TBST (TBST = 25 mM Tris⋅HCl, pH 7.4]/150 mM NaCl/0.05% Tween-20)], and were then incubated with either anti-HSC 70 or GRP 78 antibody (diluted 1:1000 in blocking solution) overnight at 4°C. After incubation with primary antibodies, membranes were washed extensively in TBST and were then incubated for 1 hr at room temperature in anti-mouse or anti-rabbit IgG-horseradish peroxidase (HRP) conjugates (diluted 1:3000 in blocking solution). After this incubation, membranes were again washed and the immune complexes were detected by ECL. The quantitative expression levels of heat shock proteins were assessed by using the NIH image 1.6 program.
Macrophage Production of TNF-α.
TNF-α ELISA (Amersham) was used for determination of TNF-α released into the medium. Briefly, a microtiter plate coated with mouse anti-TNF-α monoclonal antibody was incubated with standard peptides or with macrophage culture supernatants and a second biotinylated anti-TNF-α antibody for 2 hr at room temperature. After incubation, each well was aspirated and thoroughly washed five times with wash buffer. After the last wash, horseradish peroxidase-conjugated streptavidin was added and the mixture was incubated for 30 min at room temperature. Again, the plate was washed five times and then the chromogen (tetramethylbenzidine) solution was added. Color development was stopped by adding 0.18 M sulfuric acid, and was read with an automated ELISA plate reader/spectrophotometer (SpectraMax 250, Molecular Devices) at 450 nm. The amount of TNF-α release was determined by extrapolating the absorbance values of the culture supernatants from the absorbance and concentration of the control peptide, using the standard curve.
Modification of LDL.
Human plasma LDL was oxidized by incubating in 10 μM CuSO4 in an isotonic saline solution without EDTA at 4°C for 3 days. Oxidized LDL (Ox-LDL) was then further dialyzed in 0.3 mM EDTA in isotonic saline solution for 2 days at 4°C to remove free CuSO4. Oxidation of LDL was confirmed by measuring the level of a thiobarbituric acid-reactive substance, malonaldehyde (MDA), generated by hydrolysis of tetramethoxypropane. The degree of oxidation of LDL was determined to be >50 mmol MDA eq/mg of Ox-LDL. Ox-LDL concentration was determined by measuring the protein concentration using the BCA assay (17).
Modified-LDL Uptake.
RAW and HARM cells grown to a subconfluent state in normal medium on Lab-Tek (Nunc) chamber slides were exposed to either hypoxia or normoxia for 24 hr at 37°C. The cells were then washed once and incubated with normal medium containing acetylated LDL labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI-Ac-LDL) (10 μg/ml) at 37°C. After 4 hr of incubation, medium containing DiI-Ac-LDL was removed and cultures were washed three times with PBS. RAW and HARMs were then fixed in 3.8% formaldehyde/PBS for 30 min at room temperature and rinsed once in distilled water. Control for nonspecific uptake was done by incubating cells in culture medium without probe (DiI-Ac-LDL). Cells fixed on slides were then examined with a Bio-Rad MRC-600 confocal scanning laser microscopy (CSLM) imaging system.
Data Analysis.
Data were analyzed by determination of the percent apoptosis induced by hypoxia. Experiments were repeated five times (n = 5), and results are expressed as mean ± SD. Statistical comparisons of the percent apoptosis were performed using the paired Student t test. Significance was assessed at the P < 0.05 level of confidence.
RESULTS
Hypoxia-Induced Apoptosis in Macrophages.
We examined whether some macrophages can overcome hypoxia-induced apoptosis by using the murine macrophage-like cell line RAW. As shown in Fig. 1, when RAW cells were exposed to hypoxia for 24 hr, we observed >25% apoptosis, whereas RAW cells exposed to normoxia showed <5% apoptosis. In addition, we observed that >95% of RAW cells became detached from culture plates and exhibited viabilities of <20% (data not shown), suggesting that necrotic cell death is occurring in addition to apoptosis. The viability of attached macrophages was >90%, indicative of living cells.
