

Abstract
Introduction and Design
The introduction of the epidermal growth factor receptor inhibitors (EGFR-I) has increased the treatment options available for patients with metastatic colorectal cancer (mCRC). Two EGFR-I agents currently approved for the treatment of mCRC are the fully human monoclonal antibody panitumumab and the mouse-human chimeric monoclonal antibody cetuximab. While these agents have demonstrated activity across multiple lines of therapy, early studies suggested that clinical benefit was confined to a subset of patients treated. Mutation of the KRAS oncogene has emerged as a powerful negative predictive biomarker to identify patients with mCRC who do not benefit from EGFR-I therapy. Multiple retrospective analyses have demonstrated that clinical benefit from treatment with EGFR-I is limited to patients with tumors harboring the wild-type KRAS gene. In this review, the KRAS pathway and studies evaluating KRAS as a prognostic marker in CRC are discussed along with advances in KRAS gene mutation testing. Clinical trials evaluating the role of KRAS status in response to EGFR-I monotherapy or in combination with chemotherapy are also highlighted along with ongoing studies evaluating the role of EGFR-I treatment on curative resections rates.
Results and Conclusion
Future studies investigating EGFR-I therapy in mCRC should incorporate KRAS mutation testing into the study protocol in order to more accurately determine the patient population that will obtain clinical benefit from these novel agents.
Keywords: Bevacizumab, Cetuximab, KRAS Mutation, Panitumumab, Epidermal Growth Factor Receptor Inhibitor
Colorectal cancer (CRC) remains the fourth leading cause of cancer diagnosis and the second leading cause of cancer-related deaths in the United States.1 Treatment of patients with metastatic colorectal cancer (mCRC) has dramatically changed over the last decade. A marked advance in the treatment of patients with mCRC is represented by the monoclonal antibody epidermal growth factor receptor inhibitors (EGFR-I), including the fully human monoclonal antibody panitumumab and the mouse-human chimeric monoclonal antibody cetuximab. The small molecule inhibitors of the EGFR tyrosine kinase domain, erlotinib and gefitinib, have demonstrated activity in non-small-cell lung cancer but have not demonstrated a clinically important benefit in patients with mCRC.2,3 Both of the monoclonal antibody EGFR-I are approved for use in patients with mCRC as monotherapy, and cetuximab is also approved in combination with irinotecan.4,5 Studies with EGFR-I demonstrate that a select group of patients with mCRC exhibit clinical benefit, with response rates of approximately 10% observed across several large EGFR-I monotherapy clinical trials.6–8 Despite a lack of myelosuppression, EGFR-I therapy is associated with marked adverse effects, including skin rash, diarrhea, and hypomagnesemia.9,10 To improve quality of life and patient clinical outcomes, the selection of patients who would benefit from EGFR-I is of paramount importance, and testing of KRAS may help to improve selection of these patients.
K-ras (OMIM 190070) is a member of the Ras family of small G proteins involved in intracellular signaling.11 Activating mutations in KRAS results in the constitutive activation of downstream signaling pathways and confers resistance to inhibition of cell surface receptor tyrosine kinases, including EGFR.12
Several studies have evaluated the role of KRAS mutation as both a prognostic and predictive marker.13–27 Prognostic markers provide information on the outcome of the patient irrespective of the therapeutic intervention, while predictive markers are specific to the therapy administered to the patient. KRAS mutation occurs early in CRC carcinogenesis and was observed in 27–43% of patients with CRC (Table 1).13–19 Several older studies suggest that KRAS mutation is prognostic in CRC patients.20,21 However, recent studies continue to question the prognostic value of KRAS in mCRC.22,23
Table 1.
Incidence and rate of response of KRAS mutation within selected studies evaluating KRAS as a predictive biomarker to epidermal growth factor receptor inhibitor therapy
| Treatment | N | KRAS WT N (%) | KRAS MT N (%) | RR% KRAS WT (%) | RR% KRAS MT (%) | References |
|---|---|---|---|---|---|---|
| Cetuximab + irinotecan, cetuximab + FOLFIRI, cetuximab | 30 | 17 (57%) | 13 (43%) | 65 | 0 | Lievre et al.13 |
| Panitumumab, cetuximab + irinotecan, cetuximab | 48 | 32 (67%) | 16 (33%) | 31 | 6 | Benvenuti et al.14 |
| Cetuximab, cetuximab + irinotecan | 108 | 66 (61%) | 42 (39%) | 41 | 0 | De Roock et al.15 |
| Cetuximab + irinotecan, cetuximab, cetuximab + oxaliplatin | 81 | 49 (60%) | 32 (40%) | 27 | 6.3 | Finocchiaro et al.16 |
| Cetuximab + irinotecan, cetuximab + FOLFIRI, cetuximab | 89 | 65 (73%) | 24 (27%) | 40 | 0 | Lievre et al.17 |
| Cetuximab | 80 | 50 (62%) | 30 (38%) | 48a | 10a | Khambata-Ford et al.18 |
| Panitumumab | 62 | 38 (61%) | 24 (39%) | 11 | 0 | Freeman et al.19 |
WT wild type, RR% response rate, CR complete response, PR partial response, SD stable disease, MT mutant
aReported as percentage of disease control (CR + PR + SD)
Biomarker analysis from several recent studies demonstrated that patients with KRAS mutated tumors are resistant to monotherapy with cetuximab or panitumumab.14,23,24 The additional benefit of EGFR-I to chemotherapy is limited to patients with wild-type (WT) KRAS mCRC.25–27 However, the optimal biologic agent (bevacizumab or EGFR-I) to be combined with chemotherapy for the first- or second-line treatment of patients with WT KRAS mCRC remains to be determined.
