

Abstract
NMDA receptors (NMDARs) are critical mediators of activity-dependent synaptic plasticity, but the differential roles of NR2A- versus NR2B-containing NMDARs have been controversial. Here, we investigate the roles of NR2A and NR2B in long-term potentiation (LTP) in organotypic hippocampal slice cultures using RNA interference (RNAi) and overexpression, to complement pharmacological approaches. In young slices, when NR2B is the predominant subunit expressed, LTP is blocked by the NR2B-selective antagonist Ro25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol]. As slices mature and NR2A expression rises, activation of NR2B receptors became no longer necessary for LTP induction. LTP was blocked, however, by RNAi knockdown of NR2B, and this was rescued by coexpression of an RNAi-resistant NR2B (NR2B*) cDNA. Interestingly, a chimeric NR2B subunit in which the C-terminal cytoplasmic tail was replaced by that of NR2A failed to rescue LTP, whereas the reverse chimera, NR2A channel with NR2B tail, was able to restore LTP. Thus, expression of NR2B with its intact cytoplasmic tail is required for LTP induction, at an age when channel activity of NR2B–NMDARs is not required for LTP. Overexpression of wild-type NR2A failed to rescue LTP in neurons transfected with the NR2B–RNAi construct, despite restoring NMDA–EPSC amplitude to a similar level as NR2B*. Surprisingly, an NR2A construct lacking its entire C-terminal cytoplasmic tail regained its ability to restore LTP. Together, these data suggest that the NR2B subunit plays a critical role for LTP, presumably by recruiting relevant molecules important for LTP via its cytoplasmic tail. In contrast, NR2A is not essential for LTP, and its cytoplasmic tail seems to carry inhibitory factors for LTP.
Introduction
Long-term potentiation (LTP) is an activity-dependent form of synaptic plasticity proposed to underlie learning and memory in the brain. As best characterized at CA3–CA1 synapses of the hippocampus, conventional LTP requires calcium influx through NMDA receptors (NMDARs) (Malenka and Bear, 2004). NMDARs are tetramers composed of two NR1 subunits plus two NR2 subunits, of which there are four kinds encoded by distinct genes: NR2A, NR2B, NR2C, and NR2D (Cull-Candy et al., 2001). In CA1 pyramidal cells, NMDARs contain mainly NR2A and/or NR2B subunits in addition to NR1.
In addition to antagonists that block all NMDARs (e.g., APV), there are widely used antagonists (e.g., ifenprodil, Ro25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol]) that are selective against receptors composed of NR1 and NR2B subunits (NR2B–NMDARs) (Williams, 1993; Fischer et al., 1997). Recently, an antagonist, NVP-AAM077 ([(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4- tetrahydro-quinoxalin-5-yl)-methyl]phosphonic acid), with preference for receptors composed of NR1 and NR2A subunits (NR2A–NMDARs) (Auberson et al., 2002; Liu et al., 2004; Chen et al., 2007) has been used to probe the specific roles of NR2A–NMDAR and NR2B–NMDAR in LTP and long-term depression (LTD). Some studies in hippocampus and perirhinal cortex showed that ifenprodil and Ro25-6981 block LTD but not LTP (Liu et al., 2004; Massey et al., 2004; Fox et al., 2006; Izumi et al., 2006; Chen et al., 2007), whereas NVP-AAM077 blocks LTP but not LTD (Liu et al., 2004; Massey et al., 2004). These simple pharmacological findings suggested that activation of NR2A–NMDARs is specifically important for LTP induction, whereas NR2B–NMDARs are specifically involved in LTD induction.
However, controversy has surrounded these results. Other groups have found that LTP can be blocked or partially blocked by NR2B antagonists in cultured hippocampal slices (Barria and Malinow, 2005), acute hippocampal slices (Bartlett et al., 2007; de Marchena et al., 2008), or slices from cingulate cortex (Zhao et al., 2005). Furthermore, the relative specificity of NVP-AAM077 has come into question. Although NVP-AAM077 is reported to have a 100-fold preference for human NR2A–NMDARs over NR2B–NMDARs (Auberson et al., 2002; Liu et al., 2004), its selectivity drops to 10-fold for rodent NR2A–NMDARs (Neyton and Paoletti, 2006). At 400 nm (the concentration at which it has sometimes been used), NVP-AAM077 also significantly inhibits NR2B–NMDARs (Berberich et al., 2005; Weitlauf et al., 2005).
Here we compare molecular–genetic and pharmacological approaches in the same preparation (organotypic hippocampal slice culture) to elucidate the roles of NR2A and NR2B in LTP. Using NMDAR antagonists, we find that the NR2B dependence of LTP disappears in older slices. At this age, however, RNA interference (RNAi) suppression of NR2B still blocks LTP. This can be rescued by overexpression of RNAi-resistant NR2B but not by NR2A nor a chimeric NR2B subunit that contains the cytoplasmic tail of NR2A. Thus, for LTP induction, the physical presence of NR2B and its cytoplasmic tail are more important than the activation of NR2B–NMDARs, suggesting an essential function of NR2B as a mediator of protein interactions independent of its channel contribution. In contrast, the presence of NR2A is not essential for LTP, and, in fact, the cytoplasmic tail of NR2A seems to inhibit the induction of LTP.
Materials and Methods
Antibodies.
Antibodies used include NR2A and NR2B (gifts from A. Dunah, MassGeneral Institute for Neurodegenerative Disease, Massachusetts General Hospital and Harvard Medical School, Charlestown, MA) (described previously by Sheng et al., 1994), MAP2 (HM-2; Sigma), and NR1 (54.1; BD Pharmingen).
DNA constructs.
RNAi plasmids targeting NR2A or NR2B were constructed in pSuper vector and were described previously (Kim et al., 2005). RNAi resistant NR2B (NR2B*) was generated by mutating the NR2B RNAi target at eight different nucleotides without changing the amino acid sequence. NR2B with an NR2A C-terminal tail (NR2B*–2Atail) consists of amino acids 1-861 of NR2B* and amino acids 861-1464 of NR2A. NR2A with the C terminus deleted (NR2AΔC) consists of amino acids 1-858 of NR2A. NR2B with the C terminus deleted (NR2B*ΔC) consists of amino acids 1-861 of NR2B*. Constructs were generated by PCR. NR2A S1462A was a gift from R. Wenthold (Laboratory of Neurochemistry, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD) and has been described previously (Prybylowski et al., 2005).