Figure 1.
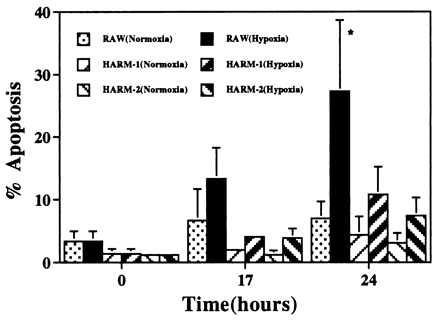
Hypoxia-induced apoptosis in macrophages. RAW and HARM cells grown in RPMI 1640 medium containing 1% FBS were exposed to hypoxia for 24 hr. Macrophage apoptosis was evaluated by fluorescence-activated cell sorting analysis after PI staining. The graph represents the results of five separate experiments. The results are expressed as mean ± SD. ∗, Data sets are different at the 95% confidence level (P < 0.05) compared with normoxic condition.
Selection of HARMs.
RAW cells were subjected to 10 cycles of exposure to hypoxic conditions. Two subpopulations of HARMs (HARM-1 and HARM-2) were eventually derived from the parental RAW cells. Fluorescence-activated cell sorting analysis of PI staining indicated that RAW cells exposed to hypoxia for up to 24 hr undergo apoptosis; 28% of RAW cells were apoptotic after 24 hr. In contrast to RAW cells, HARM cells exposed to hypoxia for 24 hr showed a significantly reduced level of apoptosis (Fig. 2A). Specifically, HARM-1 and HARM-2 exhibited only 10% and 4% apoptosis, respectively. These results demonstrate that some macrophages become resistant to apoptosis when maintained in hypoxic conditions. Because it is possible that the selection of HARM cells by repeated exposure to hypoxia may have resulted in multiple changes in RAW cells resulting in apoptosis resistance, we further selected several clones of HARM cells and examined their sensitivity to hypoxia-induced apoptosis. As shown in Fig. 2B, HARM clones 1–6 consistently exhibited resistance (<6%) to hypoxia (24 hr)-induced apoptosis as compared with the parental RAW cells (>25%).
When the morphology of HARM clones was examined, a significant difference between RAW and HARM cells was observed (Fig. 3). For the most part, HARM cells are firmly adherent and spread cells. Most of these macrophages showed elongated morphology with many filopodial processes (high-magnification image not shown). In contrast, RAW cells were mostly round with few filopodial processes. Because adhesion of macrophages is an important factor in cell activation, these results imply that HARMs/HARM clones may be more activated than are RAW cells.
Figure 3.

Photomicrograph of confluent cultures of RAW, HARM-1, and HARM-2. Cells were inoculated into culture dishes and grown to confluence in normal medium as described in the text. (Final magnification is ×100.)
Expression of Heat Shock Proteins.
To examine for a possible role of heat shock proteins in hypoxia-induced apoptosis, we analyzed the HSC 70 expression level of RAW and HARM cells exposed to either normoxia or hypoxia for 24 hr. As shown in Fig. 4A, similar expression levels of HSC 70 were observed between RAW and HARM cells exposed to normoxic conditions for 24 hr. When RAW cells were exposed to hypoxic conditions, however, a significant reduction in HSC 70 expression level was observed by 24 hr. In contrast to RAW cells, the level of HSC 70 in HARM cells was only minimally reduced by 24 hr. To see if similar changes of other heat shock proteins are induced by hypoxia, GRP 78 expression levels were examined. We observed no significant differences in GRP 78 levels between RAW and HARM cells exposed to either normoxic or hypoxic conditions (Fig. 4B). Other heat shock proteins such as HSP 90 and HSP 27 also did not change (data not shown). These results strongly suggest that HSP 70, but not GRP 78, plays a role in the resistance of HARM cells to hypoxia-induced apoptosis.
Figure 4.
