

Summary
A number of studies have been conducted recently on the model organism Drosophila to determine the function of genes involved in human disease, including those implicated in neurological disorders, cancer and metabolic and cardiovascular diseases. The simple structure and physiology of the Drosophila heart tube together with the available genetics provide a suitable in vivo assay system for studying cardiac gene functions. In our study, we focus on analysis of the role of dystrophin (Dys) in heart physiology. As in humans, the Drosophila dys gene encodes multiple isoforms, of which the large isoforms (DLPs) and a truncated form (Dp117) are expressed in the adult heart. Here, we show that the loss of dys function in the heart leads to an age-dependent disruption of the myofibrillar organization within the myocardium as well as to alterations in cardiac performance. dys RNAi-mediated knockdown in the mesoderm also shortens lifespan. Knockdown of all or deletion of the large isoforms increases the heart rate by shortening the diastolic intervals (relaxation phase) of the cardiac cycle. Morphologically, loss of the large DLPs isoforms causes a widening of the cardiac tube and a lower fractional shortening, a phenotype reminiscent of dilated cardiomyopathy. The dilated dys mutant phenotype was reversed by expressing a truncated mammalian form of dys (Dp116). Our results illustrate the utility of Drosophila as a model system to study dilated cardiomyopathy and other muscular-dystrophy-associated phenotypes. Key words: aging; arrhythmia; cardiac function; diastole; heart; heart rate; muscle fibers; muscular dystrophy; myofibrillar disarray; systole.
Introduction
Muscular dystrophy (MD) involves over 30 different inherited diseases, all causing progressive weakness and degeneration of skeletal muscle (Emery, 2002). Dystrophin (Dys) was the first mutant protein shown to cause MD. Mutations in the dystrophin gene (dys), the largest gene in the human genome, cause Duchenne MD (DMD) as well as the milder phenotype of Becker MD (BMD) (Koenig et al., 1988). Duchenne MD is caused by a nonfunctional or absent Dys protein, whereas BMD has been associated with a reduction of Dys protein levels. In humans, dys is expressed in skeletal, smooth and cardiac muscles, as well as in the brain (Holder et al., 1996). Dystrophin protein is enriched in the sarcolemma, known to link the extracellular matrix to the actin cytoskeleton via interactions with the Dys glycoprotein complex (DGC), and therefore, has an important structural role during muscle contraction and muscle stretch (Petrof et al., 1993; Straub et al., 1997). The absence of Dys destabilizes the DGC components and results in membrane fragility and disruption during contraction. Besides the mechanical role, Dys is involved in signaling cascades via their DGC protein partners (dystroglycan and syntrophin): DGC components seem to be able to interact with growth factor receptor-bound protein 2, calmodulin and neuronal nitric oxide synthase (Rando, 2001). Furthermore, Dys may be involved in the aggregation or organization of ion channels in the membrane because its absence causes abnormalities in channel function (Franco & Lansman, 1990).
The Drosophila dys gene is as complex as its mammalian counterparts. It encodes three large isoforms called Dys-like products (DLPs) and three truncated products sharing with DLPs the carboxy-terminal and cysteine-rich domains driven by separate internal promoters (Greener & Roberts, 2000; Neuman et al., 2001, 2005). The three DLPs consist of an N-terminal actin-binding domain, numerous spectrins repeats, a dystroglycan-binding domain (cysteine-rich CR domain) and a carboxy-terminal domain involved in binding to other DGC proteins. The three shorter isoforms Dp186, Dp205 and Dp117 have a unique N-terminal region appended to the common C-terminal region (Neuman et al., 2005). As in mammals, the fly dys transcripts are expressed in distinct tissue-specific patterns (Neuman et al., 2001, 2005; Dekkers et al., 2004; Van der Plas et al., 2006; Shcherbata et al., 2007).
Despite the evolutionary divergence between flies and humans, the genomic sequences indicate that around 70% of the genes involved in human disease are also found in Drosophila (Adams et al., 2000; Fortini & Bonini, 2000; Bier, 2005). Fly models have been generated for a wide spectrum of human diseases such as developmental disorders, neurological disorders, cancer, metabolic disorders and cardiovascular disease (Bonini & Fortini, 2003; Sutcliffe et al., 2003; Bier & Bodmer, 2004; Bier, 2005). Among these disease genes, a number of them are implicated in cardiac function: mutations in myosin, troponin I, tropomyosin 2 and δ-sarcoglycan cause dilated cardiomyopathy (Wolf et al., 2006; Cammarato et al., 2008), and mutations in a potassium channel α-subunit for KCNQ1 cause arrhythmias in humans as well as in flies (because of prolonged duration of contractions phases – ‘long QT’) (Ocorr et al., 2007).
In this study, we investigate the role of Dys in maintaining heart morphology and function, using the fly heart as an in vivo assay system. Using reverse transcriptase–polymerase chain reaction (RT–PCR), we determined that both the long DLPs and the short DP117 dys isoforms are expressed in the adult Drosophila heart. The dys mutant, haploinsufficiency or knockdown flies show shortened lifespan and develop age-dependent cardiac abnormalities, reminiscent of mdx mice. We characterized the cardiac properties in dys mutants by measuring changes in its dynamic parameters, including heart rate, rhythmicity, systolic and diastolic diameters and intervals and fractional shortening. The dys mutant flies have dilated and abnormally performing hearts consistent with the mammalian phenotype of dilated cardiomyopathy. Deletion of the long DLP isoforms also causes progressively disorganized myofibrillar arrangements with age that may contribute to the altered performance. We discuss the potential of the fly heart model in studying the molecular basis of MD effects in the heart.
Results
Dys protein distribution in the Drosophila heart
The dys gene products are expressed in a tissue-specific manner; the long-form DLP1 and DLP2 are predominantly found during development in the visceral mesoderm, in the gut and throughout muscle fibers, while Dp186 is present at high levels in the central nervous system (Neuman et al., 2001; Dekkers et al., 2004; Van der Plas et al., 2006; Shcherbata et al., 2007). After germ band retraction, two rows of cardiac mesoderm fuse at the dorsal midline to form the linear heart tube in Drosophila (Bodmer, 1995; Bodmer & Frasch, 1999; Haag et al., 1999). Dystrophin expression is detected in the myocardial cells shortly before fusion (data not shown) and continuously throughout embryogenesis (Fig. 1A). During metamorphosis, the heart undergoes remodeling forming a functional adult heart that contains inflow tract openings (ostia) and interval valves (Molina & Cripps, 2001; Monier et al., 2005). In the adult, the cardiac tube remains a simple linear tube that pumps the hemolymph through the organism in an open circulatory system. Dystrophin accumulates in the cell membranes outlining the membranes of the myocardial cells similar to discs large (Dlg) (Fig 1B,C). Using RT–PCR with RNA isolated from adult hearts, we detect the long DLPs dys isoforms and a short form, Dp117 (Fig. 1D). Dp186 and Dp205 appear to be absent from the adult heart.
Fig. 1.