Here, we review recent studies involving EGFR-I in advanced CRC with particular attention to incidence, prognostic value, and predictive significance of the KRAS mutation in CRC patients.
KRAS Mutation in CRC
The KRAS oncogene encodes the human cellular homolog of the transforming gene Kirsten rat sarcoma-2 virus.11 The K-ras protein is a self-inactivating signal transducer. K-ras cycles between a guanosine diphosphate (GDP) bound (“off” state) to guanosine triphosphate (GTP) bound (“on” state) in response to receptor activation. When receptor tyrosine kinases, such as EGFR, bind ligands (e.g., epidermal growth factor, transforming growth factor-α), the receptor dimerizes and undergoes a conformational change that results in phosphorylation of tyrosine residues, which leads to activation of signaling pathways that include the Ras-Raf-Mek kinase pathway. This response is usually transient as a result of the intrinsic GTPase activity of K-ras.12 However, KRAS activating mutations abolish this intrinsic GTPase activity, resulting in constitutively active K-ras proteins that activate downstream signaling pathways and can lead to oncogenesis (Fig. 1).12
Fig. 1.
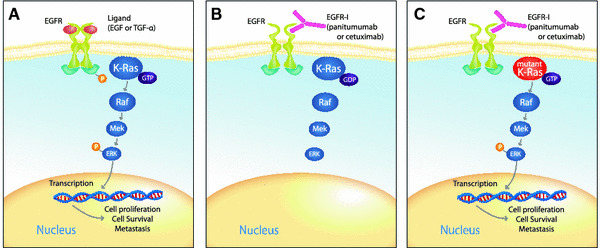
K-ras mutation governs the effects of inhibition of epidermal growth factor receptor (EGFR) on the Ras/Raf/Mek signaling pathway. a The ligands epidermal growth factor (EGF) or transforming growth factor (TGF)-α activate EGFR signaling leading to phosphorylation of the tyrosine kinase domain. K-Ras adopts a guanosine triphosphate (GTP) bound conformation and through a series of phosphorelays activates ERK. Activation of ERK leads to transcription of genes associated with cell proliferation, survival, and metastasis. b Inhibition of EGFR by the EGFR-I panitumumab or cetuximab leads to K-ras becoming guanosine diphosphate (GDP) bound, which inhibits downstream signaling. c When K-ras is mutated, it adopts a constitutively GTP bound conformation leading to activation of the Ras/Raf/Mek pathway in the presence of inhibition of EGFR. Adapted with permission from Khambata-Ford et al.18
Activating KRAS mutations are among the most common oncogenic alterations in cancer, occurring early in CRC carcinogenesis.28 In one study, 58% of colonic adenomas of >1 cm carried the KRAS mutation.29 The importance of KRAS to colorectal tumorigenesis is underscored by the finding that colon cancer cells in which a mutated KRAS gene has been removed or replaced lose their ability to form tumors in mice.30 The incidence rate of KRAS mutations is fairly consistent across several studies, and a similar incidence rate has been reported both in early-stage and metastatic disease.21,31–33
Testing for KRAS Mutations
Currently, several methods are used to detect KRAS mutations within tumors. One method that is generally used for testing KRAS mutations is an allele-specific polymerase chain reaction on DNA from tumor sections, typically obtained from a formalin-fixed, paraffin-embedded block. This assay uses an allele-specific primer extension that can detect the most common KRAS mutations located within codons 12 and 13 of the gene.
Several commercial assays are available, and currently there is no standardization. Recently, a comparability study of four commercially available KRAS tests was presented at the annual meeting of the American Association for Cancer Research.34 This analysis suggested that assays by allele-specific polymerase chain reaction or direct sequencing accurately detected the presence of mutations within the KRAS gene in a high percentage of patient tumor samples when compared to direct sequencing conducted by an internal laboratory. However, an assay that used direct hybridization technology failed to detect the presence of a mutation with the same accuracy (Table 2).34 Recently, the American Society of Clinical Oncology (ASCO) released a provisional opinion identifying allele-specific polymerase chain reaction and direct sequencing as acceptable methods for testing of KRAS mutations.35 In addition, a group of physicians from the European Union have proposed guidelines for a European Union quality assurance program to establish consistency in testing practices across Europe.36 When testing for the presence of a KRAS mutation from patient tumor samples, clinicians should ensure the validity of the assay through the use of control DNA with known mutation status.