Electrophysiology.
Recordings were performed in organotypic hippocampal slice cultures dissected from postnatal day 7 (P7) rats as described previously (Nakagawa et al., 2004). Recordings were made from CA1 pyramidal cells, either singly or in pairs consisting of one transfected cell and one untransfected cell. Postsynaptic responses were evoked at 0.2 Hz with a bipolar stimulus electrode placed in the stratum radiatum. Slices were perfused at room temperature, with a solution consisting of the following (in mm): 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 26 NaHCO3, 1 NaH2PO4, 11 glucose, 0.15 picrotoxin, and 0.004 2-chloroadenosine for slices days in vitro (DIV) 6–8 or 0.008 2-chloroadenosine for slices DIV 11–14 and DIV 18–21, pH 7.4 (gassed with 5% CO2/95% O2). Patch electrodes were filled with a solution consisting of the following (in mm): 115 cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 ATP disodium salt, 0.4 GTP trisodium salt, 10 sodium phosphocreatine, and 0.6 EGTA, pH 7.25. Slices were transfected using a biolistic gene gun (Bio-Rad) 3–4 d before recording. Gold particles were coated with 45 μg of RNAi construct (Kim et al., 2005) plus 45 μg of overexpression construct plus 10 μg of phosphorylated enhanced green fluorescent protein (pEGFP) or 90 μg of overexpression construct (Kim et al., 2005) plus 10 μg of pEGFP. AMPA–EPSCs were recorded at −70 mV, and NMDA–EPSCs were recorded at +40 mV in the presence of 0.01 mm NBQX. LTP was induced at room temperature by pairing postsynaptic depolarization to 0 mV with presynaptic stimulation consisting of 200 pulses at 2 Hz. The magnitude of LTP was measured as the mean of time points between 20 and 30 min after induction protocol.
Results
Developmental changes of NR2B and NR2A in hippocampal slice culture
Hippocampal slice cultures were prepared from P7 rats and cultured in standardized conditions. By immunoblotting, there was robust expression of NR2B protein throughout development in vitro; NR2B levels increased slightly from DIV 0 to approximately DIV 9–12, before subsiding to a moderate plateau at ∼2–3 weeks in culture (Fig. 1A). In contrast, levels of the NR2A subunit were very low at DIV 0, increased progressively to DIV 12, and then remained high. NR1 expression was more similar to NR2B, being highly expressed from DIV 0 to DIV 12, before dropping to a lower level in the third week in vitro. The developmental profile of expression of all three subunits resembles that seen in vivo (Sheng et al., 1994). These biochemical data suggest that NR2B-containing NMDARs are predominant at early stages, whereas NR2A-containing NMDARs become increasingly prominent with maturation of hippocampal slice cultures.
Figure 1.
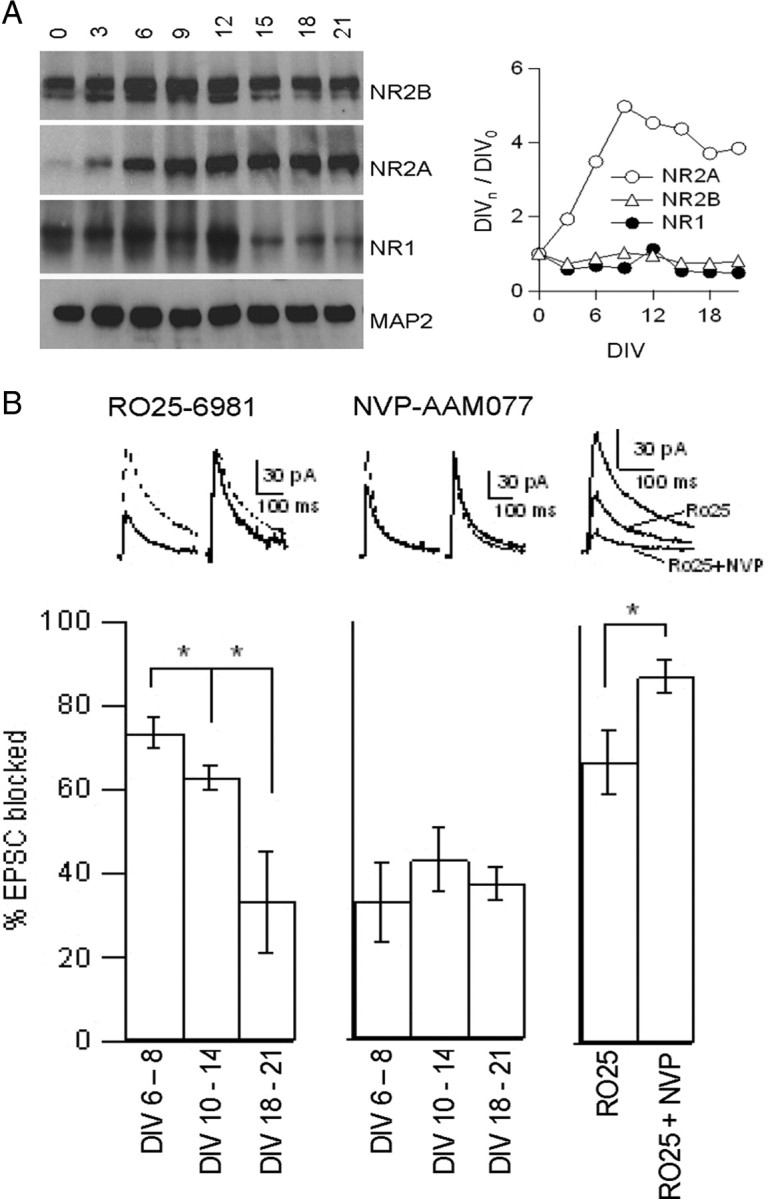
Increasing NR2A expression and falling NR2B contribution to NMDA–EPSC during development of hippocampal slice cultures. A, Immunoblot of hippocampal slices from P7 rats cultured for the indicated number of days in vitro (left). Graph showing intensity at a given DIV normalized to intensity at DIV 0 (right). B, Sample traces and histograms showing the effects of 5 μm Ro25-6981 (left), 50 nm NVP-AAM077 (middle), and 5 μm Ro25-6981 followed by 50 nm NVP-AAM077 plus 5 μm Ro25-6981 (right) on the NMDA–EPSC amplitude. Left and middle show traces from DIV 10–14 neurons taken before (dashed line) and after (solid line) drug application in both absolute value and scaled for comparison of kinetics. Right shows traces without drug (largest trace), after 5 μm Ro25-6981 (middle trace), and after 5 μm Ro25-6981 plus 50 nm NVP-AAM077 (smallest trace). Histograms show the percentage of the NMDA–EPSC amplitude that is blocked by the drugs at DIV 6–8, DIV 10–14, and DIV 18–21 (n = 12, 18, and 6, respectively, for Ro25-6981; n = 5, 5, and 6, respectively, for NVP-AAM077; n = 5 for Ro25-6981 plus NVP-AAM077.) Data are presented as mean ± SEM. *p < 0.05, Student's t test.