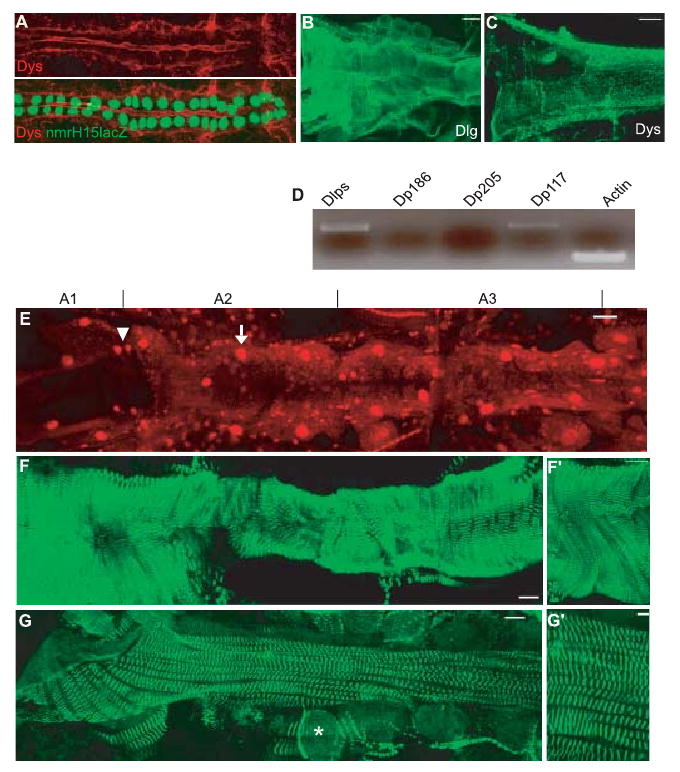
Protein distribution in Drosophila embryo and adult heart. (A) Stage 16 wild-type (wt) embryo double-labeled for Dystrophin (Dys, red) and nuclear nmrH15-lacZ (green; see Qian et al., 2005) Dystrophin (Dys) shows dorsal and ventral depositions in the myocardial cells (green nuclei) (B, C, E–G) Adult cardiac tube stained for disc large, (B) Dys, (C) protein outlining the myocardial cell membranes (bar = 25 μm). Note that Dys is localized at the Z-lines in the longitudinal muscles on the side of the myocardial cells. (D) Reverse transcriptase–polymerase chain reaction (RT–PCR) of wt adult heart using specific primers for Dys-like products (DLPs), Dp186, Dp205 and Dp117 compared to control actin (30 PCR cycles). Note that only DLPs and Dp117 are expressed in the adult heart. (E) Dmef2 expression in the adult cardiac tube (A1–A3). Dmef2 is a muscle-specific transcription factor, expressed in all Tinman (arrow, big nuclei) and Seven-up (Svp) expressing myocytes (arrowhead, smaller nuclei and localizes to the ostia) (for further details, see Molina & Cripps, 2001). (F, G) α-Actinin expression in the adult cardiac tube (abdominal segments A1–A3 are shown). (F) Sarcomeric Z-line marker α-actinin shows a spiral or transverse organization of the myofibrils of the contractile myocardium. (F′) High magnification of the myocardium shows the orientation of the myofibrils. (G) α-Actinin reveals a second type of myofibrillar organization in longitudinal non-Tinman-expressing muscles associated with the ventral part of the heart (Molina & Cripps, 2001) (* indicates pericardial cells). (G′) High magnification. For E–G, the bar = 38 μm; for F′, bar = 106 μm; and for G′ bar = 100 μm.
Staining of the cardiac tube with the sarcomeric Z-line marker, α-actinin, reveals two kinds of myofibrillar organization: the myofibrils of the contractile Dmef2-positive ‘working’ myocardium (Fig. 1E), which are also Tinman positive, are organized in a transverse or spiral fashion (Fig. 1F,F′; Molina & Cripps, 2001); and the longitudinally oriented myofibrils of syncytial muscles containing small Dmef-2 positive nuclei (without Tinman) are associated with the myocardium along the ventral side (Fig. 1G,G′).
dys Knockdown and deletion mutants decrease lifespan
To investigate dys function on organism longevity, we examined flies with reduced dys expression. A small deletion Dys8-2 has been reported to affect some of the long DLP forms, whereas Df(3R)KX43 (Dyskx43) (Shcherbata et al., 2007) and Df(3R)Exel6184 (DysExel6184) are larger deficiencies of the dys locus. We first checked the isoform expression in each transheterozygous deficiencies by RT–PCR analysis (Fig. 2A). Using specific primers for each isoform, we determined that in the transdeficiency DysExel6184/Dyskx43 flies, all isoforms were absent except the short-form Dp117, whereas the Dyskx43/dys8-2 or DysExel6184/dys8-2 combinations show a modest decrease in most isoforms (Fig. 2A). We also examined the effects of dys-RNAi transgenic flies targeting all dys isoforms (Shcherbata et al., 2007). To determine to what extent the mRNA levels of dys isoforms in the dys-RNAi line are affected, we performed quantitative RT–PCR (qRT–PCR) on heart RNA, using primers specific for all isoforms. Quantitative RT–PCR analysis shows an approximately 60% reduction in all isoforms when knockdown was achieved with the mesodermal 24B–Gal4 driver, and an approximately 40% reduction with a cardiac-specific driver GMH5–Gal4 (Fig. 2B,C; Wessells et al., 2004).
Fig. 2.

Loss of dys function decreases the lifespan in flies. (A) Whole fly reverse transcriptase–polymerase chain reaction indicates the abolishment or reduction in dys isoform transcripts. dysExel6184/dyskx43 flies abolish all but Dp117 transcripts, whereas dysExel6184/dys8-2 and dys8-2/dyskx43 mutants have moderately reduced DLPs transcript levels. (B) Adult expression pattern of GMH5-Gal4 driving with GFP expression specifically in the heart (see also Wessells et al., 2004) (bar = 19 μm). (C) Relative expression of dystrophin in cardiac tube of 1-week-old adults presented as ratio of dys to rp49 mRNA. The bar graph shows that the knockdown of all dys isoforms in dysRNAi/24B-Gal4 flies causes a reduction of ∼60%, and ∼40% with the heart-specific dysRNAi/GMH5-Gal4 knockdown. Each value represents the average ± standard error of the mean of four independent experiments. (D) Survival curves of dys mutant females. The knockdown of all dys isoforms in the mesoderm (dysRNAi/24B-Gal4) shows a moderate reduction in longevity similar to the heterozygous deficiencies dysExel6184/+ and dyskx43/+0. In contrast, the heart-specific dys knockdown does not have an effect on lifespan. Dystrophin-like-product-deficient dysExel6184/dyskx43 mutants show dramatically shortened lifespan. It is possible that this transdeficiency deletes additional genes that are contributing to a normal lifespan.
We observed a moderate lifespan reduction in the mesodermal dys knockdown flies (13%), but not when exclusively targeted to the heart (Fig. 2D). In contrast, half of the transheterozygous DysExel6184/Dyskx43 deficiency flies died at 29 days compared to 63 days in controls (Fig. 2D). Because loosing one copy of a gene usually causes a corresponding reduction in the RNA level, we also tested the single heterozygous deficiencies for dys, which showed a moderate lifespan reduction similar to the dys knockdown with 24B–Gal4. Interestingly, a 40% knockdown in dys RNA levels specifically in the heart was not sufficient to impair adult viability. This suggests that a moderate dys loss-of-function in all muscles, but not in just the heart, reduces the fly's normal lifespan. Therefore, dys influences longevity in Drosophila reminiscent of the observed reduction in lifespan of dys-deficient mdx mice (Chamberlain et al., 2007).
Age-dependent abnormalities in myofibrillar organization in dys mutant hearts
Examination of the myocardium structure in dys-deficient mdx mice shows considerable muscle degeneration that is aggravated by age (Chamberlain et al., 2007). Defects in the thoracic muscles of dys mutant flies have also been reported (Shcherbata et al., 2007). We assessed the heart structure in flies by staining with α-actinin, a structural element and component of the contractile machinery of muscles localized to the Z-bands. The α-actinin staining revealed a tight and well-aligned spiral arrangement of cardiac myofibrils in wild-type (wt) flies at 1 week and 3 weeks of age (Fig. 3A,B′,G,I). Older hearts (at 5 weeks) begin to show disruptions and abnormalities in the parallel alignment of myofibrils (Fig. 3C,C′,I). In contrast, dys deficiency flies dysExel6184/dyskx43 revealed a less organized and less compact arrangement of the myocardial myofibrils already at 1 week of age (Fig. 3D.D′,H,I). This phenotype becomes progressively more pronounced in older dys deficiency flies, in that the myocardial myofibrils are increasingly disorganized with more gaps devoid of myofibrils (Fig. 3E–F′,I). Thus, dysExel6184/dyskx43 mutants show age-dependent disruption of the heart myofibril integrity. The gaps in myofibrillar α-actinin staining were quantified by measuring the size of these areas using image J software from confocal stacks (illustrated in Fig. 3G,G′,H,H′). The percentage area devoid of myofibrils from seven to eight hearts was plotted as according to age (Fig. 3I). Wild-type flies at 5 weeks show gaps covering approximately 3% of the heart tube, whereas in dysExel6184/dyskx43 hearts the gaps cover about 13% of the total myocardium. These data suggest that dys is required for the physical integrity of the heart muscle throughout the fly's life.