Table 2.
Comparison of four commercially available KRAS mutation tests to the expected standard determined by direct sequencing conducted by an internal core laboratory 34
| Mutation | Allele-specific PCR (DxS/Histogenex) | Direct sequencing (Gentris) | Allele-specific hybridization (Invitek) | Allele-specific PCR extension (Genzyme) |
|---|---|---|---|---|
| KRAS WT detected/expected | 20/21 | 18/21 | 3/21 | 15/21 |
| KRAS mutations detected/expected | 20/19 | 14/19 | 24/19 | 20/19 |
| Inconclusivea | 0 | 8 | 13 | 5 |
PCR polymerase chain reaction, WT wild type
aKRAS tests with inconclusive, failed, or mixed results
KRAS Mutation as a Prognostic Marker
Studies evaluating the role of KRAS mutation as a prognostic marker in patients with CRC have somewhat conflicting results. A study of patients with stage II/III CRC demonstrated that KRAS mutant tumoral status was associated with an increase in the rate of recurrence of their disease (71%) compared to WT (25%).20 Another study demonstrated that the prognostic value of KRAS mutation was limited to patients with stage I/II CRC.21 Several other studies have demonstrated data in support of KRAS mutation as a negative prognostic marker, while others have concluded otherwise.31,32,37–39
In patients with mCRC, the prognostic value of KRAS also presents conflicting results. Patients with advanced disease receiving fluorouracil demonstrated no difference in survival on the basis of KRAS status.22 In addition, the NCIC CO.17 trial of patients with chemorefractory mCRC treated with cetuximab versus best supportive care (BSC) demonstrated no survival differences among patients with WT KRAS or mutant tumors receiving BSC.23 In contrast, the updated KRAS analysis of the pivotal bevacizumab with or without chemotherapy (IFL) study demonstrated lower median overall survival for mutant tumor KRAS patients versus WT in both treatment arms (IFL: 17.6 months WT vs. 13.6 months mutant; IFL with bevacizumab: 27.7 months WT vs. 19.9 months mutant), although statistical models were not powered to detect the significance of these differences.40 Therefore, the prognostic significance of KRAS in patients with CRC is still inconclusive. The most likely explanation for the disparity in the results is the heterogeneity of the patient population. Future studies should attempt to stratify patient groups by stage of disease, primary tumor site, and presence of other biomarkers.
KRAS Mutation Predicts Resistance to EGFR-I Therapy
Early small retrospective studies correlated activating mutation of KRAS with resistance or lack of response to EGFR-I in patients with mCRC, establishing the proof of concept for further investigation of this biomarker.13–19 In these studies, patients were treated with panitumumab or cetuximab monotherapy, or cetuximab in combination with irinotecan or other chemotherapeutic agents (Table 1).13–19 Mutant KRAS status in these patients was associated with a lack of response to EGFR-I-based therapy. Most patients with mutated KRAS tumors did not have an objective response when treated with EGFR-I-based therapy (Table 1). Although the role of KRAS mutation as a negative predictive marker of response to therapy was demonstrated in these studies, definitive conclusions could not be made as a result of the small patient numbers and differences in treatment.
The first large study to analyze the effect of KRAS mutational status with efficacy with EGFR-I monotherapy was conducted with patient tumor samples from the pivotal panitumumab registrational trial.24 In this randomized phase 3 trial, patients with chemorefractory mCRC were randomized to panitumumab or BSC, allowing patients to cross over from BSC to panitumumab at disease progression.8 KRAS status was determined in 427 (92%) of 463 patients (208 panitumumab and 219 BSC) with KRAS mutations identified in 184 (43%) of 427 patients (84 [40%] in the panitumumab arm).24 In the subset of patients with WT KRAS tumors, progression-free survival (PFS) was improved with panitumumab versus BSC treatment (Table 3). Median PFS time was 2.8 months for panitumumab vs. 1.7 months for BSC in the WT group (hazard ratio [HR] .45, P < .0001). No benefit was seen regardless of treatment in patients with mutant KRAS tumors. Response rates to panitumumab were 17 and 0% for the WT and mutant groups, respectively. In patients randomized to BSC who subsequently received panitumumab in the cross over study, PFS was longer in patients with WT KRAS tumors (3.8 months WT vs. 1.8 months mutant (HR .32; 95% confidence interval [95% CI] 0.22–0.46). The results of these analyses demonstrated that response to panitumumab was confined to patients with WT KRAS tumors.24
Table 3.