We next tested by electrophysiology whether there were developmental changes in properties of synaptic NMDARs that correlated with increasing NR2A/NR2B ratio. Whole-cell recordings were made from CA1 pyramidal neurons in organotypic hippocampal slice cultures, with stimulation of Schaffer collateral fibers (Fig. 1B). Example traces from DIV 10–14 neurons show that the kinetics of decay of the NMDA–EPSC were faster after application of 5 μm Ro25-6981 (solid line) than in the absence of Ro25-6981 (Fig. 1B, left, dashed line), consistent with a selective block of NR2B–NMDARs (which deactivate more slowly than NR2A receptors). Application of 5 μm Ro25-6981 blocked 74 ± 4% of NMDAR-mediated EPSCs in young slices (DIV 6–8) (Fig. 1B). The same concentration of Ro25-6981 blocked only 64 ± 3% of the NMDA–EPSC in slices at DIV 10–14 and only 34 ± 12% at DIV 18–21. This result implies that the fraction of the NMDA–EPSC carried by NR2B–NMDARs decreases with maturation of cultured slices, in line with the falling ratio of NR2B/NR2A expression levels (Fig. 1A). Even in the most mature DIV 18–21 slices, however, there is still a substantial NR2B contribution to synaptic NMDARs. The Ro25-6981 data are consistent with a decrease in the relative abundance of NR2B–NMDARs at synapses during development of cultured slices, which are similar to results found in acute slices (Kirson et al., 1999).
The possible contribution of NR2A–NMDARs to the NMDA–EPSC was probed by using the NR2A–NMDAR-preferring antagonist NVP-AAM077, which has a modest 10-fold selectivity for rat NR2A–NMDARs over NR2B–NMDARs (Neyton and Paoletti, 2006). Using a low concentration of the drug (50 nm) on DIV 10–14 slices, NVP-AAM077 decreased the NMDA–EPSC amplitude and caused a slight slowing of the decay, consistent with a preferential inhibition of NR2A–NMDARs (Fig. 1B, middle). NVP-AAM077 at 50 nm blocked NMDA–EPSCs from slices at DIV 6–8 by 33 ± 9%, similar to the amount seen by other groups (Berberich et al., 2005; Izumi et al., 2006). However, there was no significant increase in fractional block of NMDA–EPSC by 50 nm NVP-AAM007 at DIV 10–14 (43 ± 7%) or at DIV 18–21 (37 ± 4%), despite the increasing expression of NR2A. These results can be explained if (1) the accumulating NR2A subunits are mostly incorporated into triheteromeric receptors (i.e., NR2A/2B/NR1) that may be poorly blocked by NVP-AAM077 as they are by ifenprodil (Hatton and Paoletti, 2005), or (2) if NVP-AAM077 is not highly specific for NR2A–NMDARs. Even in the latter case, it would seem unlikely that 50 nm NVP-AAM077 blocked NR2B–NMDARs to a large extent.
To further test the specificity of the antagonists at the concentrations used here, we compared the effect of 5 μm Ro25-6981 versus a combination of 5 μm Ro25-6981 plus 50 nm NVP-AAM077. If the two drugs are acting on separate populations of receptors, then their effects should be additive. In DIV 10–14 slices, 67 ± 8% of the NMDA–EPSC was blocked by the initial application of 5 μm Ro25-6981. The degree of NMDA–EPSC block rose to 87 ± 4% when 50 nm NVP-AAM077 was applied in addition (Fig. 1B, right). The amount of current blocked by 50 nm NVP-AAM077 alone was 43 ± 7% (Fig. 1B, middle). The significant additive effect implies that Ro25-6981 and NVP-AAM077 act on NMDAR populations that are not completely overlapping.
Effect of Ro25-6981 on LTP
We investigated the effect of the NR2B antagonist Ro25-6981 on LTP at two different developmental stages in organotypic hippocampal slice development. At DIV 6–8, control slices showed an approximately twofold potentiation of AMPA–EPSC after a pairing protocol for inducing LTP (see Materials and Methods) (Fig. 2A). This LTP was almost completely blocked by the NR2B antagonist Ro25-6981 (5 μm) in experiments interleaved with control experiments. However, older slices (DIV 11–14) showed LTP that was completely resistant to Ro25-6981 treatment (Fig. 2B). Thus, at this later developmental stage, LTP no longer requires activation of NR2B receptors. This switch occurs at a point when the fractional contribution of NR2B–NMDARs to the NMDA–EPSC has dropped from ∼74 to ∼64%, corresponding to an ∼70% increase in the number of non-NR2B receptors (Fig. 1B).
Figure 2.

Effect of Ro25-6981 on LTP in cultured hippocampal slices over development. A, B, Time course of LTP in control conditions (open circles) and in the presence of 5 μm Ro25-6981 (filled circles) at DIV 6–8 (A; n = 10 for control; n = 6 for Ro25-6981; p < 0.05) and DIV 11–14 (B; n = 12 for control; n = 9 for Ro25-6981; p = 0.86) (Wilcoxon's test at 20–30 min) Insets show example averaged EPSCs taken before and 30 min after pairing for control cells (left, open circle) and cells in the presence of Ro25-6981 (right, filled circle).