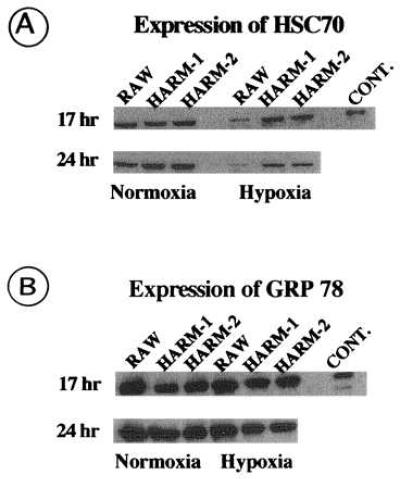
Western blot analysis of heat shock proteins in RAW and HARM cells. RAW and HARM cells exposed to either hypoxia (2% O2) or normoxia (21% O2) for 17 and 24 hr were lysed on ice and extract protein was subjected to electrophoresis in SDS/12% polyacrylamide gels. Gels were electroblotted to nitrocellulose membranes, which were immunoblotted with anti-HSC 70 (A) or anti-GRP 78 (B) antibody conjugated to horseradish peroxidase. Antibody–heat shock protein complexes were detected by chemiluminescence. Bovine brain HSC 70 and recombinant hamster GRP 78 was used as positive controls for HSC 70 and GRP 78, respectively. The figure is a fluorogram.
TNF-α Production Induced by Hypoxia.
To examine if production of TNF-α is altered in RAW and HARM cells, culture supernatants from cells exposed to hypoxia (2% O2) for 24 hr were analyzed. As shown in Fig. 5, we observed a significant difference in TNF-α release between RAW and HARM cells when cells were subjected to either normoxia or hypoxia. Under normoxic conditions, RAW cells showed a >4-fold higher level of TNF-α release than did HARM cells. When cells were exposed to hypoxic conditions, however, HARM cells showed a >3-fold higher level of TNF-α than did RAW cells. In other words, when RAW cells were exposed to hypoxia, we observed a significant decrease (70%) in TNF-α release compared with cells under normoxic conditions. In contrast, HARM cells exposed to hypoxia showed a significant increase (>3-fold) in TNF-α production compared with the control cells under normoxic conditions. These results strongly suggest that HARM cells may be active (i.e., producing cytokines such as TNF-α) under hypoxic conditions, whereas RAW cells are sensitive to hypoxia-induced apoptosis and exhibit decreased cytokine production.
Figure 5.
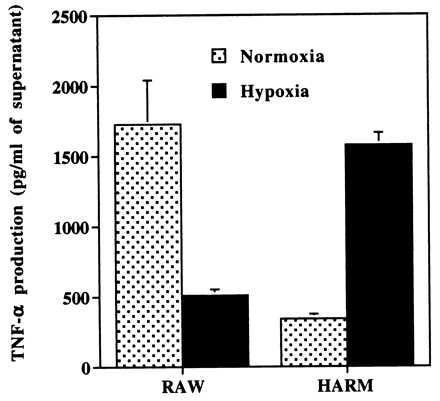
Macrophage production of TNF-α. Supernatants from RAW and HARM cells exposed to either hypoxia or normoxia for 24 hr were used for the determination of protein release by using a TNF-α ELISA kit. The graph represents the results of two experiments.
Macrophage Uptake of Modified LDL.
To examine if a macrophage function (modified-LDL uptake) is altered under hypoxic conditions, RAW and HARM cells were incubated with fluorescent dye-conjugated acetylated LDL (DiI-Ac-LDL) and exposed to either normoxic or hypoxic conditions. As shown by the representative confocal scanning laser microscopy micrographs of RAW and HARM cells in Fig. 6, RAW cells under normoxic conditions showed a high fluorescent intensity, suggesting high levels of DiI-Ac-LDL uptake. In contrast, HARM cells showed a minimal fluorescence intensity, suggesting minimal DiI-Ac-LDL uptake. Similar results were observed when RAW and HARM cells were exposed to hypoxic conditions. When an excess of oxidized LDL (87 μg/ml) was added with DiI-Ac-LDL, RAW and HARM cell fluorescence intensity was significantly reduced, suggesting that a common receptor mediates DiI-Ac-LDL and oxidized-LDL uptake.
