

Abstract
Cloned PCR products containing hepatitis C virus (HCV) genomic fragments have been used for analyses of HCV genomic heterogeneity and protein expression. These studies assume that the clones derived are representative of the entire virus population and that subsets are not inadvertently selected. The aim of the present study was to express HCV structural proteins. However, we found that there was a strong cloning selection for defective genomes and that most clones generated initially were incapable of expressing the HCV proteins. The HCV structural region (C-E1-E2-p7) was directly amplified by long reverse transcription–PCR from the plasma of an HCV-infected patient or from a control plasmid containing a viable full-length cDNA of HCV derived from the same patient but cloned in a different vector. The PCR products were cloned into a mammalian expression vector, amplified in Escherichia coli, and tested for their ability to produce HCV structural proteins. Twenty randomly picked clones derived from the HCV-infected patient all contained nucleotide mutations leading to absence or truncation of the expected HCV products. Of 25 clones derived from the control plasmid, only 8% were fully functional for polyprotein synthesis. The insertion of extra nucleotides in the region just upstream of the start codon of the HCV insert led to a statistically significant increase in the number of fully functional clones derived from the patient (42%) and from the control plasmid (72–92%). Nonrandom selection of clones during the cloning procedure has enormous implications for the study of viral heterogeneity, because it can produce a false spectrum of genomic diversity. It can also be an impediment to the construction of infectious viral clones.
Hepatitis C virus (HCV), a member of the Flaviviridae family, is the major cause of chronic liver disease worldwide (1). HCV is a positive-sense single-strand RNA virus with a genome that encodes one large polyprotein in which putative structural proteins are located at the N-terminal end, and the putative nonstructural (NS) proteins are located at the C-terminal end (1). This virus, like other RNA viruses, exhibits a significant genetic heterogeneity as a result of mutations that occur during viral replication (2). In fact, the genomes of most RNA viruses have been found to consist of a population of closely related but heterogeneous sequences (quasispecies) in a single infected individual (3, 4). The quasispecies distribution of HCV might have important biological consequences (5, 6). It has been proposed that this genetic heterogeneity allows HCV to escape immune pressure and to establish chronic infection (7–10). In addition, the existence of a heterogeneous population of HCV may influence the outcome of antiviral therapy (11–13); resistance to treatment might result from selection of minor viral populations during this therapy. Therefore, it is important to define accurately quasispecies populations of HCV.
Many analyses of viral quasispecies of HCV have been published (6). The majority of these studies have focused on the most variable part of the HCV genome, the hypervariable region 1 (HVR1) of glycoprotein E2. Most studies relied on molecular cloning of HCV genomic fragments amplified by reverse transcription–PCR (RT-PCR) but major differences were found among the studies in the degree of genomic variability and prevalence of defective viral genomes (10, 11, 14–17). This phenomenon seems not to be an isolated finding, because significant discrepancies in the prevalence of defective genomes also have been observed among different studies of HIV quasispecies (18–21). One explanation for these differences could be that the viral populations are not comparable in different individuals. However, it is important to remember that the procedures used to analyze the quasispecies populations in these studies differed to varying degrees, and therefore, different subpopulations might have been sampled inadvertently.
In this study, we have analyzed some of the shortcomings of molecularly cloning a virus that circulates as a quasispecies (HCV) and have demonstrated that severely biased selection can take place during plasmid DNA amplification in Escherichia coli. This selection can lead to dramatic variation in the types of clones obtained and can prevent the recovery of functional clones.
METHODS
Plasmid Construction.
The vector pcDNA3.1(+) (Invitrogen) was used to construct expression plasmids of the structural region of HCV. This vector contains a T7 promoter for in vitro transcription and a human CMV immediate-early promoter/enhancer for high-level protein expression in mammalian cells. The structural region of HCV plus a short fragment of the NS2 region (nucleotides 1–2646 of the ORF) was amplified by RT-PCR from plasma of patient H (strain H77) (22) by using primers containing a restriction enzyme site for convenient cloning (Table 1).
Table 1.