Response rate and progression-free survival of patients treated in phase 3 epidermal growth factor receptor inhibitor monotherapy trials by KRAS mutational status 23,24
| KRAS | Panitumumab | BSC | HR (P) | Cetuximab | BSC | HR (P) |
|---|---|---|---|---|---|---|
| WT KRAS | ||||||
| Response rate | 17% | 0% | 12.8% | 0% | ||
| Median PFS | 2.8 moa | 1.7 moa | .45 (<.0001) | 3.7 mo | 1.9 mo | .40 (<.001) |
| Mutant KRAS | ||||||
| Response rate | 0% | 0% | 1.2% | 0% | ||
| Median PFS | 1.7 moa | 1.7 moa | .99 (NR) | 1.8 mo | 1.8 mo | .99 (.96) |
NR not reported, PFS progression free survival, BSC best supportive care, HR hazard ratio for progression-free survival
aReported as weeks in the original publication
Similar findings have been demonstrated in a large randomized phase 3 trial of patients treated with cetuximab monotherapy.23 In this study, 572 patients with chemorefractory mCRC were randomized to cetuximab versus BSC. The crossover of BSC patients to the cetuximab arm of the trial was not mandated by the study protocol.7 Tumor specimens were available for KRAS mutational testing from 394 patients (69%) with mutations detected in 42% of these patients. Among patients treated with cetuximab, the objective response rate for WT KRAS patients was 12.8%, compared with a 1.2% response rate for patients in the KRAS mutant group. Cetuximab monotherapy was associated with significant improvements in PFS (3.7 vs. 1.9 months, P < .001) and overall survival in WT KRAS patients treated with cetuximab (9.5 months for cetuximab vs. 4.8 months for BSC, P < .001) (Table 3). However, increased overall survival was not observed in patients with KRAS-mutated tumors (4.5 months for cetuximab vs. 4.6 months for BSC, P = .89).
The data from these studies strongly support the hypothesis that the activating mutation in KRAS confers resistance to EGFR-I. The clinical benefit from single-agent panitumumab and cetuximab is restricted to patients with WT KRAS. In WT KRAS patients, EGFR-I treatment is associated with marked objective response, and improvements in PFS and overall survival over BSC in patients with chemorefractory mCRC.
Benefit from the Addition of EGFR-I to Chemotherapy is Limited to WT KRAS Patients
Several recent studies have evaluated the addition of cetuximab or panitumumab to standard chemotherapy in first- or second-line setting in patients with mCRC. Data from these studies have demonstrated an association between KRAS mutational status and response to the addition of EGFR-I to standard chemotherapy.
Two studies presented at the 2009 ASCO annual meeting examined the safety and efficacy of panitumumab in combination with irinotecan-based chemotherapy in patients with mCRC whose disease previously failed to respond to oxaliplatin-based therapy. In the PRECEPT trial, the role of KRAS is being prospectively evaluated in patients with mCRC treated with panitumumab with FOLFIRI; the STEPP study also evaluated the efficacy of panitumumab in combination with FOLFIRI or irinotecan with KRAS mutational status available for 92% of patients.25,41 In both of these studies, clinical end points for efficacy of panitumumab with chemotherapy were in favor of patients with WT KRAS tumors.25,41
A more favorable efficacy profile was reported among patients with WT KRAS mCRC receiving cetuximab in combination with FOLFOX in a randomized phase 2 trial evaluating FOLFOX with or without cetuximab as first-line treatment (OPUS).26 Efficacy results of the WT KRAS population demonstrated that response rate greatly increased when cetuximab was added to FOLFOX (61 vs. 37%, P = .011). Conversely, patients with mutant KRAS had a nonsignificant decrease in response rate when cetuximab was added to FOLFOX (33 vs. 49%, P = .106). These data suggest that clinical benefit from addition of cetuximab to FOLFOX is limited to patients with WT KRAS.
In the CRYSTAL trial, previously untreated patients with mCRC were randomized to FOLFIRI with or without cetuximab.27 Of the 1198 patients analyzed in the original intention-to-treat population, 540 (45%) were included in the KRAS analysis and 36% had a KRAS mutation. The addition of cetuximab to FOLFIRI in patients with WT KRAS tumors resulted in a nonsignificant increase in median PFS compared to patients receiving FOLFIRI alone (9.9 vs. 8.7 months, P = .07). In contrast, the median PFS of mutant tumor KRAS patients receiving cetuximab with FOLFIRI was lower (7.6 months) compared with patients receiving FOLFIRI alone (8.1 months, HR 1.07, 95% CI 0.71–1.61). A nonsignificant increase in overall survival was observed in WT KRAS patients treated with cetuximab with FOLFIRI compared to patients treated with FOLFIRI alone (24.9 vs. 21 months, P = .44).27 These data demonstrate that the benefit observed from the addition of cetuximab to chemotherapy is limited to patients with WT KRAS tumors; however, future studies will be needed to determine the optimal clinical settings for the use of cetuximab in combination with FOLFIRI in the first-line treatment of mCRC.