NR2B Expression is required for LTP
Although pharmacological antagonists indicate which receptor subtypes need to conduct current for induction of LTP, molecular manipulation can indicate which subunits or parts of subunits need to be physically present. Thus, to complement the pharmacologic experiments, we used overexpression and RNAi knockdown of NR2 subunits for 3–4 d to assess the specific roles of NR2A versus NR2B in LTP. These are relatively acute manipulations compared with germ-line genetics and should be less likely to induce compensatory mechanisms.
We previously developed an RNAi construct that specifically suppresses expression of NR2A in heterologous cells and in cultured hippocampal neurons, without affecting NR2B (Kim et al., 2005). We had also developed several specific RNAi constructs against NR2B that decrease NR2B expression without affecting NR2A expression in HEK293 cells. However, all of these NR2B–RNAi constructs inhibit NR2A expression in neurons, perhaps because NR2B–NMDARs are required to support NR2A expression (Kim et al., 2005). This problem could be circumvented by overexpressing NR2A together with NR2B–RNAi (NR2B–RNAi plus NR2A) in neurons, which results in NR2A-dominant NMDARs (Kim et al., 2005) (see below).
RNAi and overexpression constructs of NR2 subunits were transfected into CA1 pyramidal neurons of hippocampal slice cultures. To assess the effects of these constructs on the composition of synaptic NMDARs, we measured the amplitudes and kinetics of NMDA–EPSCs in transfected neurons compared with neighboring untransfected cells in the same slice.
NR2B–RNAi alone caused a drastic reduction of NMDA–EPSC amplitude (to 25 ± 2% of control) (Fig. 3Aa). This marked loss of NMDA–EPSC by NR2B–RNAi appeared to result from loss of not only NR2B but also NR2A–NMDARs, because the half-width of NMDA–EPSCs was only slightly reduced (0.9 ± 0.05) (Fig. 3Ab, p < 0.05). These results are consistent with our previous immunostaining data showing that NR2B–RNAi reduces expression of both NR2A and NR2B subunits at synapses (Kim et al., 2005). Our findings are also consistent with a recent report that a prolonged (6–8 d) exposure to NR2B–RNAi virtually eliminated total NMDA–EPSCs in dissociated cortical neurons (Hall et al., 2007).
Figure 3.

Effect of altering NR2B expression on LTP. A, Histograms showing the average ratio of the NMDA–EPSC (n = 13, 14, and 6) (a), the ratio of the half-width (HW) (n = 13, 14, and 6) (b), and the ratio of the AMPA–EPSC in transfected cells to that of untransfected cells (n = 11, 13, and 10) (c), respectively, for NR2B–RNAi, NR2B* plus NR2B–RNAi, and NR2B). B–D, Time course of LTP for untransfected cells (open circles) and neighboring cells transfected with NR2B–RNAi (n = 10; p < 0.04) (B), NR2B* plus NR2B–RNAi (n = 6; p = 0.7) (C), or NR2B (n = 6; p = 0.9) (D) (Wilcoxon's test at 20–30 min). Insets show example averaged EPSCs taken before and 30 min after pairing. *p < 0.05.
Cells transfected with NR2B–RNAi showed no significant change in AMPA–EPSC amplitude (1.1 ± 0.2) (Fig. 3Ac), but the magnitude of LTP was greatly diminished compared with untransfected cells (1.0 ± 0.1-fold, NR2B–RNAi; 1.7 ± 0.4-fold, control) (Fig. 3B). The loss of LTP is not surprising given the profound inhibition of NMDA–EPSC amplitude in neurons transfected with NR2B–RNAi (Fig. 3A).
The effects of NR2B–RNAi could be largely rescued by an RNAi-resistant NR2B cDNA (NR2B*), in which silent mutations were introduced into the RNAi targeted site. When NR2B* was cotransfected with NR2B–RNAi, the NMDA–EPSC amplitude was restored to 76 ± 18% of control (untransfected cell) amplitude (Fig. 3Aa). Moreover, the half-width of NMDA–EPSC was increased relative to NR2B–RNAi alone (1.1 ± 0.06) (Fig. 3Ab). Thus, the effect of NR2B–RNAi on the NMDA–EPSC amplitude was largely rescued by the NR2B* construct. The mean basal AMPA–EPSC amplitude in NR2B* plus NR2B–RNAi-transfected cells was similar to untransfected cells (1.1 ± 0.3) (Fig. 3Ac). Importantly, coexpression of NR2B* restored LTP to normal magnitude (1.5 ± 0.1-fold, NR2B* plus NR2B–RNAi; 1.6 ± 0.3-fold, untransfected) (Fig. 3C).
As an additional control, we overexpressed NR2B in the absence of any RNAi. Cells expressing NR2B displayed an NMDA–EPSC that was not statistically different from neighboring untransfected neurons (0.9 ± 0.2) (Fig. 3Aa). The half-width of transfected cells was similarly unaffected (0.9 ± 0.01) (Fig. 3Ab). These results are consistent with previous findings indicating that, although exogenously expressed NR2B receptors are incorporated into synapses, it appears to be at the expense of endogenous NR2B receptors, because the total NR2B content of the synapse does not change (Barria and Malinow, 2002). LTP in NR2B transfected cells was unchanged (1.8 ± 0.5-fold, NR2B; 2.1 ± 0.6-fold, untransfected) (Fig. 3D).
NR2A expression is not required for LTP
We next examined the effects of altered NR2A expression on LTP. In neurons transfected with an NR2A–RNAi construct, the amplitude of the NMDA–EPSC was reduced to 0.71 ± 0.05 that of untransfected neighboring cells (Fig. 4Aa). The half-width of NMDA–EPSC in transfected cells was increased relative to untransfected cells by 1.4 ± 0.8-fold (Fig. 4Ab); the slowed kinetics of deactivation is consistent with a reduction in the fraction of NR2A–NMDARs at synapses. RNAi knockdown of NR2A had little effect on basal AMPA–EPSC amplitude relative to untransfected cells (0.94 ± 0.1) (Fig. 4Ac). Despite that the mean NMDA–EPSC amplitude was inhibited ∼30% by NR2A–RNAi (Fig. 4A), LTP was still robustly inducible in NR2A–RNAi cells by the pairing protocol (Fig. 4B). In fact, the magnitude of LTP showed a tendency to be higher when NR2A was suppressed by RNAi compared with untransfected cells (2.2 ± 0.3-fold, NR2A–RNAi; 1.7 ± 0.2-fold, control) (Fig. 4B). These data indicate that induction of LTP is not impaired by a substantial reduction of synaptic NR2A.