Primers used to amplify and clone the structural region of HCV H77 strain
| Restriction enzyme site | |
|---|---|
| Sense primers | |
| A0 (45-mer) 5′-ACGCGTAAGCTTATGAGCACGAATCCTAAACCTCAAAGAAAAACC-3′ | HindIII |
| A0 (38-mer) 5′-ACGCGTAAGCTTATGAGCACGAATCCTAAACCTCAAAG-3′ | HindIII |
| A + 1 5′-ACGCGTAAGCTTCATGAGCACGAATCCTAAACCTCAAAG-3′ | HindIII |
| A + 2 5′-ACGCGTAAGCTTCCATGAGCACGAATCCTAAACCTCAAAG-3′ | HindIII |
| A + 3 5′-ACGCGTAAGCTTCCCATGAGCACGAATCCTAAACCTCAAAG-3′ | HindIII |
| Antisense primer | |
| B 5′-TTCAGAGAATTCCTACGGGTGTACTACACACGTGAGTAAG-3′ | EcoRI |
Restriction enzyme sites are underlined; the HCV initiation codon is shown in italic characters; the extra nucleotides upstream of the HCV initiation codon are shown in bold characters.
Amplification was carried out by “long” RT-PCR (23). Briefly, RNA was extracted from 10 μl of plasma with the TRIzol Reagent (GIBCO/BRL). The RNA pellet was resuspended in 100 μl of RNase-free water containing 10 mM DTT (Promega) and 5% RNasin (20–40 units/μl) (Promega). An RNA aliquot of 10 μl (105 genome equivalents of H77) was incubated for 2 min at 65°C. To the RNA, 4 μl of 5× First Strand Synthesis Buffer (GIBCO/BRL), 1 μl of 100 mM DTT, 1 μl of a 10 mM stock solution of dNTPs (Pharmacia), 2.5 μl of 10 μM antisense primer solution, 0.5 μl of RNasin, and 1 μl (200 U) of Superscript II reverse transcriptase (GIBCO/BRL) were added. After 1 hr incubation at 42°C, 1 μl of RNase T1 (900–3,000 units/μl) (GIBCO/BRL) and 1 μl of RNase H (1–4 units/μl) (GIBCO/BRL) were added and the reaction was incubated at 37°C for 20 min. Long PCR was performed with a high-fidelity DNA polymerase mixture (Advantage KlenTaq polymerase mix, CLONTECH). Briefly, to 3 μl of cDNA, 5 μl of 10× KlenTaq PCR buffer (CLONTECH), 1.25 μl of a 10 mM stock solution of dNTPs, 1 μl of 10 μM sense primer, 1 μl of 10 μM antisense primer, 1 μl of 50× Advantage KlenTaq Polymerase mix, and water to a final volume of 50 μl were added. The PCR was performed in a Robocycler thermal cycler (Stratagene) for 35 cycles with denaturation at 99°C for 35 sec, annealing at 67°C for 30 sec, and elongation at 68°C for 3 min and 30 sec.
The PCR products digested with HindIII and EcoRI (New England Biolabs) were inserted into the digested expression vector pcDNA3.1(+) by using T4 ligase (Promega). E. coli DH5 alpha library-competent cells (GIBCO/BRL) were transformed and plated in Luria–Bertani agar containing ampicillin (100 μg/ml) (Sigma). DNA was prepared from 100 ml of bacterial cultures, grown at 37°C in the presence of ampicillin (100 μg/ml), with the modified alkaline lysis method by using the Qiagen plasmid Maxi kit.
As a control, the same fragment was amplified from 0.1 ng of a plasmid containing the full-length sequence of H77 (24). This plasmid encoded the consensus amino acid sequence of the structural region of H77. Cloning of the amplification products and DNA preparation were performed as described above.
Sequence Analysis.
Both strands of plasmid DNA were sequenced with the ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit by using AmpliTaq DNA polymerase (Perkin–Elmer) and H77 specific primers. Multiple sequence alignments were performed by using the computer software package geneworks (Oxford Molecular Group) (2).
Protein Expression.
One microgram of nonlinearized plasmid was used for in vitro transcription and translation in 25 μl of the TNT Coupled Reticulocyte Lysate System (Promega) containing [S35]methionine (Amersham). Synthesis was at 30°C for 90 min. Total translation products were separated in 12% SDS/PAGE and identified by autoradiography.
Statistical Analysis.
Quantitative values are expressed as mean ± SD. Categorical variables were compared with the Fisher’s exact test, and quantitative variables were compared with a nonparametric test (Mann–Whitney). For calculation of the estimated DNA polymerase error rate and the numbers of silent and nonsilent mutations, the primer regions were not included in the analysis.
RESULTS
Inability to Express the Complete Structural Region of HCV from an Expression Vector.