An important finding from the CRYSTAL trial was the observation that the surgical resection rate of metastases was higher in patients treated with cetuximab with FOLFIRI compared to patients treated with FOLFIRI alone (7.0 vs. 3.7%).27 In addition, the R0 resection rate with curative intent was statistically higher in favor of patients receiving cetuximab (4.8 vs. 1.7%; odds ratio for cetuximab with FOLFIRI group, 3.02; 95% CI, 1.45–6.27; P = .002).27 However, the relationship of these findings from the CRYSTAL trial to KRAS status of the tumor is currently unknown.
Recently, at the 2009 Gastrointestinal Cancers Symposium, Folprecht et al. presented preliminary data from a randomized multicenter study of cetuximab with FOLFOX or FOLFIRI in neoadjuvant treatment of nonresectable colorectal liver metastasis (CELIM study).42 A confirmed overall response rate of 70% was observed in patients with WT KRAS tumors treated with FOLFOX or FOLFIRI with cetuximab.42 Among all patients with disease that was initially deemed nonresectable, 25% were classified as resectable after treatment. Overall, 34% of patients enrolled onto the CELIM study underwent R0 resection and demonstrated no evidence of residual disease.42 Perioperative morbidity and mortality were comparable to historical data. These preliminary data are encouraging and warrant further studies of evaluating EGFR-I in combination with chemotherapy as a neoadjuvant therapy in patients with KRAS WT tumors with liver metastasis.
Two current phase 3 studies are evaluating the influence of KRAS on response to panitumumab with chemotherapy in patients with mCRC. The PRIME study is evaluating FOLFOX with or without panitumumab in the first-line setting, while the 20050181 study is evaluating FOLFIRI with or without panitumumab for second-line treatment.43,44 Tumor sample availability was required for enrollment onto these studies, and KRAS status will be incorporated into the primary analysis to test the role of KRAS in the combination therapy setting. Future trials seeking to evaluate the role of a biomarker such as KRAS may benefit from the incorporation of biomarker evaluation into the study design.
Optimal Biologics in WT KRAS Is Still Unknown
The studies mentioned above support the beneficial effect of the addition of EGFR-I to standard chemotherapy in patients with WT KRAS mCRC. However, the optimal biologics to combine with chemotherapy in first- and second-line setting remains unclear.
Retrospective analysis of the pivotal trial of bevacizumab with IFL demonstrated no statistically significant correlation between KRAS mutation and the benefit from bevacizumab.40 The median PFS for chemotherapy alone versus chemotherapy with bevacizumab was 7.4 vs. 13.5 months (HR .44; P < .0001) in WT KRAS and 5.5 and 9.3 months (HR .41; P < .0008) in KRAS mutant groups.
Two recent trials have evaluated the safety and efficacy of the addition of EGFR-I to standard chemotherapy and bevacizumab in treatment-naive patients with mCRC. The first study to evaluate this combination was the PACCE trial.45 In this phase 3 study, patients were randomized to receive chemotherapy and bevacizumab with or without panitumumab. The chemotherapeutic regimen was either FOLFOX or FOLFIRI, based on the choice of the investigator. Efficacy results demonstrated that patients with WT or mutant KRAS tumors experienced a decrease in PFS when panitumumab was added to FOLFOX with bevacizumab versus FOLFOX with bevacizumab alone (Table 4). Similar results were demonstrated in patients treated in the FOLFIRI cohort (Table 4).45
Table 4.