Figure 4.

Effect of altering NR2A expression on LTP. A, Histograms showing the average ratio of the NMDA–EPSC (n = 21, 6, and 6) (a), the ratio of the half-width (HW) (n = 21, 6, and 6) (b), and the ratio of the AMPA–EPSC in transfected cells to that of untransfected cells (n = 13, 13, and 10) (c), respectively, for NR2A–RNAi, NRB RNAi plus NR2A, or NR2A. B–D, Time course of LTP for untransfected cells (open circles) and neighboring cells transfected with NR2A–RNAi (n = 11; p = 0.2) (B), NR2A plus NR2B–RNAi (n = 8; p < 0.007) (C), and NR2A (n = 7; p < 0.05) (D) (Wilcoxon's test at 20–30 min). Insets show example averaged EPSCs taken before and 30 min after pairing. *p < 0.05.
We next investigated the effects of replacing the endogenous NR2 subunits with NR2A. Overexpression of NR2A with NR2B–RNAi restored NMDA–EPSC amplitude to a level similar to that observed with cotransfection of NR2B* (0.75 ± 0.05 of untransfected cells) (Fig. 4Aa). NMDA–EPSCs showed a greatly reduced half-width relative to control cells or cells transfected with NR2B–RNAi alone (0.5 ± 0.02) (Fig. 4Ab), indicating a much higher fraction of current being carried by NR2A-containing channels. The basal AMPA–EPSC amplitude was slightly but significantly reduced to 0.81 ± 0.1 relative to untransfected cells (Fig. 4Ac). However, unlike coexpression of NR2B* (Fig. 3C), coexpression of NR2A failed to rescue LTP in cells transfected with NR2B–RNAi (1.1 ± 0.1-fold, NR2A plus NR2B–RNAi; 1.9 ± 0.2-fold, control) (Fig. 4C), although the mean NMDA–EPSC amplitude was no more impaired than by NR2A–RNAi alone (Fig. 4Aa), application of 5 μm Ro25-6981 (Fig. 1B), or by NR2B* coexpression (Fig. 3Aa), all of which had no effect on LTP.
Although overexpression of NR2B alone has been shown to have no effect on the NR2A/NR2B ratio at synapses, overexpression of NR2A alone significantly increases this ratio (Barria and Malinow, 2002). We therefore examined the effects of overexpressing NR2A in the absence of NR2B–RNAi. Cells overexpressing NR2A alone displayed an increased NMDA–EPSC (1.6 ± 0.15) (Fig. 4Aa) and a decreased half-width (0.7 ± 0.02) (Fig. 4Ab) relative to untransfected cells. These cells also displayed a decreased AMPA–EPSC amplitude relative to untransfected cells (0.8 ± 0.1) (Fig. 4Ac). Like cells transfected with NR2B–RNAi plus NR2A, cells transfected with NR2A alone displayed diminished LTP (1.2 ± 0.3-fold, NR2A; 1.9 ± 0.3-fold, untransfected) (Fig. 4D) despite the increased NMDA conductance. In summary, in DIV 11–14 slice cultures, LTP induction requires the normal expression of NR2B but not of NR2A. Together, the data indicate that NR2B expression is important for LTP and cannot be substituted by NR2A.
The NR2B cytoplasmic tail is important for LTP
It is intriguing that RNAi knockdown of NR2B together with NR2A overexpression [which results in NR2A-predominant synaptic NMDA receptors (Fig. 4A)] prevented LTP in DIV 11–14 slices, whereas pharmacological blockade of NR2B–NMDAR channels at the same age did not (Fig. 2B). This apparent discrepancy could be explained if the NR2B subunit needs to be present structurally at synapses for LTP to be induced, but the opening of NR2B–NMDAR channels is not absolutely necessary to induce LTP. The C-terminal cytoplasmic tail of NR2B, which represents almost half of the length of the protein, could provide a scaffold for recruiting critical molecules to the synapse (Barria and Malinow, 2005). This scaffolding function would be suppressed by RNAi but not by drug antagonists. To explore this idea further, we generated two chimeric constructs: (1) an RNAi-resistant NR2B construct (termed NR2B*–2Atail), in which the entire C-terminal cytoplasmic tail of NR2B* is replaced with that of NR2A; (2) the converse chimera (NR2A–2Btail), which contains the entire N-terminal extracellular regions and channel-forming transmembrane portions of NR2A fused to the entire cytoplasmic tail of NR2B (NR2A–2Btail).
In neurons transfected with NR2B–RNAi, the NR2B*–2Atail chimera rescued the basal NMDA–EPSC amplitude to 65 ± 16% of untransfected neurons (Fig. 5Aa), which is indistinguishable from the effectiveness of wild-type NR2B* (Fig. 3Aa). Additionally, cells transfected with NR2B*–NR2Atail plus NR2B–RNAi displayed a slower half-width than controls (1.2 ± 0.1) (Fig. 5Ab), also similar to NR2B* (Fig. 3Ab). In contrast to the wild-type NR2B*, however, NR2B*–2Atail failed to restore LTP induction in neurons transfected with NR2B–RNAi (1.1 ± 0.3-fold, NR2B*–NR2Atail plus NR2B–RNAi; 2.2 ± 0.6-fold, untransfected controls) (Fig. 5B). Baseline AMPA–EPSC was not significantly affected by NR2B*–2Atail (Fig. 5Ac). These data indicate that the NR2A tail cannot support LTP.
Figure 5.

Effect of the of NR2A and NR2B chimeric constructs on LTP. A, Histograms showing the average ratio of the NMDA–EPSC (n = 11, 5, and 7) (a) and the ratio of the half-widths (HW) (n = 11, 5, and 7) (b) in transfected and untransfected cells for NR2B–RNAi alone and NR2B–RNAi plus NR2B*–NR2Atail or NR2A–NR2Btail, the ratio of the AMPA–EPSC (n = 13 and 7) (c), respectively, for NR2B–RNAi plus NR2B*–NR2Atail or NR2A- NR2Btail. B, C, The time course of LTP in untransfected cells (open circles) and neighboring cells transfected with NR2B–RNAi plus either NR2B*–NR2Atail (n = 7; p < 0.05) (B) or NR2A–NR2Btail (n = 7; p = 0.7) (C) (Wilcoxon's test at 20–30 min).*p < 0.05, **p < 0.01.