We amplified the structural region of HCV plus a small fragment of the NS2 region to analyze the quasispecies population of strain H77 and to study protein expression. For this purpose, a region encompassing nucleotides 1–2646 of the ORF of H77 was amplified in a single round of long RT-PCR with primers A0 (45-mer) and B (Table 1) and cloned into the expression vector pcDNA3.1(+).
Eight randomly picked clones amplified from the patient showed a high degree of genetic variability. Although the consensus sequence deduced from these eight clones was the same as the consensus sequence of H77 (24), no two of the analyzed clones were identical (Fig. 1). Compared with the consensus sequence of H77, the mean number of nucleotide substitutions per clone (excluding the primer regions) was 17.6 ± 12, ranging from 8 to 40. Of a total of 141 nucleotide substitutions, 77 (55%) were silent and 64 (45%) resulted in changes in the deduced amino acid sequence. Unexpectedly, all eight clones appeared defective for polyprotein synthesis because one clone had a mutation that created an in-frame stop codon and seven clones each had a single nucleotide deletion that introduced an in-frame stop codon a few codons after the deletion. Interestingly, in five of the latter clones the deletion was located within the sense primer region (in three different positions).
Figure 1.

Sequence analysis of eight clones derived from the patient. Amplification was performed with primer A0 (45-mer). Black boxed regions indicate putative translated HCV products. Continuous lines indicate nonsilent mutations, and discontinuous lines indicate silent mutations, compared with the consensus sequence of HCV H77 (24).
In vitro transcription–translation of these clones confirmed that they were defective. Indeed, none of the eight clones produced a protein of the desired size (≈85 kDa). However, truncated products of a size predicted from the location of the stop codon were obtained (data not shown).
As a control for the fidelity of the PCR and cloning procedures, we amplified the same region of HCV from a plasmid that contained a homogeneous and viable full-length sequence of HCV (24). Given the high prevalence of deletions in the sense primer region, and to exclude an oligonucleotide synthesis problem, primers A0 (45-mer) and B were resynthesized and gel-purified in an independent laboratory. Amplification and cloning were performed exactly as described above for the sample derived from the patient. A short region encompassing the sense primer region was sequenced in 10 random clones. Five (50%) of the 10 clones obtained with the new primers also presented a deletion in this region, indicating that a defective lot of primers was not responsible for the previous results. Sequence analysis of the entire product of two of the clones without deletion in the primer region identified a mutation that created a stop codon and a nucleotide deletion that caused a frameshift, respectively (data not shown). Once again, full-length polypeptide products were not obtained by in vitro transcription–translation in these two clones. These results suggested that translationally defective clones were being preferentially selected by the cloning procedure.
Modification of the Region Immediately Upstream of the HCV Initiation Codon Increased the Proportion of Functional Clones.
We hypothesized that spurious translation of HCV sequences was producing a protein toxic to E. coli. Therefore, we repeated the amplification of the structural region of HCV from the control plasmid as described above, except that we used sense primers that introduced extra nucleotides just upstream of the HCV initiation codon in an attempt to prevent translation of HCV sequences. For convenience, we synthesized a shorter sense primer (A0, 38-mer) and introduced one (primer A+1), two (primer A+2), or three (primer A+3) extra nucleotides (Table 1). The restriction enzyme site of the sense primers, as well as the sequence and the restriction enzyme site of the antisense primer, were the same as those used in the previous experiments. Conditions for long PCR and cloning were the same in all experiments.
We sequenced eight randomly selected clones, obtained after amplification of the control template with primer A0 (Fig. 2A). The mean number of nucleotide changes per clone (excluding mutations in the primer regions) when compared with the template sequence was 3.4 ± 1.8, with a range of from 0 to 5. Therefore, based on the product length and the 35 cycles of amplification, the estimated polymerase error rate was 3.7 × 10−5/nt per cycle. Of a total of 27 mutations, 8 (30%) were silent whereas 19 (70%) led to amino acid changes. Although amplification was performed from a functional cDNA template, all eight clones appeared defective for polyprotein synthesis. One of the eight analyzed clones had an in-frame stop codon, and six other clones each had a single nucleotide deletion (three of them in the sense primer region, in two different positions), which were predicted to terminate translation (Fig. 2A). The final clone had a unique mutation that changed the AUG start codon to GUG (in the sense primer region). Transcription–translation analysis showed that none of these clones, with one exception, encoded a full-length 85-kDa protein; the clone having a GUG start codon yielded an 85-kDa product but only in minute amounts.