Median progression-free survival of patients treated in trials evaluating the addition of epidermal growth factor receptor inhibitor therapy to fluoropyrimidine-based regimens in combination with bevacizumab
| KRAS | PACCE oxaliplatin cohorta | ||
|---|---|---|---|
| FOLFOX + bevacizumab (months) | Panitumumab + FOLFOX + bevacizumab (months) | HR (95% CI) | |
| WT KRAS | 11.5 | 9.8 | 1.36 (1.04–1.77) |
| KRAS MT | 11.0 | 10.4 | 1.25 (0.91–1.71) |
| KRAS | PACCE irinotecan cohorta | ||
|---|---|---|---|
| FOLFIRI + bevacizumab (months) | Panitumumab + FOLFIRI + bevacizumab (months) | HR (95% CI) | |
| WT KRAS | 12.5 | 10.0 | 1.50 (0.82–2.76) |
| KRAS MT | 11.9 | 8.3 | 1.19 (0.65–2.21) |
| KRAS | CAIRO2b | ||
|---|---|---|---|
| CAPEOX + bevacizumab (months) | Cetuximab + CAPEOX + bevacizumab (months) | P value | |
| WT KRAS | 10.6 | 10.5 | .30 |
| KRAS MT | 12.5 | 8.1 | .003 |
The CAIRO2 trial evaluated the combination of capecitabine, oxaliplatin, and bevacizumab, with or without cetuximab.46 For the total patient group, the addition of cetuximab was associated with a statistically significant decrease in median PFS and increased grade 3/4 adverse events. In patients with WT KRAS, there was no statistically significant change in median PFS by the addition of cetuximab (10.6 vs. 10.5 months, P = .3) (Table 4). However, patients with mutant KRAS tumors treated with cetuximab with chemotherapy and bevacizumab had a statistically significant decrease in median PFS compared to mutant tumor KRAS patients not treated with a cetuximab-containing regimen (12.5 vs. 8.1 months, P = .003) (Table 4). The results of the PACCE and CAIRO2 studies demonstrate that the addition of EGFR-I to chemotherapy and bevacizumab for first line treatment of mCRC does not improve clinical outcomes for patients with mCRC, regardless of KRAS mutational status, and may decrease clinical outcome. Furthermore, these data strongly suggest a class effect of EGFR-I when used in combination with bevacizumab and fluoropyrimidine-based chemotherapy.
Two ongoing studies will continue to evaluate the optimal biologics to combine with chemotherapy in patients with WT KRAS mCRC. In the CALGB 80405 study, patients will be randomized to chemotherapy with bevacizumab alone or cetuximab alone, or in combination in the first-line setting.47 This study has been amended to include only patients with WT KRAS tumors. In the phase 2 SPIRITT study, patients with WT KRAS mCRC whose disease progressed while receiving oxaliplatin-based chemotherapy and bevacizumab will be randomized to FOLFIRI with either bevacizumab or panitumumab.48 These studies will provide further information about the optimal biologic to combine with standard chemotherapy in patients with WT KRAS mCRC.
Future directions in Biomarker Research to EGFR-I-Based Therapy
Mutation of the KRAS gene has emerged as a powerful biomarker of resistance to EGFR-I-based therapy. Although most studies focus on mutations within codons 12 and 13 of the KRAS gene, a recent analysis demonstrated that mutations within codons 61 and 146 also confer resistance to EGFR-based therapy.49 Recent studies have demonstrated that in addition to KRAS, other biomarkers including BRAF (OMIM 164757) and PIK3CA (OMIM 171834) can increase the predictive ability of response to EGFR-I in patients with WT KRAS tumoral status.49–51 However, data from another study suggest that PIK3CA does not predict for response to EGFR-I therapy, and further studies will be needed to validate PIK3CA as a predictive biomarker.52 In addition, a retrospective analysis suggested that patients treated on the NCIC CO.17 clinical trial with WT KRAS tumors and high mRNA expression of the EGFR ligand epiregulin (OMIM 602061) may derive the greatest benefit from cetuximab therapy.53 Continued evaluation of these biomarkers and others will allow for a personalized approach for the treatment of patients with mCRC.
Conclusions
The desired goal in cancer therapy is to individualize treatment according to the underlying predictive factors to minimize the unnecessary adverse effects and complications from cancer treatment. Recent data on the presence of a KRAS mutation and lack of response to EGFR-I represent an important step toward achieving that goal. Great progress has been made in the treatment of patients with mCRC over the last 10 years. The availability of newer chemotherapy and biologics has increased treatment options for patients and clinicians.
The current data strongly support the hypothesis that patients with KRAS mutation with mCRC have disease resistant to EGFR-I therapy. The beneficial effects of EGFR-I monotherapy or in combination with chemotherapy are limited to patients with WT KRAS tumors. Preliminary data suggest that EGFR-I in combination with chemotherapy can result in improvements in tumor responses and can potentially lead to downstaging of patients with nonresectable KRAS WT liver metastases. Testing for KRAS mutation should be strongly considered in patients with mCRC before the initiation of any EGFR-I. The current data do not support the addition of anti-EGFR monoclonal antibodies to standard chemotherapy with bevacizumab in the first-line setting. Ongoing trials will address the optimal biologics to combine with chemotherapy in WT KRAS patients.
Acknowledgment
We thank William Fazzone, PhD, of MediTech Media Ltd. for writing and editorial assistance with funding from Amgen Inc.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Note added in proof
During the review of this paper, the results of the PRIME and 20050181 phase 3 clinical trials discussed above were presented at the 2009 Joint ECCO 15—ESMO 34th Multidisciplinary Congress in Berlin, Germany. The results of both of these studies demonstrated significant increases in median PFS of patients with WT KRAS mCRC who received chemotherapy with panitumumab compared with patients receiving chemotherapy alone. (Peeters M, et al. European Journal of Cancer Supplements, Vol. 7, No 3, September 2009, Page 9, Abstract 14LBA; Douillard J, et al. European Journal of Cancer Supplements, Vol. 7, No 3, September 2009, Page 6, Abstract 10LBA) A similar benefit to panitumumab was not observed in patients with KRAS mutant tumors. These data provide further support to the conclusions made in this manuscript.