The converse chimera NR2A–NR2Btail rescued the NMDA–EPSC amplitude to 0.6 ± 0.2-fold control (Fig. 5Aa) and significantly decreased the half-width of the NMDA–EPSC (0.5 ± 0.1) (Fig. 5Ab). In contrast to NR2B*–2Atail, however, the NR2A–2Btail construct was able to restore LTP (1.7 ± 0.4-fold, NR2A–NR2Btail; 1.8 ± 0.2-fold, control) (Fig. 5C), with no effect on the basal AMPA–EPSC (1.1 ± 0.3) (Fig. 5Ac). Thus, an intact NR2B cytoplasmic tail is required for NR2B to support LTP, and this cannot be substituted by the tail of NR2A. In contrast, the extracellular and channel-forming portion of either 2A or 2B is able to support LTP when fused to the tail of NR2B.
To further explore the roles of the C-terminal tails of NR2A and NR2B in LTP, we constructed C-terminal deletions. We began by making an NR2B* construct lacking the entire C-terminal tail after the fourth transmembrane domain (NR2B*ΔC). This construct did not significantly alter the AMPA–EPSC amplitude (Fig. 6Ac). Coexpression of NR2B*ΔC with NR2B–RNAi rescued the NMDA–EPSC to 80 ± 7% of control untransfected cells (Fig. 6Aa). Despite restoring the NMDA–EPSC to such a degree, the NR2BΔC construct was unable to rescue LTP (1.0 ± 0.2-fold, NR2B*ΔC plus NR2B–RNAi; 1.8 ± 0.4-fold, untransfected controls) (Fig. 6B), consistent with the idea that the NR2B tail is critical for LTP induction.
Figure 6.

Effect of removing the C-terminal tails of NR2A or NR2B on LTP. A, Histograms showing the average ratio of the NMDA–EPSC (n = 11, 10, and 6) (a) and the ratio of the half-widths (HW) (n = 11, 10, and 6) (b) in transfected and untransfected cells for NR2B–RNAi alone and NR2B–RNAi plus NR2AΔC or NR2B*ΔC, the ratio of the AMPA–EPSC (n = 15 and 9) (c), respectively, for NR2B–RNAi plus NR2AΔC or NR2B*ΔC. B, C, The time course of LTP in untransfected cells (open circles) and neighboring cells transfected with NR2B–RNAi plus either NR2B*ΔC (n = 6; p < 0.05) (B) or NR2AΔC (n = 10; p = 0.1) (C) (Wilcoxon's test at 20–30 min). *p < 0.05, **p < 0.01.
We next created an NR2A truncation mutant that lacks its entire C-terminal tail after the fourth transmembrane domain (NR2AΔC). This construct had no significant effect on baseline AMPA–EPSC (Fig. 6Ac). In neurons transfected with NR2B–RNAi, coexpression of the NR2AΔC construct restored the basal NMDA–EPSC amplitude to 56 ± 11% of untransfected neighboring controls (Fig. 6Aa), which is lower than the level reached with coexpression of wild-type NR2A. Remarkably, however, this tail-less NR2A construct fully rescued LTP (2.3 ± 0.8-fold, NR2AΔC; 1.6 ± 0.4-fold, control) (Fig. 6C), whereas wild-type NR2A was unable to do so (Fig. 4C). This surprising result implies that the C-terminal tail of NR2A carries a determinant(s) that inhibits induction of LTP.
We hypothesized that the NR2A tail binds to and recruits a protein that antagonizes LTP. To narrow down the area of the tail in which this protein might be binding, we constructed an NR2A mutant lacking the last C-terminal one-third of the cytoplasmic tail from amino acid 1204 to the end (NR2AΔC1204). This NR2A truncation mutant restored the NMDA–EPSC to 0.8 ± 0.18-fold of control. Similarly to NR2AΔC, NR2AΔC1204 was able to restore LTP in cells cotransfected with NR2B–RNAi (1.7 ± 0.2-fold, NR2AΔC1204; 1.8 ± 0.1-fold, control) (Fig. 7B), and it did so despite significantly increasing the basal AMPA–EPSC amplitude (1.9 ± 0.3-fold, control) (Fig. 7Ac). These results are consistent with the idea that an inhibitory molecule for synaptic potentiation may bind to the C-terminal one-third of the NR2A tail.
Figure 7.

Effect of mutations in the C-terminal tail of NR2A on LTP. A, Histograms showing the average ratio of the NMDA–EPSC (n = 11, 9, and 8) (a) and the ratio of the half-widths (HW) (n = 11, 9, and 8) (b) in transfected and untransfected cells for NR2B–RNAi alone and NR2B–RNAi plus NR2AΔC1204 or NR2A–S1462A), the ratio of the AMPA–EPSC (n = 9 and 15) (c), respectively, for NR2B–RNAi plus NR2AΔC1204 or NR2A–S1462A. B, C, The time course of LTP in untransfected cells (open circles) and neighboring cells transfected with NR2B–RNAi plus either NR2AΔC1204 (n = 7; p < 0.9) (B) or NR2A–S1462A (n = 7; p < 0.05) (C) (Wilcoxon's test at 20–30 min). *p < 0.05, **p < 0.01.
An attractive possibility is that this inhibitory protein interacts with the very C terminus of NR2A, which terminates in a PDZ (postsynaptic density-95/Discs large/zona occludens-1) binding sequence with affinity for PSD-95 family proteins. However, an NR2A construct with a point mutation in the extreme C-terminal PDZ binding motif (NR2A–S1462A) failed to restore LTP in cells cotransfected with NR2B–RNAi (1.1 ± 0.3-fold, NR2A-S1462A plus NR2B–RNAi; 1.9 ± 0.4-fold, control) (Fig. 7C), although it boosted NMDA–EPSC to levels similar to NR2AΔC and also had no significant effect on baseline AMPA–EPSCs (Fig. 7Ac). This finding argues that the putative inhibitory factor recruited by NR2A likely binds to the last one-third of the cytoplasmic tail, but it is not PSD-95 or a PSD-95-related protein.