Figure 6.
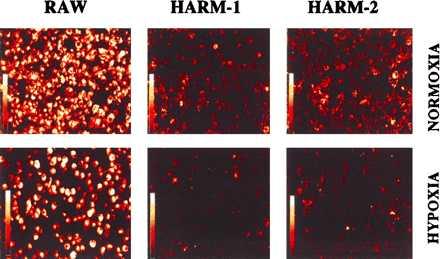
Confocal images of macrophage uptake of modified LDL under hypoxic or normoxic conditions. RAW and HARM cells grown to subconfluent state were exposed to either hypoxia or normoxia for 24 hr. Cells were then incubated with acetylated-LDL conjugated with a fluorescent probe (DiI-Ac-LDL) for an additional 4 hr. A confocal scanning laser microscopy imaging system was used to examine macrophage uptake of modified LDL. The intensity of fluorescence represents the level of modified-LDL uptake by cells.
DISCUSSION
Our study demonstrates that macrophage apoptosis is induced by hypoxia and that subpopulations of macrophages became resistant to apoptosis (HARM cells) after chronic exposure to hypoxia. This differential response to hypoxia indicates that macrophages are multifunctional cells, able to modify their response to hypoxic stress. It is possible that HARM cells are either a selected subpopulation of macrophages that have adapted to hypoxic stress or are genetically predetermined to be resistant to hypoxia.
Regardless of which alternative is true, different macrophage subpopulations in inflammatory sites may have distinct roles. Previous studies have shown that heterogeneous populations of macrophages are recruited to inflammatory sites (14, 18). A small population of macrophages that are not transformed into foam cells is observed in atherosclerotic lesions (14). Additionally, Tidball and St. Pierre (18) demonstrated that ED1 or ED2 antigen-expressing macrophages are specifically recruited to muscle inflammatory sites and cleared through apoptosis during the resolution of inflammation. These studies suggest that specific subtypes of macrophages recruited to inflammatory sites in vivo might have distinct effects on the outcome of inflammation. HARM cells may mimic the action of macrophages that are not transformed into foam cells in atherosclerotic lesions. Because apoptosis mediates the reduction of macrophage populations in vivo during the resolution of inflammation (18, 19), the persistent presence of HARM-like cells in inflammatory sites may play a critical role in perpetuation of inflammatory lesions, as they can overcome hypoxia-induced apoptotic signals.
When HARM cells were exposed to hypoxic stress, the expression level of HSC 70 was maintained, whereas RAW cells showed a 2-fold reduction in the level of HSC 70. This appears to be specific to HSC 70, because GRP 78 (and other heat shock proteins such as HSP 90 and HSP 27) expression was not altered in RAW and HARM cells exposed to either hypoxic or normoxic conditions. These results strongly suggest that HSP expression may play a role in hypoxia-induced resistance to apoptosis. This idea is further supported by previous studies which demonstrated that heat shock protein expression may determine cell resistance or tolerance to stresses that are normally lethal (20–23). For example, we have previously reported that nitric oxide-induced apoptosis-resistant macrophages show a 2-fold increase in the expression of HSC 70 (20, 21). Heat shock protein-transfected cells have also been shown to become more resistant to the cytotoxic effects of anticancer drugs that induce apoptosis (22). Additionally, Mailhos et al. (23) have demonstrated that cells subjected to heat shock are protected from apoptosis induced by growth factor withdrawal, suggesting a possible heat shock-induced stress protein role in apoptosis resistance. Because RAW cells exposed to hypoxic conditions exhibit diminished HSC 70 expression coincident with induction of apoptosis, it is possible that HSC 70 may play a role in hypoxia-induced apoptosis resistance. This possible mechanism of apoptosis resistance could be further analyzed by overexpression of HSC 70 in the parent RAW cells to see if this would produce resistance to hypoxia-induced apoptosis. HSC 70 may protect against apoptosis, by inhibiting degradation of proteins essential to macrophage survival under hypoxic conditions.