Fig. 3.
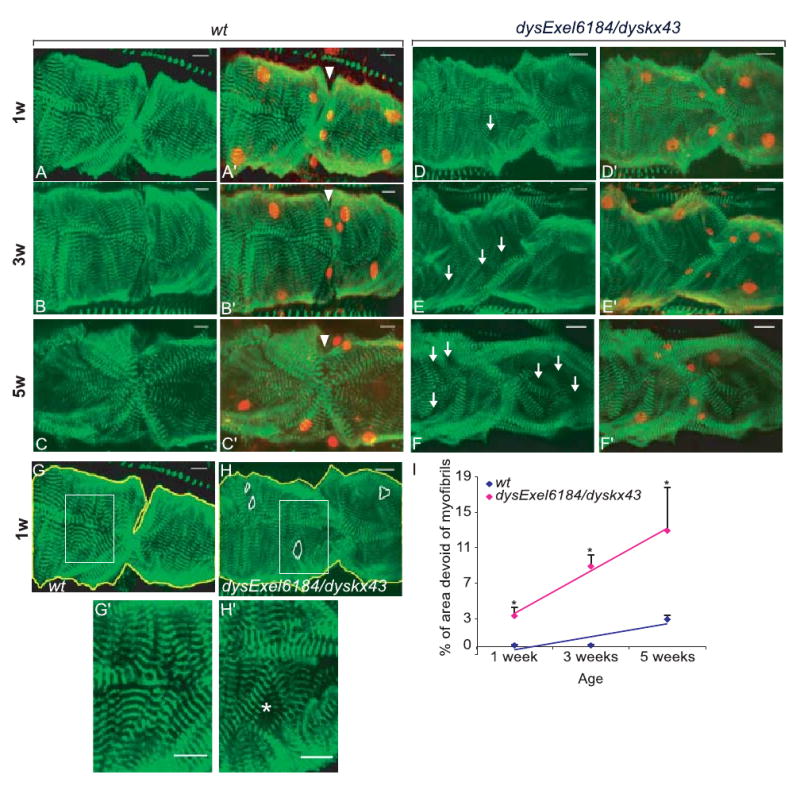
dysExel6184/dyskx43 Mutant flies exhibit age-dependent abnormalities in myofibrillar organization. Representative confocal stacks of adult hearts (posterior A2-anterior A3 segment) stained with α-actinin (green) and anti-Dmef2 (red, right panels) antibodies revealing detail of heart structure. (A-C′) Wild-type (wt) heart structure shows myofibrillar organization in a spiral fashion shown at progressively older different age (A, A′ 1-week-old: 1w; B, B′ 3-week-old: 3w; C, C′ 5-week-old hearts: 5w). The wt hearts at 5w reveal some disruptions of the myocardial myofibrils (C, C′). Ostia at the segmental boundaries of the myocardium were identified as an opening in the heart wall, associated with two pairs of smaller Dmef2-positive nuclei (A′–F′, arrowhead). (D–F′) The dysExel6184/dyskx43 mutant heart structure at 1w (D, D′), 3w (E, E′) and 5w (F, F′), respectively, shows age-dependent disruption of the heart myofibril integrity (D–F arrow pointing out to more spacement between myofibrils). (G) Wild-type and (H) dysExel6184/dyskx43 mutant hearts showing outlined areas devoid of myofibrils in the confocal stacks (H, white circulated area). Areas without myofibrils were measured and normalized to the total area of heart examined (outlined in yellow) using Image J software. See G′ and H′ for a close-up of the myofibril arrangement and that in H′ the asterisk points to a gap between myofibrils (I) Plot of quantification by age of areas devoid of myofibrils (dark areas not stained with α-actinin). Note that dysExel6184/dyskx43 mutant hearts show much more disorganization by this measure than wt. Each data point was from six to eight hearts. P < 0.005 at 1 week; P < 0.0001 at 3 weeks; P < 0.01 at 5 weeks between wt and dysExel6184/dyskx43 mutants. Scale bar = 12 μm.
Characterization of cardiac function in dys mutants
While the development of the Drosophila heart involves similar genetic pathways and molecular mechanisms as the vertebrate system, the fly has not yet been extensively exploited in heart physiology studies, with notable recent exceptions (Johnson et al., 2002; Wessells et al., 2004; Wolf et al., 2006; Ocorr et al., 2007). To study heart function, we dissected adult flies in artificial hemolymph to record cardiac contractions with a high-speed digital video camera (Ocorr et al., 2007); see Materials and Methods. M-mode traces of movie clips provided details in the heart wall edge positions (y-axis) over time (x-axis), illustrating the rhythmicity and the dynamics of heart contractions. The exposed and largely denervated heart of wt Drosophila shows regular rhythmic contractions (Fig. 4A, 1 week wt), which become progressively more irregular with age (Fig. 4A–C; Supplementary Fig. S1). Drosophila dys-deficient hearts lacking DLPs function (dysExel6184/dyskx43) also become more irregular with age (Fig. 4A–C), but in addition exhibit a significantly increased heart rate that corresponds to a reduced heart period (HP, defined diastolic plus systolic interval) at 3 weeks and 5 weeks of age compared to wt (Fig. 4B). The distribution of all the measured heart periods for all flies of a specific genotype and age is represented in histogram format (Fig. 4B). The majority of the wt flies show a relatively tight clustering of the heart period lengths at 1–3 weeks, and this distribution broadens in 5-week-old flies because of the increased variability in heart period with age (see also Ocorr et al., 2007).
Fig. 4.
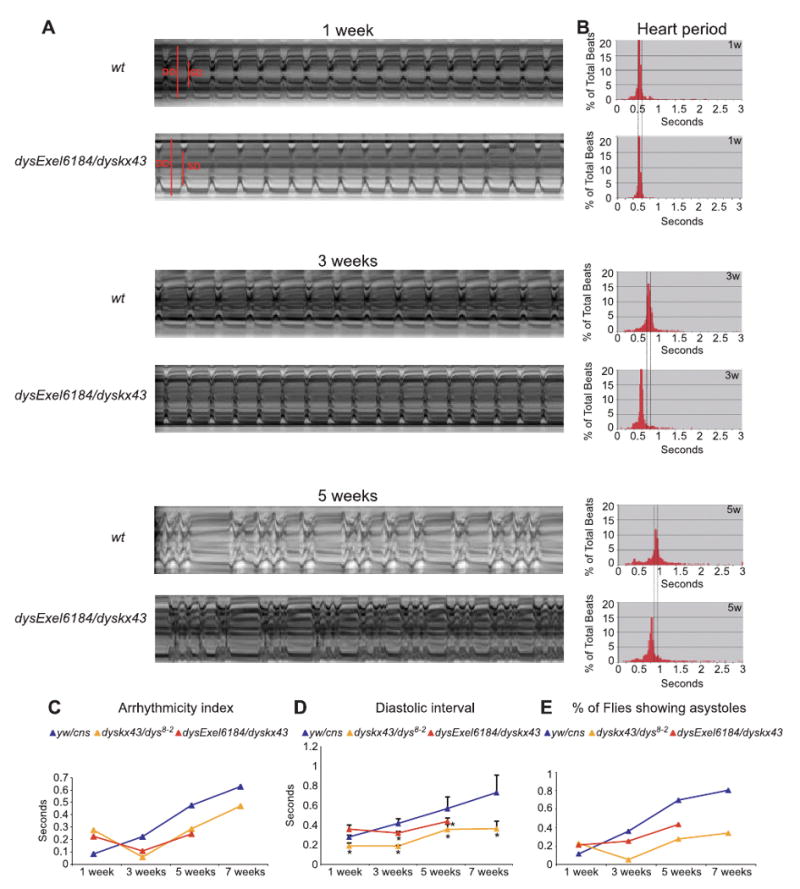
Reduced level or loss of the long dystrophin (Dys)-like product isoforms of Dys results in faster heart rate by shortening the diastolic intervals. (A) Representative M-mode traces (10 s) from high-speed movies of semi-intact flies. Wild-type (wt) flies show rhythmic heart beating at 1-week-old to 3-week-old of age, but moderate arrhythmicity at 5 weeks. dysExel6184/dyskx43 hearts show increased heart rate in 3-week-old and 5-week-old flies. (B) Heart period histograms obtained from 1 min movies plotted as individual data points illustrating the variability of the heart period within a group of flies for controls and dysExel6184/dyskx43 mutants. At 1 week, the mean heart period for wt and dysExel6184/dyskx43 flies is about 0.5 s, which increases in wt to 1 s at 5 weeks. In contrast, aging dysExel6184/dyskx43 shows less of an increase, to only 0.8 s at 5 weeks (bottom panel in B). (C) Standard deviation of the heart period was used as a measure of irregularity in heart rhythm (‘arrhythmicity index’). All time points (except 1 week) show less arrhythmicity for dys mutants compared to wt flies. Differences were estimated by t-test P < 0.05 and are indicated by *. (D) Mean diastolic interval (± SEM) for wt and dys mutant flies obtained from 1 min high-speed movies at the indicated ages. Significant differences were determined by two-tailed independent samples t-test. P values < 0.05 were considered significant (*P < 0.01). (E) Percentage of total flies showing asystoles (prolonged diastolic phases of more than 1 s) in 1-min movie clips. Differences were estimated by t-test P < 0.05 and are indicated by *.