Discussion
The development of the cultured hippocampal slice mimics that of the hippocampus in vivo in terms of increasing NR2A/NR2B ratio and decreasing abundance of synaptic NR2B–NMDARs. Our pharmacological results show that there is a developmental shift in the subunit dependence of LTP induction that correlates with the developmental increase in NR2A. Slices start from an initial phase when LTP induction requires activation of NR2B receptor channels, to a mature state when activation of NR2B–NMDARs become dispensable, presumably because NR2A-containing NMDARs take over more of the synaptic NMDAR function.
By using both pharmacological and molecular–genetic approaches in the same preparation, we discovered an interesting paradox. In DIV 11–14 slices, the RNAi data imply that NR2B protein expression is required for LTP, yet the Ro25-6981 data in the same-age slices demonstrate that activation of NR2B–NMDARs is not required for LTP. The apparent contradiction can be explained by the fact that NR2B, in addition to forming part of a ligand-gated channel, also has a long cytoplasmic tail that binds (directly or indirectly) to a variety of postsynaptic signaling molecules (Köhr et al., 2003; Kim and Sheng, 2004; Barria and Malinow, 2005; Al-Hallaq et al., 2007). On the one hand, pharmacological antagonists inform about which receptor subtypes need to be activated for induction of LTP, speaking to the channel or ion conducting role of the subunit. On the other hand, RNAi suppresses protein expression and informs about which subunit needs to be physically present. Together, the pharmacological and molecular–genetic data argue that NR2B plays a structural role in LTP that appears to be more critical than its channel role, insofar as LTP is inducible when NR2B receptors are blocked but not when NR2B expression is suppressed.
If indeed it is the tail of NR2B that is important for LTP and not the channel, then it should be possible to induce LTP in the absence of the NR2B channel as long as the C-terminal tail is present. Indeed, an NR2A–NR2Btail chimera restored LTP when neither the converse construct (NR2B*–NR2A) nor wild-type NR2A could, indicating that the identity of the channel portion of the receptor is not what controls LTP but rather that the tail portion is critical. These experiments provide additional evidence that the structural role of the different subunits shapes the type of plasticity mediated by NMDA receptor subtypes, beyond the contribution of the subunits to the channel function of NMDA receptors.
Our results are consistent with Barria and Malinow (2005) who found that a segment of the tail of NR2B that binds to CaMKII is required for LTP. Our findings raise the possibility that, in mature synapses in which NR2A and NR2B coexist, the two NMDAR subunits cooperate to induce LTP–NR2A as part of the conducting channel and NR2B as a structural scaffold for recruiting proteins important for LTP. Our data cannot distinguish whether these collaborating NR2A and NR2B subunits are in the same (triheteromeric) NMDAR complex or in different NMDARs (NR2A–NMDARs and NR2B–NMDARs) that lie near each other.
We emphasize here the bipartite functions of NR2B as channel and scaffold for signaling molecules because it can offer a potential reconciliation between pharmacological experiments that suggest that NR2B–NMDARs are not important for LTP (Liu et al., 2004; Massey et al., 2004; Izumi et al., 2006) and genetic experiments that suggest that a higher NR2B/NR2A ratio is more favorable for LTP. For instance, LTP is increased in the NR2B transgenic mouse (Tang et al., 1999), perhaps because the greater abundance of NR2B in synapses promotes the recruitment of NR2B-binding proteins such as CaMKII (Leonard et al., 1999; Strack et al., 2000; Mayadevi et al., 2002; Barria and Malinow, 2005). A similar dissociation between the channel and “structural” roles of NMDARs has been observed in assays of spine stability, in which the C-terminal tail of NR1 is crucially important but blocking NMDARs pharmacologically has no effect (Alvarez et al., 2007).
Unlike overexpression of NR2B*, overexpression of wild-type NR2A could not rescue LTP in neurons transfected with NR2B–RNAi. Strikingly, however, a tail-less deletion mutant of NR2A (NR2AΔC) gained the ability to rescue LTP, although it restored NMDA–EPSC amplitudes less effectively than wild-type NR2A (Figs. 4Aa, 6Aa). The simplest explanation of this result is that the NR2A tail also plays a structural role, but, in contrast to that of NR2B, the cytoplasmic tail of NR2A recruits a negative regulator of LTP to the synapse.
If NR2B is essential for LTP, why does NR2AΔC overexpression rescue LTP in cells transfected with NR2B–RNAi? NR2B–RNAi leaves ∼25% of endogenous NMDAR function intact (Fig. 3A) including NR2B; in other words, NR2B–RNAi is not completely effective in suppressing NR2B expression, which is not unexpected. In this context, restoring the NMDA–EPSC with NR2AΔC is sufficient to rescue LTP because the remaining endogenous NR2B provides enough structural function. However, full-length NR2A does not restore LTP, presumably because of recruitment of the NR2A-associated negative regulator.
What might be the putative LTP-inhibitory protein that binds to the NR2A tail? An immediate candidate would be PSD-95, which binds to the C terminus of NR2A (Kornau et al., 1995; Niethammer et al., 1996). Indeed, the genetic disruption of PSD-95 has been shown to enhance LTP and impair LTD, and mutations in PSD-95 can specifically block LTD (Migaud et al., 1998; Kim et al., 2007; Xu et al., 2008). However, a point mutant of NR2A (NR2A–S1462A), which should be unable to bind to PSD-95 family PDZ proteins (Prybylowski et al., 2005), was also unable to rescue LTP, suggesting that the negative determinants for LTP lie upstream of the PDZ binding motif of NR2A. Additional truncation of the C-terminal one-third of the NR2A tail did restore LTP, suggesting that the suppressing factor binds to this region. Proof of this hypothesis will first require identification of the protein(s) that interact with the cytoplasmic tail of NR2A. The inhibitory role of the NR2A tail in LTP is consistent with the trend toward larger magnitude of LTP that we observed with RNAi knockdown of NR2A.