The differential reactivity of HARM cells is further demonstrated by an increased release of TNF-α in response to hypoxia exposure as compared with RAW cells, which showed a significant decrease in TNF-α release. Because RAW cells are sensitive to hypoxic stress resulting in apoptotic cell death, a decrease in TNF-α release may reflect decreased numbers of RAW cells capable of cytokine synthesis. The differential response of macrophages to hypoxic stress is also demonstrated by the morphological differences observed between RAW and HARM cells. HARM cells appeared to be more firmly adhered and spread, whereas RAW cells were rounded and weakly attached to the surface. Because attachment of macrophages to a substrate is important in the process of cell activation (24), HARM cells with a greater degree of spreading and many filopodial processes may represent more activated macrophages, which exhibit increased cytokine production, as compared with RAW cells.
HARM cells also differ from RAW cells with respect to another marker of macrophage function, uptake of modified LDL. HARM cells take up minimal levels of Ac-LDL, as indicated by low fluorescent intensity, whereas RAW cells show maximal uptake. Because macrophages are a major scavenger of modified (or oxidized) LDL, minimal uptake of Ac-LDL by HARM cells was unexpected. Interestingly, low uptake of modified LDL by HARM cells is associated with increased TNF-α release in response to hypoxia. A similar phenotype (macrophages that are not transformed into foam cells) has been observed in human atherosclerotic lesions (14). The production of IL-1α, IL-1β, and TNF-α was associated only with the macrophages that were not transformed into foam cells. HARM cells might be similar to the subpopulations of macrophages found in atherosclerotic lesions that are not transformed into foam cells. Furthermore, our results suggest that subtypes of macrophages such as HARM cells may participate in inflammation rather than in the scavenger uptake of the modified LDL. Because the thickened intima or inner media of lesions could produce hypoxia within the vascular wall, it is likely that macrophages recruited to the inflammatory sites are exposed to hypoxic conditions (25, 26). Therefore, macrophages such as HARM cells, that are resistant to apoptosis induced by pathologic hypoxic conditions, may play an important role in perpetuation of secretive and proliferative events involved in plaque formation and development.
Acknowledgments
This work was supported by a generous grant to E.G.L. from the Cleveland Foundation and by the Department of Medicine at University Hospitals, Cleveland, OH.
ABBREVIATIONS
- RAW
murine macrophage-like RAW 264.7 cells
- HARM
hypoxia-induced apoptosis-resistant macrophage
- HSC 70
constitutive heat shock protein 70
- GRP 78
glucose-regulated protein 78
- TNF-α
tumor necrosis factor α
- IL
interleukin
- LDL
low density lipoprotein
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- PI
propidium iodide
References
- 1.Allen J, Winterford C, Axelsen R A, Gobe G C. Renal Failure. 1992;14:453–460. doi: 10.3109/08860229209047652. [DOI] [PubMed] [Google Scholar]
- 2.Muschel R, Bernhard E J, Garza L, McKenna W G, Koch C J. Cancer Res. 1995;55:995–998. [PubMed] [Google Scholar]