The variability in the heart periodicity can be quantified using the heart period standard deviation as an ‘arrhythmicity index’ (Fig. 4C, AI; Ocorr et al., 2007). Wild-type flies show a low value for this AI at 1 week, and this value increased with age (Fig. 4C, see also Ocorr et al., 2007). The AI for dys mutants is elevated at 1 week compared to wt, but is reduced at progressively older ages in dys mutants (Fig. 4C). The shorter heart period/increased heart rate is mainly because of a shorter diastolic interval (relaxation period; Fig. 4D). This elevated heart rate may explain the more regular heartbeat observed in older dys flies. Another parameter of rhythm abnormalities studied in dys mutants is the distribution of asystoles within the 1-min movies. An asystolic event is a prolonged relaxation we defined as a diastolic interval longer than 1 s. We find that dyskx43/dys8-2 and dysExel6184/dyskx43 flies show a reduced incidence of asystolic events with age than do the control flies (Fig. 4E). These results suggest that partial or total lack of DLP function causes shorter diastolic intervals, resulting in increased heart rates and fewer asytoles, and producing a more regular heartbeat with age than wt.
We also tested the overall cardiac performance of dys mutants by pacing the heart to a higher rate (6 Hz) using external electrical pacing (as described in Wessells et al., 2004). dys-RNAi/24B–Gal4 or heterozygous dysExel6184/+ deficiency flies display a similar age-dependent increase in the heart failure rate immediately after pacing compared to wt (Wessells et al., 2004) or the age-matched dysRNAi/+control without the driver (Supplementary Fig. S1E). The reason why a reduced dys function does not cause an elevated susceptibility to electrical pacing may be that they already have an increase in heart rate, making them less sensitive to this stress.
dys Mutant flies exhibit a ‘dilated cardiomyopathy’ phenotype
Our image analysis of heart contractions also provides cardiac chamber parameters, including end-diastolic and end-systolic diameters. In addition, the proportional decrease in heart wall diameter during contraction, referred to as the fractional shortening, provides an indication of the cardiac output. Wild-type flies display an average diastolic diameter of about 60 μm and a systolic diameter of 40 μm, which do not change much with age (Fig. 5A–D), and the fractional shortening in wt flies was 35–40% (Fig. 5E). In contrast, dyskx43/dys8-2 and dysExel6184/dyskx43 have a significantly wider diastolic (80–90 μm) and systolic (60 μm) diameter (Fig. 5A–D; see also Supplementary Fig. S1). In addition, the dys mutant fractional shortening was also reduced compared to wt (25–30%; Fig. 5E). Interestingly, some of the heterozygous dys mutants also show enlarged tube diameters (see Supplementary Fig. S2A,B). Three situations of phenotype are found according to the mutant severity: the dys mutant heterozygotes dysExel6184/+ and Dyskx43/+ show a dilated phenotype without reducing a fractional shortening, dyskx43/dys8-2 in addition have reduced fractional shortening later in life and dysExel6184/dyskx43 already from week 1 on (Fig. 5; Suppl. movies). This phenotype of cardiac chamber enlargement and impaired systolic function in dys mutants recapitulates the dilated cardiomyopathy phenotype observed in DMD patients or the mouse model mdx remarkably well (Quinlan et al., 2004; Wehling-Henricks et al., 2005).
Fig. 5.

Reduced or no dystrophin-like products (DLPs) causes dilated cardiomyopathy in the adult Drosophila heart. (A) Two-segment image of a 1-week-old wild-type (wt) heart in systole (top) and diastole (bottom). (B) 1-week-old dysExel6184/dyskx43 heart in systole and diastole. Note that both diastolic and systolic diameters are wider in the DLPs-deficient mutants compared to wt. Arrowheads indicate the heart wall in both phases of the cardiac cycle. (C–E) Heart parameters in wt and dys mutants. (C,D) dysExel6184/dyskx43 and dys8-2/dyskx43 mutants have a larger systolic (C) and diastolic diameters (D) than the wt at all ages. Significant differences were determined by t-test. P values < 0.05 were considered significant (*P < 0.0001). (E) Plot of fractional shortening for the wt and dys mutants. dysExel6184/dyskx43 shows lower fractional shortening at all ages, whereas dys8-2/dyskx43 mutants only at 3 weeks or older (*P < 0.001, except dys8-2/dyskx43 at 7 weeks is P < 0.05). Movies were taken from 15 to 20 flies for each genotype. (F, G) Rescue of dysExel6184/dyskx43 mutants by the mammalian short dys isoform Dp116. Mesodermal expression of Dp116 restores the dilated systolic diameter (F) and the reduced fractional shortening to near normal (G). The Dp116 transgene was driven by the mesodermal driver 24B-GAL4 in the dys mutant background (Dp116/+; dyskx43/dysExel6184,24B). Cardiac function evaluated in 1-week-old rescue dys mutants shows a normalization of the systolic dysfunction (F, G) (*P < 0.01 by t-test assuming equal variances). For the genotype Dp116/+; dyskx43/dysExel6184,24B measurements (± standard error) were derived from movies of 20 flies. Similar rescue as with 1-week-old flies was observed in 3-week-old flies (data not shown).
A truncated mammalian Dys isoform (Dp116) rescues the dilated phenotype of Drosophila dys mutants
Next, we attempted to rescue the dilated cardiomyopathy phenotype characteristic of DLP-deficient dys mutant flies (dysExel6184/dyskx43) by expressing a truncated version of mouse dys. We sought to determine if any of the shorter isoforms of Dys, which assemble and localize the DGC but do not bind to the actin cytoskeleton (Yue et al., 2003; Judge et al., 2006), might be able to rescue any of the abnormalities in the fly hearts. Thus, we first expressed Dp116, a short C-terminal isoform of mammalian dys (Judge et al., 2006), in a wt background under the control of the mesodermal 24B–GAL4 driver and found no difference compared to wt (data not shown). In contrast, when we expressed mouse Dp116 in the mesoderm of dysExel6184/dyskx43 flies, we observed a smaller systolic diameter and a higher fractional shortening than dysExel6184/dyskx43 flies, which was comparable to wt and the other controls (Fig. 5F,G). This result suggests that introducing the short mammalian Dp116 transgene to the heart of dys mutant flies significantly restores the wt heart diameters and fractional shortening, and hence ameliorates abnormal systolic function. This would also be consistent with the idea that the functionally important Dys domains that rescue the dynamic cardiac properties include two spectrin repeats, WW-, CR- and C-terminal domains, and human exon 55S unique to Dp116 (Judge et al., 2006). The ability to rescue the abnormalities observed in dysExel6184/dyskx43 mutants with a dys transgene indicates strongly that dys is required for normal heart function.