Figure 2.

Sequence analysis of clones derived from the control plasmid. (A) Eight clones derived from the control plasmid with primer A0 (38-mer). (B) Ten clones derived from the control plasmid with primer A+1. Black boxed regions indicate putative translated HCV products. Continuous lines indicate nonsilent mutations, and discontinuous lines indicate silent mutations, compared with the control plasmid sequence (24). The last clone of A contains the mutation within the start codon (AUG to GUG). This mutation, theoretically, could result in a low level of translation.
In the same experiment we used the modified sense primer A+1 to amplify the structural region from the control plasmid. A significant increase in the cloning efficiency was apparent: in two independent experiments, the number of transformed colonies was at least five times greater following amplification with primer A+1 than with primer A0. We found a total of 29 mutations in the nonprimer region in 10 randomly selected clones amplified with the A+1 primer (Fig. 2B); thus, the estimated polymerase error rate was 3.2 × 10−5/nt per cycle. The mean number of nucleotide changes per clone when compared with the template sequence was 2.9 ± 2.2, with a range of from 1 to 7. Therefore, the estimated polymerase error rate was very similar to that observed when the A0 primer was used. What was strikingly different was that 7 of the 10 clones appeared, on the basis of sequence, to be functional for polyprotein synthesis, and only 3 clones were presumed to be nonfunctional because they contained a stop codon (two clones) or a single nucleotide deletion (one clone). As predicted, transcription–translation analysis showed that the seven clones presumed to be functional were indeed able to produce a full-length 85-kDa product. Thus, whereas none of the 8 clones obtained by using primer A0 for amplification was functional, 7 of 10 obtained with primer A+1 were functional. This difference was statistically significant (P = 0.004).
We also compared clones amplified from the patient with primer A0 (20 clones) and primer A+1 (24 clones). None of the 20 clones obtained after amplification with primer A0 was functional for polyprotein synthesis in the in vitro transcription–translation system, whereas 10 of 24 (42%) of the clones obtained after amplification with primer A+1 expressed a full-length polyprotein of 85 kDa (P < 0.001) (Table 2).
Table 2.
Analysis by in vitro transcription–translation of clones obtained from the patient or from a control plasmid
| Functional clones | Defective clones | Total | |
|---|---|---|---|
| Clones derived from the patient | |||
| Sense primer A0* | 0 (0%) | 20 (100%) | 20 |
| Sense primer A + 1*† | 10 (42%) | 14 (58%) | 24 |
| Clones derived from the control plasmid | |||
| Sense primer A0‡ | 2 (8%) | 23 (92%) | 25 |
| Sense primer A + 1†‡ | 18 (72%) | 7 (28%) | 25 |
| Sense primer A + 2‡ | 23 (92%) | 2 (8%) | 25 |
| Sense primer A + 3‡ | 22 (88%) | 3 (12%) | 25 |
The difference between the proportion of defective clones obtained from the patient with sense primers A0 and A + 1 was statistically significant (P < 0.001).
The difference between the proportion of defective clones obtained with sense primer A + 1 from the patient and from the control plasmid was statistically significant (P = 0.045).
The differences in the proportion of defective clones obtained with sense primer A0 compared with the proportions of defective clones obtained with sense primers A + 1, A + 2, and A + 3, respectively, were statistically significant (P < 0.001 in all three cases).
Finally, clones obtained after amplification of the control plasmid with primers A0, A+1, A+2, and A+3, respectively, were analyzed with the in vitro transcription–translation system. In total, only 2 of 25 (8%) of the clones obtained with primer A0 were fully functional for polyprotein synthesis, whereas 18 of 25 (72%), 23 of 25 (92%), and 22 of 25 (88%) were functional when primers A+1, A+2, and A+3 were used, respectively (Table 2; proteins expressed from representative clones obtained with primers A0 and A+3, respectively, are depicted in Fig. 3). These differences were statistically significant (P < 0.001).
Figure 3.
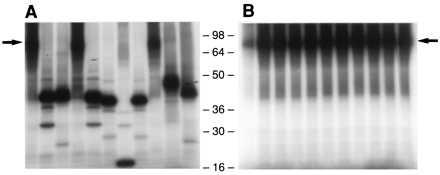
Transcription–translation of clones derived from the control plasmid. Amplification with primer A0 (38-mer) (A) or primer A+3 (B). First lane, positive control (a clone with the consensus amino acid sequence of H77). Remaining lanes, representative clones. Arrow, position of the expected full-length product (≈85 kDa).