- 3.Shimizu S, Eguchi Y, Kamiike W, Itoh Y, Hasegawa J-I, Yomabe K, Otsuki Y, Matsuda H, Tsujimoto Y. Cancer Res. 1996;56:2161–2166. [PubMed] [Google Scholar]
- 4.Graeber T G, Osmaniah C, Jacks T, Housman D E, Koch C J, Lowe S W, Giaccia A J. Nature (London) 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 5.Hannah S, Mecklenburgh K, Rahman I, Bellingan G J, Greening A, Haslett C, Chilvers E R. FEBS Lett. 1995;372:233–237. doi: 10.1016/0014-5793(95)00986-j. [DOI] [PubMed] [Google Scholar]
- 6.Scannell G, Waxman K, Kaml G J, Ioli G, Gatanage T, Yamamoto R, Granger G A. Ann Surg Res. 1993;54:281–285. doi: 10.1006/jsre.1993.1044. [DOI] [PubMed] [Google Scholar]
- 7.Ghezzi P, Dinarello C A, Bianchi M, Rosandich M E. Cytokine. 1991;3:189–194. doi: 10.1016/1043-4666(91)90015-6. [DOI] [PubMed] [Google Scholar]
- 8.Albina J E, Henry W L, Mastrofrancesco B, Martin B-A, Reichner J S. J Immunol. 1995;155:4391–4396. [PubMed] [Google Scholar]
- 9.Björkerud S, Björkerud B. Am J Pathol. 1996;149:367–380. [PMC free article] [PubMed] [Google Scholar]
- 10.Han D K, Haudenschild C C, Hong M K, Tinkle B T, Leon M B, Liau G. Am J Pathol. 1995;147:267–277. [PMC free article] [PubMed] [Google Scholar]
- 11.Geng Y-J, Libby P. Am J Pathol. 1995;147:252–266. [PMC free article] [PubMed] [Google Scholar]
- 12.Tipping P G, Hancock W W. Am J Pathol. 1993;142:1721–1728. [PMC free article] [PubMed] [Google Scholar]
- 13.Apostolopoulos J, Davenport P, Tipping P G. Arterioscler Thromb Vasc Biol. 1996;16:1007–1012. doi: 10.1161/01.atv.16.8.1007. [DOI] [PubMed] [Google Scholar]
- 14.Nagornev V A, Maltseva S V. Atherosclerosis. 1996;121:245–251. doi: 10.1016/0021-9150(95)05726-9. [DOI] [PubMed] [Google Scholar]
- 15.Telford W G, King L E, Fraker P J. Cytometry. 1992;13:137–143. doi: 10.1002/cyto.990130205. [DOI] [PubMed] [Google Scholar]
- 16.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz M A, Lassota P, Traganos F. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- 17.Smith P K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 18.Tidball J G, St. Pierre B A. J Leukocyte Biol. 1996;59:380–388. doi: 10.1002/jlb.59.3.380. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen K B, McCombe P A, Pender M P. J Autoimmun. 1994;7:145–152. doi: 10.1006/jaut.1994.1011. [DOI] [PubMed] [Google Scholar]
- 20.Hirvonen M-R, Brune B, Lapetina E G. Biochem J. 1996;315:845–849. doi: 10.1042/bj3150845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brüne B, Götz C, Messmer U K, Sandau K, Hirvonen M-R, Lapetina E G. J Biol Chem. 1997;272:7253–7258. doi: 10.1074/jbc.272.11.7253. [DOI] [PubMed] [Google Scholar]
- 22.Oesrerreich S, Weng C, Qiu M, Hilsenbeck S G, Osborne C K, Fuqua S A W. Cancer Res. 1993;53:4443–4448. [PubMed] [Google Scholar]
- 23.Mailhos C, Howard M K, Latchman D S. Neuroscience. 1993;55:621–627. doi: 10.1016/0306-4522(93)90428-i. [DOI] [PubMed] [Google Scholar]
- 24.Chen C, Mrksich M, Huang S, Whitesides G M, Ingber D E. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 25.Crawford D W, Blankenhorn D H. Atherosclerosis. 1991;89:97–108. doi: 10.1016/0021-9150(91)90049-9. [DOI] [PubMed] [Google Scholar]
- 26.Mandel P, Poirel G, Simard-Duquesne N. J Atheroscler Res. 1966;6:463–466. doi: 10.1016/s0368-1319(66)80073-x. [DOI] [PubMed] [Google Scholar]