References
- 1.American Cancer Society. Cancer facts and figures. http://www.cancer.org/downloads/STT/2008CAFFfinalsecured.pdf. Accessed March 25, 2009.
- 2.Veronsese ML, Sun W, Giantonio B, et al. A phase II trial of gefitinib with 5-fluorouracil, leucovorin, and irinotecan in patients with colorectal cancer. Br J Cancer. 2005;92:1846–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Townsley CA, Major P, Siu LL, et al. Phase II study of erlotinib (OSI-774) in patients with metastatic colorectal cancer. Br J Cancer. 2006;94:1136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erbitux (cetuximab) [prescribing information]. Branchburg, NJ: ImClone Systems Inc; revised November 2008.
- 5.Vectibix (panitumumab) [prescribing information]. Thousand Oaks, CA: Amgen Inc.; revised February 2009.
- 6.Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. [DOI] [PubMed] [Google Scholar]
- 7.Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. [DOI] [PubMed] [Google Scholar]
- 8.Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell EP, Perez-Soler R, Van Cutsem E, Lacouture ME. Clinical presentation and pathophysiology of EGFRI dermatologic toxicities. Oncology (Williston Park). 2007;21(11 Suppl 5):4–9. [PubMed] [Google Scholar]
- 10.Tejpar S, Piessevaux H, Claes K, et al. Magnesium wasting associated with epidermal-growth-factor receptor-targeting antibodies in colorectal cancer: a prospective study. Lancet Oncol. 2007;8:387–94. [DOI] [PubMed] [Google Scholar]
- 11.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. [DOI] [PubMed] [Google Scholar]
- 12.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. [DOI] [PubMed] [Google Scholar]
- 13.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. [DOI] [PubMed] [Google Scholar]
- 14.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–8. [DOI] [PubMed] [Google Scholar]
- 15.De Roock W, Piessevaux H, De Schutter J, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–15. [DOI] [PubMed] [Google Scholar]
- 16.Finocchiaro G, Cappuzzo F, Janne PA, et al. EGFR, HER2 and Kras as predictive factors for cetuximab sensitivity in colorectal cancer. J Clin Oncol. 2007;25:168s. [Google Scholar]
- 17.Lievre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–9. [DOI] [PubMed] [Google Scholar]
- 18.Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–7. [DOI] [PubMed] [Google Scholar]
- 19.Freeman DJ, Juan T, Reiner M, et al. Association of K-ras mutational status and clinical outcomes in patients with metastatic colorectal cancer receiving panitumumab alone. Clin Colorectal Cancer. 2008;7:184–90. [DOI] [PubMed] [Google Scholar]
- 20.Benhattar J, Losi L, Chaubert P, et al. Prognostic significance of K-ras mutations in colorectal carcinoma. Gastroenterology. 1993;104:1044–8. [DOI] [PubMed] [Google Scholar]
- 21.Lee JC, Wang ST, Lai MD, et al. K-ras gene mutation is a useful predictor of the survival of early stage colorectal cancers. Anticancer Res. 1996;16:3839–44. [PubMed] [Google Scholar]
- 22.Etienne-Grimaldi MC, Formento JL, Francoual M, et al. K-Ras mutations and treatment outcome in colorectal cancer patients receiving exclusive fluoropyrimidine therapy. Clin Cancer Res. 2008;14:4830–5. [DOI] [PubMed] [Google Scholar]
- 23.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. [DOI] [PubMed] [Google Scholar]
- 24.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. [DOI] [PubMed] [Google Scholar]
- 25.Cohn AL, Smith DA, Neubauer A, et al. Results from panitumumab (pmab) regimen evaluation in colorectal cancer to estimate primary response to treatment (PRECEPT): second-line treatment with pmab and FOLFIRI by tumor KRAS status. J Clin Oncol. 2009;27:184s. [Google Scholar]
- 26.Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–71. [DOI] [PubMed] [Google Scholar]
- 27.Van Cutsem E, Kohene C-H, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. [DOI] [PubMed] [Google Scholar]
- 28.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. [DOI] [PubMed] [Google Scholar]
- 29.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. [DOI] [PubMed] [Google Scholar]
- 30.Rak J, Mitsuhashi Y, Bayko L, et al. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995;55:4575–80. [PubMed] [Google Scholar]
- 31.Esteller M, Gonzalez S, Risques RA, et al. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J Clin Oncol. 2001;19:299–304. [DOI] [PubMed] [Google Scholar]
- 32.Westra JL, Schaapveld M, Hollema H, et al. Determination of TP53 mutation is more relevant than microsatellite instability status for the prediction of disease-free survival in adjuvant-treated stage III colon cancer patients. J Clin Oncol. 2005;23:5635–43. [DOI] [PubMed] [Google Scholar]
- 33.Samowitz WS, Holden JA, Curtin K, et al. Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. Am J Pathol. 2001;158:1517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juan T, Suggs S, Wolf M, et al. A comparability study of 4 commercial KRAS tests (abstract 1811). Paper presented at: 99th AACR Annual Meeting; 2008; San Diego, CA.