Discussion
Drosophila has been used extensively to study cardiac development (Bodmer, 1995; Bodmer & Frasch, 1999; Prall et al., 2002; Zaffran & Frasch, 2002). More recently, the Drosophila model has also been adopted to investigate cardiac physiology, performance and aging (Wessells et al., 2004; Sanyal et al., 2006; Wolf et al., 2006; Ocorr et al., 2007; Mery et al., 2008) using assays to determine the contractility, rhythmicity and other performance parameters, for example, stress assay, in which the cardiac performance is tested by using external electrical pacing of the heart rate (Paternostro et al., 2001; Wessells & Bodmer, 2004), ultrasound-like image analysis of the heart in intact flies [using optical coherence tomography (OCT); Wolf et al., 2006], digital high-speed video imaging of semi-intact adult fly heart, which in addition allows characterizing the contractility and rhythmicity of the heart (Ocorr et al., 2007). Using these assays, it was found that the fly homologue of the KCNQ1 potassium channel gene, which is associated with human cardiac arrhythmias and long QT, causes heart rhythm abnormalities also when mutated in flies, and they become more severe with age (Ocorr et al., 2007). Moreover, mutations of genes encoding sarcomeric contractile proteins (tropomyosin II, troponin I, δ-sarcoglycan) showed dilated cardiomyopathy in flies (Wolf et al., 2006). Our studies of dys-dependent heart function abnormalities further validate the utility of Drosophila for studying the genetic basis of cardiac physiology.
The data described here explore the function of the cytoskeletal protein Dys in the fly heart. We find that: (i) mesodermal expression of Drosophila Dys is required for ‘normal’ lifespan; (ii) is necessary for the integrity of the heart structure (dilated heart phenotype and myofibril disorganization in dys mutants); and (iii) plays a physiologically important function in directly or indirectly modulating heart rate, rhythmicity and pumping efficiency (systolic dysfunction in dys mutants). These phenotypes are reminiscent of the ‘dystrophic’ heart phenotypes in mammals (Quinlan et al., 2004; Wehling-Henricks et al., 2005).
Lifespan analyses and age-dependent heart muscle disruption
Model organisms have been used extensively for studying the MD pathology (mdx-mouse, dog, hamster, Caenorhabditis elegans and zebra fish (see Gieseler et al., 2000; Watchko et al., 2002; Bassett & Currie, 2004). These animal models of muscular dystrophies have been complicated by gene redundancy, which can substitute at least in part for the deleted or mutated gene (e.g. dys-related utrophin; Grady et al., 1997). The single dys gene in Drosophila simplifies studies of functional requirements, and together with the fly's efficient genetic tools makes Drosophila a powerful model for probing dystrophic mechanisms. To study the effect of dys function on the fly's lifespan, we conducted longevity assays with dys mutants, including mesodermal and heart-specific dys knockdown. We found that mesodermal knockdown (dys-RNAi/24B–Gal4) reduces lifespan similar to heterozygote deficiencies (dysExel6184/+ and dyskx43/+), by about 13%. The decrease in lifespan of dys-RNAi/24B–Gal4 flies is likely a consequence of skeletal muscle deterioration (Shcherbata et al., 2007), as opposed to cardiac abnormalities, because (moderate) cardiac dysRNAi knockdown did not affect longevity.
Interestingly, a recent study shows that mdx mice show an approximately 20% reduction in lifespan compared to wt mice (Chamberlain et al., 2007). This mild reduction in mdx mice lifespan could be attributed to a partially compensatory up-regulation of the dys relative utrophin (reviewed in Blake et al., 2002). In old mdx mice, muscle histopathology is also more extensive and pronounced than what has been observed in young mdx mice (Chamberlain et al., 2007), which is consistent with our observations in the fly heart: dysExel6184/dyskx43 mutants show a strongly age-dependent disorganization of cardiac myofibrils. In mdx mice, fibrous connective tissue is observed in cardiomyocytes that is likely the consequence of the damage (Quinlan et al., 2004; Williams & Allen, 2007). In contrast to the vertebrate mdx heart, the fly myocardium does not show an increase in collagen gene expression (collagen types IV and XVIII), as determined by RT–PCR (data not shown).
Drosophila as a model for dilated cardiomyopathy
The lack of Dys in cardiac muscle leads to progressive cardiomyocyte degeneration and fibrosis (Rudge & Duncan, 1988; Williams & Allen, 2007). In the late phase of the disease, when extensive fibrosis is present, the conduction system is also perturbed. Among the DMD patients, 50% will have cardiac abnormalities by 18 years of age (Nigro et al., 1990). A progressive dilated cardiomyopathy occurs in all boys with DMD (Nigro et al., 1990). In BMD, cardiac disease is seen in over 70% of men by the age of 40 (Nigro et al., 1990; Saito et al., 1996). A severe dilated cardiomyopathy occurs in BMD with relatively preserved skeletal muscle function (Grain et al., 2001). Duchenne MD gene therapy has been mainly focused on correcting the skeletal muscle pathology in mdx mice (Chamberlain, 2002), with little progress to treat the cardiomyopathy (Koh et al., 1995; Yue et al., 2003). A notable exception is the recent finding that adenoviral-mediated delivery of a micro-Dys to mdx mice ameliorated the hemodynamic abnormalities and pump dysfunction of the heart under stress (Townsend et al., 2007). The paucity of cardiac gene therapy attempts in the mdx mice is mainly caused by our incomplete understanding of the role of Dys in this tissue. Using physiological assay in Drosophila may thus facilitate the role of cardiac dys function.
Analyzing the effects of dys mutations (reduced or complete loss of the long DLP form), we observed that loss of DLP function causes dilated hearts and lower fractional shortening, accompanied by increasing myofibril disorganization with age. dyskx43/dys8-2 Mutants show dilated heart, but the systolic function was not affected in young flies, only at more advanced ages. In contrast, the mutant DLP-deficient dysExel6184/dyskx43 flies exhibit a more severe phenotype already in young flies. Heterozygous dysExel6184/+ flies have been reported to also show dilated cardiomyopathy, using OCT (Wolf et al., 2006), which is consistent with our results using high-speed transmitted light imaging (Supplementary Fig. S2A,B). It is important to note that in our semi-intact preparation, heart physiology is recorded under drastically reduced neuronal or hormonal influences, thus assessing the myogenic properties of the heart. The observed phenotypes suggest a haploinsufficiency for dys in cardiac physiology even in the absence of obvious structural abnormalities, supporting the idea of a conserved role for dys in maintaining heart function from flies to humans (this study; Nigro et al., 1990; Goodwin & Muntoni, 2005).
We also observed an increased heart rate with age in dys mutant flies, which is mainly because of shorter diastolic phases, and is accompanied by a more regular heart rhythm and less asystolic episodes. Interestingly, a recent report shows that mdx mice have increased heart rate apparently because of a shortened PR interval (Bostick et al., 2008). Moreover, the inactivation of the C. elegans dys-1 gene does not show muscle degeneration, perhaps because of their short lifespan, but these animals display body wall muscle hypercontraction and are hyperactive (Bessou et al., 1998; Carre-Pierrat et al., 2006). This phenotype has been linked to the BK (Slo) calcium-activated large conductance potassium channels. The hyperactivity phenotype in C. elegans dys-1 is because of the down-regulation of the SLO-1 channel activity in muscles but not in neurons (Carre-Pierrat et al., 2006). Further studies are required to determine whether the increased heart rate in flies has a similar cause as the worm hypercontractility. Interestingly, other Dys models also seem to exhibit ion channel misregulation (e.g. slo, Kir4.1 and calcium channels; Mallouk & Allard, 2000; Connors & Kofuji, 2002). Abnormalities in calcium handling could also be a mechanism that contributes to the observed increase in heart rate in flies, hypercontractility in worms and hypercontraction and death of mouse mdx myocardial cells (see Yasuda et al., 2005). Furthermore, mdx hearts have been shown to have defects in Ca2+ handling proteins, including decreased cardiac sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) mRNA and delayed normalization of intracellular Ca2+ concentration (Rohman et al., 2003; Williams & Allen, 2007).