Distinction Between True and Artificial Genetic Heterogeneity.
Distinction between true and artificial genetic heterogeneity is important for understanding the biology of HCV. We analyzed the pattern of mutations in the clones obtained with primer A0 from the patient and from the control plasmid. When compared with the consensus sequence of H77, the number of nucleotide substitutions per clone was substantially higher in the clones obtained from the patient (Fig. 1) than in those obtained from the control (Fig. 2A) (17.6 ± 12 vs. 3.4 ± 1.8, respectively; P < 0.001). These data indicated that there was true genetic heterogeneity in the structural gene region of the viruses circulating in the patient. We also compared the number of changes in the deduced amino acid sequence of clones derived from the patient and of clones derived from the cDNA control. Approximately two-thirds of the random nucleotide changes produced by polymerase mistakes are expected to introduce an amino acid change. In fact, 19 of 27 (70%) of the mutations in the clones derived from the control plasmid did indeed introduce coding changes, whereas only 64 of 141 (45%) of the substitutions in clones amplified from the patient led to amino acid changes (P = 0.02). This difference suggests that there was a biological selection in the patient against coding mutations.
There was also a significant difference between the proportion of defective clones obtained from the patient and from the control when amplification of the cloned region was performed with primer A+1. With primer A+1, 14 of 24 (58%) of clones obtained from the patient were defective for polyprotein synthesis, whereas only 7 of 25 (28%) of the clones obtained from the control plasmid were defective (P = 0.045) (Table 2). These results strongly suggest that some of the viruses circulating in the patient are indeed defective for polyprotein synthesis.
DISCUSSION
Although there is a general impression that some genes are harder than others to clone in bacterial plasmids, the scope and magnitude of this problem may have been underestimated. It was a surprise to find that all of the clones of HCV that we isolated initially contained stop codons or frameshift mutations and thus were defective for translation. The failure to recover a cDNA clone that could express the HCV structural proteins suggested that either the majority of HCV virions circulating in the patient were defective or that cDNA sequences that could encode the HCV proteins of interest were eliminated during the cloning procedure and the genomic sequences obtained were not representative of the viral population. The former hypothesis has major implications for understanding the biology of the virus whereas the latter could invalidate some quasispecies analyses. The first hypothesis seemed unlikely because there was only a 10-fold difference between PCR titer and infectivity titer for this sample (22, 25). It seemed more probable that the recovery of HCV cDNA clones was strongly biased toward nonfunctional clones.
One hypothesis accounting for selection of defective clones was that HCV proteins were being translated during plasmid amplification in bacteria and that these proteins were toxic for E. coli. Translation could initiate within E. coli sequences and continue into HCV sequences, or it could initiate directly within the HCV portion of a transcript. Because there were amber and ocher stop codons in the polylinker preceding the HCV insert, it seemed unlikely that an E. coli–HCV fusion protein was causing the problem.
We considered it possible that spurious translation was initiating within the HCV sequence. In E. coli, the translation efficiency is in part determined by the Shine–Dalgarno (SD) interaction (the base pairing of the 3′ end of the 16S ribosomal RNA to complementary nucleotides located upstream of the initiation codon in the messenger RNA). We analyzed the sequence upstream of the initiating AUG codon of HCV and found sequences complementary to the 16S ribosomal RNA (SD sequences) in positions −5 to −7, and −11 to −13, which might conceivably function in initiation complex formation and allow translation of the HCV sequences in E. coli. When we introduced a frameshift mutation of 1 or 2 extra nucleotides just upstream of the HCV translation–initiation codon, the proportion of translationally competent clones dramatically increased. If this were entirely a result of elimination of a fusion protein, introduction of the third nucleotide would have restored the original reading frame and defective clones should have been selected again. In contrast, when the third nucleotide was inserted, the proportion of functional clones remained high, confirming that the fusion-protein theory was not the likely explanation. Modifications of the spacing between the SD sequence and the gene start codon, as well as changes in the secondary structure of this region, have been shown to affect significantly the translation efficiency of some genes (26–31). Therefore, we suggest that selection of certain clones occurred because translation initiated at an HCV codon to produce a toxic protein and incorporation of 1–3 additional nucleotides between the SD-like sequences and the first AUG in the HCV region interfered with this initiation step.