- 35.Allegra CJ, Jessup JM, Sommerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–6. [DOI] [PubMed] [Google Scholar]
- 36.van Krieken JH, Jung A, Kirchner T, et al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch. 2009;453:417–31. [DOI] [PubMed] [Google Scholar]
- 37.Ahnen DJ, Feigl P, Quan G, et al. Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study. Cancer Res. 1998;58:1149–58. [PubMed] [Google Scholar]
- 38.Russo A, Bazan V, Agnese V, et al. Prognostic and predictive factors in colorectal cancer: Kirsten Ras in CRC (RASCAL) and TP53CRC collaborative studies. Ann Oncol. 2005;16(Suppl 4):44–9. [DOI] [PubMed] [Google Scholar]
- 39.Bouzourene H, Gervaz P, Cerottini JP, et al. p53 and K-ras as prognostic factors for Dukes’ stage B colorectal cancer. Eur J Cancer. 2000;36:1008–15. [DOI] [PubMed] [Google Scholar]
- 40.Hurwitz HI, Yi J, Ince W, et al. The clinical benefit of bevacizumab in metastatic colorectal cancer is independent of K-ras mutation status: analysis of a phase III study of bevacizumab with chemotherapy in previously untreated metastatic colorectal cancer. Oncologist. 2009;14:22–8. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell EP, LaCouture M, Shearer H, et al. Final STEPP results of prophylactic versus reactive skin toxicity (ST) treatment (tx) for panitumumab (pmab)-related ST in patients (pts) with metastatic colorectal cancer (mCRC). J Clin Oncol. 2009;27:805s. [Google Scholar]
- 42.Folprecht G, Gruenberger T, Hartmann JT, et al. Cetuximab plus FOLFOX6 or cetuximab plus FOLFIRI as neoadjuvant treatment of nonresectable colorectal liver metastases: a randomized multicenter study (CELIM-study). Paper presented at: 2009 Gastrointestinal Cancers Symposium; January 15–17, 2009; San Francisco, CA.
- 43.Siena S, Tabernero J, Burkes RL, et al. Phase III study (PRIME/20050203) of panitumumab (pmab) with FOLFOX compared with FOLFOX alone in patients (pts) with previously untreated metastatic colorectal cancer (mCRC): pooled safety data. J Clin Oncol. 2008;26:186s. [Google Scholar]
- 44.Peeters M, Wilson G, Ducreux M, et al. Phase III study (20050181) of panitumumab (pmab) with FOLFIRI versus FOLFIRI alone as second-line treatment (tx) in patients (pts) with metastatic colorectal cancer (mCRC): pooled safety results. J Clin Oncol. 2008;26:194s. [Google Scholar]
- 45.Hecht JR, Mitchell E, Chidiac T, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27:672–80. [DOI] [PubMed] [Google Scholar]
- 46.Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–72. [DOI] [PubMed] [Google Scholar]
- 47.A phase III trial of irinotecan/5-FU/leucovorin or oxaliplatin/5-FU/leucovorin with bevacizumab, or cetuximab (C225), or with the combination or bevacizumab and cetuximab for patients with untreated metastatic adenocarcinoma of the colon or rectum. http://www.clinicaltrials.gov/ct2/show/NCT00265850. Accessed April 18, 2009.
- 48.SPIRITT-Q2W FOLFIRI regimen plus panitumumab or a Q2W FOLFIRI regimen plus bevacizumab for 2nd-line mCRC. http://www.clinicaltrials.gov/ct2/show/NCT00418938. Accessed April 18, 2009.
- 49.Loupakis F, Ruzzo A, Cremolini C, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1857–7. [DOI] [PubMed] [Google Scholar]
- 51.Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. [DOI] [PubMed] [Google Scholar]
- 52.Prenen H, De Schutter J, Jacobs B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15:3184–8. [DOI] [PubMed] [Google Scholar]
- 53.Jonker DJ, Karapetis C, Harbison C, et al. High epiregulin (EREG) plus K-ras (WT) status as predictors of cetuximab benefit in the treatments of advanced colorectal cancer (ACRC): results from NCIC CTG CO.17—a phase III trial of cetuximab versus best supportive care (BSC). J Clin Oncol. 2009;27:172s. [Google Scholar]