Drosophila has been proven to be a valuable model to dissect the DMD pathogenicity (Shcherbata et al., 2007), including that of the heart (this study). Importantly, we have shown that a truncated human form of dys (Dp116) is able to rescue the dilated cardiac phenotype of DLP-deficient fly mutants. This is in contrast to the finding that expression of Dp116 in skeletal vertebrate muscles acts as a dominant negative, aggravating the dystrophic phenotype (Judge et al., 2006). Even though Dp116 expression does not rescue dystrophic skeletal muscles of mdx mice, it does ameliorate the severe dystrophin:utrophin double mutant phenotype (L. Judge and J.S.C., unpublished). The rescue of the fly heart function by Dp116 may thus be caused by the assembling and the restoration of higher levels of the DGC and increased signaling functions of the DGC, as recently shown in skeletal muscles of dystrophin:utrophin double knockout mice (L. Judge and J.S.C., unpublished). Notably, both flies and humans naturally have a short C-terminal dys isoform expressed in the heart (Dp71 in mammals and Dp117 in flies; Muntoni et al., 2003; present data). Thus, it may be that Dp116 has a positive effect on muscle functionality in the heart but not in skeletal muscle. Alternatively, the fly system may be simpler and thus be able to use this truncated form to ameliorate (heart) muscle function, which in mammals may only become evident if both dys and utrophin are compromised. In any case, our data indicate that Drosophila can be used as a genetic tool to probe for suppressors or enhancers of (dilated) ‘cardiomyopathy’, thus allowing us to elucidate the mechanism of Dys-dependent disease.
Materials and methods
Drosophila strains
Exelixis stock dysExel6184 was obtained from the Drosophila Stock Center (Bloomington, IN, USA; http://flystocks.bio.indiana.edu). Df(3R)Dl-X43 referred to as dyskx43 and the dys8-2 deletion mutant flies were a generous gift from Hannele Ruohola-Baker (University of Washington, Seattle, WA, USA) (Shcherbata et al., 2007). dys RNAi transgenic Drosophila against all isoforms was generously provided by Uri Nudel (Weizmann Institute of Science, Rehovot, Israel; Shcherbata et al., 2007). The heart-specific driver GMH5–Gal4 is a 900 bp heart enhancer fragment 73 from the tinman gene (Bodmer, 1993; Venkatesh et al., 2000) that was cloned into the P{GaWB} vector upstream of the Gal4 sequences. This driver was enhanced with multiple copies of a UAS–Gal4 element allowing stronger myocardial expression when activated in late embryonic or during adult stages (detailed description in Wessells et al., 2004). 24B–GAL4, also known as P{GawB}how[24B], which drives expression in all muscles was described previously in Brand & Perrimon (1993) (see also Zaffran et al., 1997 and full description on the flybase Web site http://flystocks.bio.indiana.edu/Browse/misc-browse/gal4.htm).
Micro-Dys Dp116 transgenic flies
The mouse short C-terminal isoform Dys Dp116 (containing the first exon from the human Dp116 isoform) was generated by Judge et al. (2006). Dp116 contains two spectrins repeats, the ww domain (dystroglycan-binding domain) and a carboxy-terminal domain involved in binding to other DGC proteins like syntrophin and dystrobrevin (for details of the construct, see Judge et al., 2006). For the generation of transgenic Dys flies, Dp116 cDNA construct was subcloned into the Gal4-inducible vector pUAST at the NotI site. The UAS–Dp116 construct was injected into w1118 embryos, and transgenic lines (two) were established (according to standard procedures). UAS–Dp116 transgenic flies were crossed to the heterozygous deficient flies dyskx43/+ to generate a stock Dp116/cyo, dyskx43/TM3. A stock of 24B–Gal4, dysExel6184/TM3 was generated by recombining the driver 24B–Gal4 and the deficiency line dysExel6184/TM3 in the same line. Thus, the flies Dp116/cyo, dyskx43/TM3 were crossed to 24B–Gal4, dysExel6184/TM3 and Dp116/+; dyskx43/24BGal4-dysExel6184 were generated and tested for heart function.
Immunostaining of embryos and adult Drosophila hearts
The embryo staining was performed as described previously (Kosman et al., 2004). The following primary antibodies were used: rabbit anti-Dys antibody was a gift from Andreas Wodarz (University of Göttingen, Göttingen, Germany) used at 1/1000 (see Schneider et al., 2006), mouse anti β-galactosidase 1/500 (Sigma, St Louis, MO, USA), mouse anti α-actinin (a gift from J. Saide; Saide et al., 1989) used at 1/40, rabbit anti-Dmef2 at 1/100 (Lilly et al, 1995) and mouse anti-Dlg 1/100 (Hybridoma Bank, University of Iowa, Iowa City, IA, USA). The secondary antibodies donkey anti-rabbit and anti-mouse conjugated with Alexa Fluor 555 dye and Alexa Fluor 488 dye used at 1/500 (Molecular Probes, Eugene, OR, USA), respectively. Adut flies were dissected to expose the heart, fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) for 20 min, washed three times in PBT (PBS/0.1% Triton X-100). The preparations were incubated with primary antibodies in PBT/Western blocking reagent 1× [Roche (Roche Applied Science, Indianapolis, IN, USA) #1921673] for 2 h, washed three times 10′ and then incubated with secondary antibodies for 1 h at room temperature. After washing in PBT/Western blocking reagent 1× three times 10′, they were mounted onto slides in DABCO/Tris/glycerol [2.5% DABCO (Sigma #D-2522)/50 mm Tris pH 8.0/90% glycerol] and analyzed using a single-photon laser scanning microscope. The quantification of the areas devoid from myofibrils was performed by measuring the dark areas not stained with α-actinin with mage J software. The average and the standard error was calculated by Microsoft Excel software.
Reverse transcription reaction and qRT–PCR analysis
Total RNA was extracted from whole flies or isolated hearts using Trizol reagent (Promega, Madison, WI, USA). The samples were treated with DNAase (Invitrogen, Carlsbad, CA, USA) to eliminate any remaining DNA. First-strand cDNA synthesis was performed using SuperScript III First-Strand Synthesis System for RT–PCR (Invitrogen #18080-051). Polymerase chain reaction was performed with actin or ribosomal protein RP49 as controls, and dys-specific primers for Dlps, Dp186, Dp205 and Dp117. The oligonucleotides used were as follows: actin forward ATCCGCAAGGATCTGTATGC, actin reverse ACATCTGCTGGAAGGTGGAC; Rp49 forward GACGCTTCAAGGGACAGTATCTG, Rp49 reverse AAACGCGGTTCTGCATGAG; 281 bp Dlps product forward AGCGTTGGCCGGCAGCTTCG, Dlps reverse CCGCTGAACAGTGCGGTGCT; 294 bp product of Dp186 forward AGGAACTTCCGGTTCGCAGC, Dp186 reverse AGCTGGGTTTCGTCGTGCGA; 263 bp Dp205 product forward CAAGTGCTCGGAGGCCCTGC, Dp205 reverse ACAGCCGCATGCGATCGTCG; 263 bp product for Dp117 forward TCGCTGAAGCGTCGCAGTCG, Dp117 reverse TGCGAGTAACTCAGGCTCAG. To determine the extent of RNAi knockdown of all dys transcripts, we used primers in the common sequence for all isoforms and outside the dsRNA sequence: forward primer CAAGTGGCCTAGTGACCGTAA; reverse primer CGTCGTCGTCGTGGTGTTCGT corresponding to a 104 bp fragment. For qPCR, we used the LightCycler FastStart DNA MasterPLUS SYBR Green I kit (Roche Applied Science).