Four pieces of data support the theory that synthesis of HCV proteins in E. coli resulted in selection of translation-defective clones. First, all of the clones originally isolated had stop codons or frameshift mutations that were not randomly distributed but were located within the first half of the HCV sequence. The majority of these mutations would have prevented translation of virtually the entire HCV insert. Second, a clone containing a single mutation that changed the AUG start codon to GUG was isolated. Translation efficiency usually is reduced significantly by a GUG start codon (28), and this mutation therefore would have the same practical effect as a stop codon but only if translation initiated at this site. Third, only opal stop codons were identified although amber or ocher stop codons should have occurred with similar frequency. We do not have an explanation for the absence of ocher codons. However, the absence of amber stop codons seems best explained by the fact that the amber suppressor allele supE44 is present in the DH5 alpha strain of E. coli we used, and therefore, an amber stop codon would not have prevented protein synthesis (32). Fourth, minor modifications of the vector close to the putative site of translation–initiation decreased or eliminated the number of clones encoding prematurely terminated proteins. Because it currently is not possible to predict which peptides or proteins might be toxic to E. coli, the possibility of biased recovery of clones should be considered whenever foreign genes are biologically amplified as plasmids.
The recovery of a majority of translationally functional HCV clones after modification of the vector has important implications. First, it suggests that studies identifying a very high proportion of quasispecies clones with stop codons or frameshifts may have been flawed by a similar selection during amplification in E. coli. However, when the A+1 primer, which decreased the number of defective clones, was used, we did recover a significantly higher proportion of translation-defective clones from the patient than from the control. This result demonstrated that there are HCV particles that do indeed contain a defective genome. The question remains, however, as to what their true level is and whether they are perpetuated by helper viruses or generated de novo during each round of replication. The answer to this question is crucial to understanding the dynamics of HCV replication.
The data also confirmed that there was sequence diversity in the structural regions of circulating viruses. The eight clones derived with primer A0 from the patient had 5.2 times more mutations (141 vs. 27) than did the eight comparable clones derived from the homogeneous control plasmid. These excess mutations almost certainly represent real genetic variants of HCV. However, as reviewed by Smith et al. (17), there is no simple way to determine which mutations in clones are present in circulating virus and which are artifacts of the PCR amplification; therefore, the true sequence of the quasispecies in the patient could not be unequivocally identified.
Our results suggested that silent mutations were more likely to be retained in the virus population than nonsilent mutations. Of the eight clones derived with primer A0 from the control plasmid, 30% of the mutations were silent mutations, close to the 33% predicted by random mutation, and eight translation-defective clones derived from the patient had 55% of the mutations as silent mutations. A probable explanation for the difference in percentage of silent mutations is that coding mutations were often detrimental to virus replication and viruses containing them failed to thrive in the patient, leading to preferential accumulation of silent mutations in the circulating virus population.
These data suggested a number of explanations for the difficulties previously encountered in expressing HCV glycoproteins (33) and in obtaining an infectious cDNA clone of HCV. Both laboratories that succeeded in producing an infectious clone did so by constructing a clone with a sequence almost identical to the consensus sequence (24, 34). The apparent preferential accumulation in circulating viruses of silent mutations compared with coding mutations noted above suggests that the structural proteins of HCV (with the obvious exception of the hypervariable region) may not be very plastic, and thus, random coding mutations introduced into the cDNA by polymerase errors might have diminished or abolished the infectivity of any nonconsensus cloned genome. However, an even more intriguing possibility should be considered. What if production of HCV toxic proteins could be circumvented not only by preventing translation of the HCV sequence but also by selecting for coding mutations, which altered the HCV protein so drastically that it was no longer toxic for the bacterium? Such a severe alteration almost certainly would destroy the ability of the protein to function in HCV replication also. If such cloning selection of lethal mutations occurred, the vectors and conditions for amplifying plasmids in E. coli could have more impact than polymerase errors on diminishing the recovery of infectious clones. Additionally, this type of scenario would complicate quasispecies analysis; the common appearance of a particular mutation might not reflect a major quasispecies but rather highlight a particularly potent inactivating mutation that was well tolerated by E. coli. This caveat has the corollary that the best way to ensure that a consensus sequence is valid may be to determine it by direct sequencing of PCR products rather than to deduce it from the sequence of a limited number of cloned products.