We hypothesize that the NR2A subunit also has a dual function. On one hand, it acts as a channel that facilitates LTP by conducting calcium. On the other hand, it acts as a scaffold that presumably recruits a protein to the synapse that inhibits LTP. Such a protein could act by antagonizing the activation of LTP signaling pathways [e.g., synaptic Ras-GTPase-activating protein (Kim et al., 2005)] or by stimulating the LTD signaling pathways [such as Rap and p38 mitogen-activated protein kinase) (Thomas and Huganir, 2004; Zhu et al., 2005; Li et al., 2006)].
What could be alternative explanations for the behavior of our NR2A tail mutants in LTP induction? Electrophysiological data suggest that NR2A-containing receptors are localized more synaptically, whereas NR2B-containing receptors are localized more perisynaptically or extrasynaptically (Stocca and Vicini, 1998; Tovar and Westbrook, 1999; Townsend et al., 2003), although there is no doubt that both subtypes contribute to synaptic NMDA receptor currents. It is possible that the NR2A constructs with truncated tails are less restricted to the synapse than the full-length NR2A as a result of loss of anchoring to PSD-95. If straying more extrasynaptically, these truncation mutants may have greater effect on opening of L-type Ca2+ channels, which may facilitate LTP (Grover and Teyler, 1990; Zhao et al., 2006). Arguing against this idea is the NR2A–S1462A mutant, which should not interact with PSD-95 (Lim et al., 2002; Prybylowski et al., 2005) and thus act similarly to the other NR2A truncation mutants, also does not restore LTP. However, we cannot exclude the possibility that NR2A–S1462A may localize more synaptically than the truncation mutants. Additional investigation of the exact mechanism by which the tail of NR2A interferes with LTP using additional mutants and investigation of interacting proteins will be necessary.
The result of the opposing actions of NR2A and NR2B tails on LTP inducibility is that the ratio of NR2A/NR2B protein expression becomes an important factor for determining synaptic plasticity. Higher relative levels of NR2B should favor induction of LTP, whereas increasing relative levels of NR2A should act against it. Consistent with this idea, changes in plasticity in the visual cortex are correlated with a change in the NR2A/NR2B ratio. Light deprivation lowers the threshold of induction for LTP and is associated with a decrease in synaptic NR2A (Quinlan et al., 1999a,b; Philpot et al., 2001, 2007). Also, during postnatal maturation of the brain, the ability to induce LTP wanes, and this change occurs concomitantly with a decrease in the fraction of NR2B-containing NMDARs as evidenced by a decreased ifenprodil sensitivity (Barth and Malenka, 2001). The developmental change in NMDA–EPSC kinetics, however, occurs after the decline of LTP, suggesting that the change in plasticity might be attributable to the different structural features of NR2A and NR2B subunit tails rather than, or in addition to, their distinct channel properties.
Footnotes
This work was supported by National Institutes of Health Grants F32NS055495-01 (K.A.F.) and MH076936 (M.S.). M.S. was an Investigator of the Howard Hughes Medical Institute.
References
- Al-Hallaq RA, Conrads TP, Veenstra TD, Wenthold RJ. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci. 2007;27:8334–8343. doi: 10.1523/JNEUROSCI.2155-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez VA, Ridenour DA, Sabatini BL. Distinct structural and ionotropic roles of NMDA receptors in controlling spine and synapse stability. J Neurosci. 2007;27:7365–7376. doi: 10.1523/JNEUROSCI.0956-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4:235–236. doi: 10.1038/85070. [DOI] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52:60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype specificity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, He S, Hu XL, Yu J, Zhou Y, Zheng J, Zhang S, Zhang C, Duan WH, Xiong ZQ. Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent brain-derived neurotrophic factor gene regulation and limbic epileptogenesis. J Neurosci. 2007;27:542–552. doi: 10.1523/JNEUROSCI.3607-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- de Marchena J, Roberts AC, Middlebrooks PG, Valakh V, Yashiro K, Wilfley LR, Philpot BD. NMDA receptor antagonists reveal age-dependent differences in the properties of visual cortical plasticity. J Neurophysiol. 2008;100:1936–1948. doi: 10.1152/jn.90290.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Fox CJ, Russell KI, Wang YT, Christie BR. Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus. 2006;16:907–915. doi: 10.1002/hipo.20230. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Hall BJ, Ripley B, Ghosh A. NR2B signaling regulates the development of synaptic AMPA receptor current. J Neurosci. 2007;27:13446–13456. doi: 10.1523/JNEUROSCI.3793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005;46:261–274. doi: 10.1016/j.neuron.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Auberson YP, Zorumski CF. Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J Neurosci. 2006;26:7181–7188. doi: 10.1523/JNEUROSCI.1258-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kirson ED, Schirra C, Konnerth A, Yaari Y. Early postnatal switch in magnesium sensitivity of NMDA receptors in rat CA1 pyramidal cells. J Physiol. 1999;521:99–111. doi: 10.1111/j.1469-7793.1999.00099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby Ø. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tian X, Hartley DM, Feig LA. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci. 2006;26:1721–1729. doi: 10.1523/JNEUROSCI.3990-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayadevi M, Praseeda M, Kumar KS, Omkumar RV. Sequence determinants on the NR2A and NR2B subunits of NMDA receptor responsible for specificity of phosphorylation by CaMKII. Biochim Biophys Acta. 2002;1598:40–45. doi: 10.1016/s0167-4838(02)00315-1. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M. Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron. 2004;44:453–467. doi: 10.1016/j.neuron.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–169. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-d-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A. 1999a;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999b;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Yoshii A, Mishina M, Constantine-Paton M. Developmental loss of miniature N-methyl-d-aspartate receptor currents in NR2A knockout mice. Proc Natl Acad Sci U S A. 2003;100:1340–1345. doi: 10.1073/pnas.0335786100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitlauf C, Honse Y, Auberson YP, Mishina M, Lovinger DM, Winder DG. Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci. 2005;25:8386–8390. doi: 10.1523/JNEUROSCI.2388-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JP, Phillips MA, Constantine-Paton M. Long-term potentiation in the juvenile superior colliculus requires simultaneous activation of NMDA receptors and L-type Ca2+ channels and reflects addition of newly functional synapses. J Neurosci. 2006;26:12647–12655. doi: 10.1523/JNEUROSCI.3678-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, Velamoor V, Auberson YP, Osten P, van Aelst L, Sheng M, Zhu JJ. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]