Lifespan analysis
Procedures for lifespan studies were as described in Wessells et al. (2004). Flies were kept at 25 °C transferred to fresh food on alternate days and scored the deaths after each transfer.
Heart physiological analysis
Anesthetized flies with fly nap (Carolina Biol., Corp., Burlington, NC, USA) for 2 min were aligned dorsal down on a dish and cutting off the head and the ventral parts of the body (thorax + abdomen). These semi-intact preparations of flies were cleaned from fat and other tissues except the heart, and kept for 20 min equilibration before taking movies. Movies were taken at rates 100–130 frames per second by using Simple PCI software (Compix, Sewickley, PA, USA). Cardiac parameter measurements were obtained as output from the MatLab-based program (Mathworks, Natick, MA, USA). For further description, see Ocorr et al. (2007).
External electrical pacing of the fly's heart rate was carried out as previously described (Wessells & Bodmer, 2004; Wessells et al., 2004).
Supplementary Material
Fig. S1 Cardiac parameters in wild-type and dys mutants at 1 week, 3 weeks, 5 weeks and 7 weeks of age. (A) Mean heart period in seconds (s) [±standard error of the mean (SEM)] illustrating shorter heart period for dys mutants compared to age-matched controls yw/CantonS flies obtained from 1-min movies at the indicated ages; n = 15–20 flies per data point. Note that dys8-2/dyskx43 flies show shorter heart period at 1–7 weeks old compare to age-matched controls, and dysExel6184/dyskx43 from 3-week-old on. Significant differences were determined by t-test; P values < 0.05 were considered significant (*). (B) Standard deviation of the heart period is used as a measure of the arrhythmicity (‘arrhythmicity index’). Note all time points (except 1-week-old) dys mutants are more rhythmical than the controls. (C) Mean diastolic intervals (±SEM) for dys mutants are shorter than the controls at different ages. Significant differences were determined by sample t-test. P values < 0.05 were considered significant (*P < 0.01). (D) Percentage of total flies showing asystolic episodes (diastolic intervals > 1.3 s) in 60-s movies. Significant differences were estimated by t-test, *P < 0.05. (E) Mesodermal knockdown of the dys long-form dystrophin-like products does not exhibit elevated heart failure rates in response to electrical pacing compared with controls dysRNAi/CantonS. The hetezygous dys flies dysExel6184/cantons do not show either an increase in heart failure compared to controls.
Fig. S2 (A, B, C) Heart parameters in wild-type and dys mutants. (A, B) Dystrophin-like product mutants dysExel6184/dyskx43 and dys8-2/dyskx43, as well as the heterozygotes dysExel6184/cantonS and dyskx43/cantonS have a larger diastolic (A) and systolic diameters (B) than age-matched controls. Significant differences were determined by t-test. P values < 0.05 were considered significant (*P < 0.0001; P < 0.001 for dysExel6184/cantons at 1-week-old and 7-week-old). (C) Plot of fractional shortening for the controls and dys mutants. The dys Exel6184/dyskx43 shows lower fractional shortening from younger age (1 week) to 5-week-old, whereas the mutant dys8-2/dyskx43 illustrates lower fractional shortening from 3-week-old. P values < 0.05 were considered significant (*P < 0.001 for 1 week; P < 0.0001 for 3 weeks and 5 weeks; dys8-2/dyskx43 at 7 weeks P < 0.05). Note the dilated hearts for dysExel6184/CantonS and dyskx43/CantonS do not show impairment in systolic function. All data are expressed as mean ± standard error of the mean.
Suppl. movies: View of a beating adult cardiac tube for wild type and dys mutants flies (1 week old).
Acknowledgments
We thank Martina Schneider and Hannele Ruohola-Baker for generously providing dys RNAi transgenic, dys8-2 and dyskx43 flies. We are grateful to Judith Saide for sending anti α-actinin antibodies, and to Andrea Wodarz for sending anti-Dys antibodies. We thank Katherine Kling for helping with the preparation of the manuscript, Anthony R. Cammarato for the myofibril quantification and Laurent Perrin for critically reading the manuscript. This research was supported by grants from NIA and NHLBI of the National Institutes of Health. Uri Nudel and David Yaffe receive support from AFM, MDA and the Israel Science Foundation.
Footnotes
Supplementary material: The following supplementary material is available for this article:
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1474-9726.2008.00367.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, et al. The genome sequence of Drosophila melanogaster. Science. 2000;5461:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Bassett D, Currie PD. Identification of a zebrafish model of muscular dystrophy. Clin Exp Pharmacol Physiol. 2004;8:537–540. doi: 10.1111/j.1440-1681.2004.04030.x. [DOI] [PubMed] [Google Scholar]
- Bessou C, Giugia JB, Franks CJ, Holden-Dye L, Segalat L. Mutations in the Caenorhabditis elegans dystrophin-like gene dys-1 lead to hyperactivity and suggest a link with cholinergic transmission. Neurogenetics. 1998;2:61–72. doi: 10.1007/s100480050053. [DOI] [PubMed] [Google Scholar]
- Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet. 2005;1:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- Bier E, Bodmer R. Drosophila, an emerging model for cardiac disease. Gene. 2004;342:1–11. doi: 10.1016/j.gene.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–729. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- Bodmer R. Heart development in Drosophila and its relationship to vertebrate systems. Trends Cardiovasc Med. 1995;5:21–27. doi: 10.1016/1050-1738(94)00032-Q. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Frasch M. Genetic determination of Drosophila heart development. In: Rosenthal N, Harvey R, editors. Heart Development. San Diego: Academic Press; 1999. pp. 65–90. [Google Scholar]
- Bonini NM, Fortini ME. Human neurodegenerative disease modeling using Drosophila. Annu Rev Neurosci. 2003;26:627–656. doi: 10.1146/annurev.neuro.26.041002.131425. [DOI] [PubMed] [Google Scholar]
- Bostick B, Yue Y, Long C, Duan D. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res. 2008;102 doi: 10.1161/CIRCRESAHA.107.162982. Epub. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cammarato A, Dambacher CM, Knowles AF, Kronert WA, Bodmer R, Ocorr K, Bernstein SI. Myosin transducer mutations differentially affect motor function, myofibril structure and the performance of skeletal and cardiac muscles. Mol Biol Cell. 2008;2:553–562. doi: 10.1091/mbc.E07-09-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carre-Pierrat M, Grisoni K, Gieseler K, Mariol MC, Martin E, Jospin M, Allard B, Segalat L. The SLO-1 BK channel of Caenorhabditis elegans is critical for muscle function and is involved in dystrophin-dependent muscle dystrophy. J Mol Biol. 2006;358:387–395. doi: 10.1016/j.jmb.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Chamberlain JS. Gene therapy of muscular dystrophy. Hum Mol Genet. 2002;11:2355–2362. doi: 10.1093/hmg/11.20.2355. [DOI] [PubMed] [Google Scholar]