In summary, our data clearly show that under certain common cloning conditions plasmids containing stop codons or frameshift mutations in HCV proteins (and presumably other proteins) can be preferentially amplified in E. coli. The data do not rule out the possible selection of lethal or debilitating mutations by a similar mechanism, and this would be even harder to recognize. It is necessary to consider such selection pressure when studying quasispecies or cloning genes for expression. A low efficiency of transformation may be a warning that negative selection pressure is operating. If selection is detected, it may be possible to decrease or eliminate it by changing or modifying the vector or insert. The data emphasize the fact that we still do not understand all of the variables that can affect the apparent quasispecies distribution of a virus and suggest that the most powerful techniques currently used to analyze virus genomes in particular and other genes in general may be generating more artifacts than is commonly recognized.
Acknowledgments
We thank Ms. L. Rasmussen and other staff members at Science Applications International Corporation, Frederick, MD, for assistance in sequence analysis. We also thank Dr. T. Heller, Ms. Y. Huang, and Dr. M. Yanagi for their helpful advice. This study was supported in part by National Institutes of Health Contract N01-CO-56000. X.F. was the recipient of a grant from “La Caixa” Fellowship Program.
ABBREVIATIONS
- HCV
hepatitis C virus
- RT
reverse transcription
References
- 1.Houghton M, Weiner A, Han J, Kuo G, Choo Q-L. Hepatology. 1991;14:381–388. [PubMed] [Google Scholar]
- 2.Bukh J, Miller R H, Purcell R H. Sem Liver Dis. 1995;15:41–63. doi: 10.1055/s-2007-1007262. [DOI] [PubMed] [Google Scholar]
- 3.Holland J J, De la Torre J C, Steinhauer D A. Curr Top Microbiol Immunol. 1992;176:1–20. doi: 10.1007/978-3-642-77011-1_1. [DOI] [PubMed] [Google Scholar]
- 4.Domingo E, Martínez-Salas E, Sobrino F, de la Torre J C, Portela A, Ortin J, López-Galindez C, Pérez-Breña P, Villanueva N, Nájera R, VandePol S, Steinhauer D, DePolo N, Holland J. Gene. 1985;40:1–8. doi: 10.1016/0378-1119(85)90017-4. [DOI] [PubMed] [Google Scholar]
- 5.Martell M, Esteban J I, Quer J, Genescà J, Weiner A, Esteban R, Guardia J, Gómez J. J Virol. 1992;66:3225–3229. doi: 10.1128/jvi.66.5.3225-3229.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farci P, Bukh J, Purcell R H. Springer Sem Immunopathol. 1997;19:5–26. doi: 10.1007/BF00945022. [DOI] [PubMed] [Google Scholar]
- 7.Weiner A J, Geysen H M, Christopherson C, Hall J E, Mason T J, Saracco G, Bonino F, Crawford K, Marion C D, Crawford K A, Brunetto M, Barr P J, Miyamura T, McHutchinson J, Houghton M. Proc Natl Acad Sci USA. 1992;89:3468–3472. doi: 10.1073/pnas.89.8.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato N, Ootsuyama Y, Sekiya H, Ohkoshi S, Nakazawa T, Hijikata M, Shimotohno K. J Virol. 1994;68:4776–4784. doi: 10.1128/jvi.68.8.4776-4784.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Doorn L-J, Capriles I, Maertens G, DeLeys R, Murray K, Kos T, Schellekens H, Quint W. J Virol. 1995;69:773–778. doi: 10.1128/jvi.69.2.773-778.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, Shimizu Y, Shapiro M, Alter H J, Purcell R H. Proc Natl Acad Sci USA. 1996;93:15394–15399. doi: 10.1073/pnas.93.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okada S-I, Akahane Y, Suzuki H, Okamoto H, Mishiro S. Hepatology. 1992;16:619–624. doi: 10.1002/hep.1840160302. [DOI] [PubMed] [Google Scholar]
- 12.Enomoto N, Kurosaki M, Tanaka Y, Marumo F, Sato C. J Gen Virol. 1994;75:1361–1369. doi: 10.1099/0022-1317-75-6-1361. [DOI] [PubMed] [Google Scholar]
- 13.Koizumi K, Enomoto N, Kurosaki M, Murakami T, Izumi N, Marumo F, Sato C. Hepatology. 1995;22:30–35. [PubMed] [Google Scholar]
- 14.Higashi Y, Kakumu S, Yoshioka K, Wakita T, Mizokami M, Ohba K, Ito Y, Ishikawa T, Takayanagi M, Nagai Y. Virology. 1993;197:659–668. doi: 10.1006/viro.1993.1641. [DOI] [PubMed] [Google Scholar]
- 15.Kao J-H, Chen P-J, Lai M-Y, Wang T-H, Chen D-S. J Infect Dis. 1995;172:261–264. doi: 10.1093/infdis/172.1.261. [DOI] [PubMed] [Google Scholar]
- 16.Yoshioka K, Aiyama T, Okumura A, Takayanagi M, Iwata K, Ishikawa T, Nagai Y, Kakumu S. J Infect Dis. 1997;175:505–510. doi: 10.1093/infdis/175.3.505. [DOI] [PubMed] [Google Scholar]
- 17.Smith D B, McAllister J, Casino C, Simmonds P. J Gen Virol. 1997;78:1511–1519. doi: 10.1099/0022-1317-78-7-1511. [DOI] [PubMed] [Google Scholar]
- 18.Meyerhans A, Cheynier R, Albert J, Seth M, Kwok S, Sninsky J, Morfeldt-Månson L, Asjö B, Wain-Hobson S. Cell. 1989;58:901–910. doi: 10.1016/0092-8674(89)90942-2. [DOI] [PubMed] [Google Scholar]
- 19.Balfe P, Simmonds P, Ludlam C A, Bishop J O, Leigh Brown A J. J Virol. 1990;64:6221–6233. doi: 10.1128/jvi.64.12.6221-6233.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vartanian J-P, Meyerhans A, Henry M, Wain-Hobson S. AIDS. 1992;6:1095–1098. doi: 10.1097/00002030-199210000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz D H, Viscidi R, Laeyendecker O, Song H, Ray S C, Michael N. Immunol Lett. 1996;51:3–6. doi: 10.1016/0165-2478(96)02547-3. [DOI] [PubMed] [Google Scholar]
- 22.Feinstone S M, Alter H J, Dienes H P, Shimizu Y, Popper H, Blackmore D, Sly D, London W T, Purcell R H. J Infect Dis. 1981;144:588–598. doi: 10.1093/infdis/144.6.588. [DOI] [PubMed] [Google Scholar]
- 23.Tellier R, Bukh J, Emerson S U, Miller R H, Purcell R H. J Clin Micobiol. 1996;34:3085–3091. doi: 10.1128/jcm.34.12.3085-3091.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanagi M, Purcell R H, Emerson S U, Bukh J. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimizu Y K, Iwamoto A, Hijikata M, Purcell R H. Proc Natl Acad Sci USA. 1992;89:5477–5481. doi: 10.1073/pnas.89.12.5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thanaraj T A, Pandit M W. Nucleic Acids Res. 1989;17:2973–2985. doi: 10.1093/nar/17.8.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sprengart M L, Fatscher H P, Fuchs E. Nucleic Acids Res. 1990;18:1719–1723. doi: 10.1093/nar/18.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gualerzi C O, Pon C L. Biochemistry. 1990;29:5881–5889. doi: 10.1021/bi00477a001. [DOI] [PubMed] [Google Scholar]
- 29.de Smit M H, van Duin J. J Mol Biol. 1994;235:173–184. doi: 10.1016/s0022-2836(05)80024-5. [DOI] [PubMed] [Google Scholar]
- 30.Miller J H. In: A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Miller J H, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. pp. 20.1–20.4. [Google Scholar]
- 31.Ringquist S, Shinedling S, Barrick D, Green L, Binkley J, Stormo G D, Gold L. Mol Microbiol. 1992;6:1219–1229. doi: 10.1111/j.1365-2958.1992.tb01561.x. [DOI] [PubMed] [Google Scholar]
- 32.Eggertsson G, Söll D. Microbiol Rev. 1988;52:354–374. doi: 10.1128/mr.52.3.354-374.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yi M, Nakamoto Y, Kaneko S, Yamashita T, Murakami S. Virology. 1997;231:119–129. doi: 10.1006/viro.1997.8516. [DOI] [PubMed] [Google Scholar]
- 34.Kolykhalov A A, Agapov E V, Blight K J, Mihalik K, Feinstone S M, Rice C M. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]