- Chamberlain JS, Metzger J, Reyes M, Townsend D, Faulkner JA. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007;9:2195–2204. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- Connors NC, Kofuji P. Dystrophin Dp71 is critical for the clustered localization of potassium channels in retinal glial cells. J Neurosci. 2002;22:4321–4327. doi: 10.1523/JNEUROSCI.22-11-04321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers LC, van der Plas MC, van Loenen PB, Dunnen JT, van Ommen GJ, Fradkin LG, Noondermeer JN. Embryonic expression patterns of the Drosophila dystrophin-associated glycoprotein complex orthologs. Gene Expr Patterns. 2004;2:153–159. doi: 10.1016/j.modgep.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Fortini ME, Bonini NM. Modeling human neurodegenerative diseases in Drosophila: on a wing and a prayer. Trends Genet. 2000;4:161–167. doi: 10.1016/s0168-9525(99)01939-3. [DOI] [PubMed] [Google Scholar]
- Franco A, Jr, Lansman JB. Stretch-sensitive channels in developing muscle cells from a mouse cell line. J Physiol. 1990;427:361–380. doi: 10.1113/jphysiol.1990.sp018176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieseler K, Grisoni K, Segalat L. Genetic suppression of phenotypes arising from mutations in dystrophin-related genes in Caenorhabditis elegans. Curr Biol. 2000;18:1092–1097. doi: 10.1016/s0960-9822(00)00691-6. [DOI] [PubMed] [Google Scholar]
- Goodwin FC, Muntoni F. Cardiac involvement in muscular dystrophies: molecular mechanisms. Muscle Nerve. 2005;32:577–588. doi: 10.1002/mus.20352. [DOI] [PubMed] [Google Scholar]
- Grady RM, Merlie JP, Sanes JR. Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol. 1997;136:871–882. doi: 10.1083/jcb.136.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grain L, Cortina-Borja M, Forfar C, Hilton-Jones D, Hopkin J, Burch M. Cardiac abnormalities and skeletal muscle weakness in carriers of Duchenne and Becker muscular dystrophies and controls. Neuromuscul Disord. 2001;11:186–191. doi: 10.1016/s0960-8966(00)00185-1. [DOI] [PubMed] [Google Scholar]
- Greener MJ, Roberts RG. Conservation of components of the dystrophin complex in Drosophila. FEBS Lett. 2000;1–2:13–18. doi: 10.1016/s0014-5793(00)02018-4. [DOI] [PubMed] [Google Scholar]
- Haag TA, Haag NP, Lekven AC, Hartenstein V. The role of cell adhesion molecules in Drosophila heart morphogenesis: faint sausage, shotgun/DE-cadherin, and laminin A are required for discrete stages in heart development. Dev Biol. 1999;1:56–69. doi: 10.1006/dbio.1998.9188. [DOI] [PubMed] [Google Scholar]
- Holder E, Maeda RD, Bies RD. Expression and regulation of the dystrophin Purkinge promotor in human skeletal muscle, heart and brain. Hum Genet. 1996;2:232–239. doi: 10.1007/BF02265272. [DOI] [PubMed] [Google Scholar]
- Johnson E, Sherry T, Ringo J, Dowse H. Modulation of the cardiac pacemaker of Drosophila: cellular mechanisms. J Comp Physiol [B] 2002;3:227–236. doi: 10.1007/s00360-001-0246-8. [DOI] [PubMed] [Google Scholar]
- Judge LM, Haraguchiln M, Chamberlain JS. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J Cell Sci. 2006;119:1537–1546. doi: 10.1242/jcs.02857. [DOI] [PubMed] [Google Scholar]
- Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–226. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- Koh GY, Soonpaa MH, Klug MG. Stable fetal cardiomyocyte grafts in the hearts of dystrophic mice and dogs. J Clin Invest. 1995;96:2034–2042. doi: 10.1172/JCI118251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosman D, Mizutani CM, Lemons D, Cox WG, McGinnis W, Bier E. Multiplex detection of RNA expression in Drosophila embryos. Science. 2004;305:846. doi: 10.1126/science.1099247. [DOI] [PubMed] [Google Scholar]
- Lilly B, Zhao B, Ranganayakulu G, Paterson BM, Schulz RA, Olson EN. Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science. 1995;267:688–693. doi: 10.1126/science.7839146. [DOI] [PubMed] [Google Scholar]
- Mallouk N, Allard B. Stretch-induced activation of Ca(2+)-activated K(+) channels in mouse skeletal muscle fibers. Am J Physiol Cell Physiol. 2000;278:C473–C479. doi: 10.1152/ajpcell.2000.278.3.C473. [DOI] [PubMed] [Google Scholar]
- Mery A, Taghli-Lamallem O, Clark KA, Beckerle MC, Wu X, Ocorr K, Bodmer R. The Drosophila muscle LIM protein, Mlp84B, is essential for cardiac function. J Exp Biol. 2008;(Pt 1):15–23. doi: 10.1242/jeb.012435. [DOI] [PubMed] [Google Scholar]
- Molina MR, Cripps RM. Ostia, the inflow tracts of the Drosophila heart, develop from a genetically distinct subset of cardial cells. Mech Dev. 2001;1:51–59. doi: 10.1016/s0925-4773(01)00509-3. [DOI] [PubMed] [Google Scholar]
- Monier B, Astier M, Semeriva M, Perrin L. Steroid-dependent modification of Hox function drives myocyte reprogramming in the Drosophila heart. Development. 2005;23:5283–5293. doi: 10.1242/dev.02091. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- Neuman S, Kaban A, Volla T, Yeffe D, Nudel U. The dystrophin/utrophin homologues in Drosophila and sea urchin. Gene. 2001;263:17–29. doi: 10.1016/s0378-1119(00)00584-9. [DOI] [PubMed] [Google Scholar]
- Neuman S, Kovlio M, Yaffe D, Nudel U. The Drosophila homologue of the dystrophin gene – introns containing promoters are the major contributors to the large size of the gene. FEBS Lett. 2005;579:5365–5371. doi: 10.1016/j.febslet.2005.08.073. [DOI] [PubMed] [Google Scholar]
- Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-g. [DOI] [PubMed] [Google Scholar]
- Ocorr K, Reeves N, Wessells R, Fink M, Chen HSV, Akasaka T, Yasuda S, Metzger JM, Giles W, Posakony J, Bodmer R. KNCQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci USA. 2007;104:3943–3948. doi: 10.1073/pnas.0609278104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternostro G, Vignola C, Bartsch DU, Omens JH, McCulloch AD, Reed JC. Age-associated cardiac dysfunction in Drosophila melanogaster. Circ Res. 2001;10:1053–1058. doi: 10.1161/hh1001.090857. [DOI] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stress developed during muscle contractions. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prall OW, Elliott DA, Harvey RP. Developmental paradigms in heart disease: insights from tinman. Ann Med. 2002;34:148–156. [PubMed] [Google Scholar]
- Qian L, Liu J, Bodmer R. Neuromancer Tbx20-related genes (H15/midline) promote cell fate specification and morphogenesis of the Drosophila heart. Dev Biol. 2005;279:509–524. doi: 10.1016/j.ydbio.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Rando TA. Role of nitric oxide in the pathogenesis of muscular dystrophies: a ‘two hit’ hypothesis of the cause of muscle necrosis. Microsc Res Tech. 2001;4:223–235. doi: 10.1002/jemt.1172. [DOI] [PubMed] [Google Scholar]
- Rohman MS, Emoto N, Takeshima Y, Yokoyama M, Matsuo M. Decreased mAKAP, ryanodine receptor, and SERCA2a gene expression in mdx hearts. Biochem Biophys Res Commun. 2003;310:228–235. doi: 10.1016/j.bbrc.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Rudge MF, Duncan CJ. Ultrastructural changes in the cardiomyopathy of dystrophic hamsters and mice. Tissue Cell. 1988;20:249–253. doi: 10.1016/0040-8166(88)90046-8. [DOI] [PubMed] [Google Scholar]
- Saide JD, Chin-Bow S, Hogan-Sheldon J, Busquets-Turner L, Vigoreaux JO, Valgeirsdottir K, Pardue ML. Characterization of components of Z-bands in the fibrillar flight muscle of Drosophila melanogaster. J Cell Biol. 1989;109:2157–2167. doi: 10.1083/jcb.109.5.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Kawai H, Akaike M, Adachi K, Nishida Y, Saito S. Cardiac dysfunction with Becker muscular dystrophy. Am Heart J. 1996;132:642–647. doi: 10.1016/s0002-8703(96)90250-1. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Jennings T, Dowse H, Ramaswami M. Conditional mutations in SERCA, the sarco-endoplasmic reticulum Ca(2+)-ATPase, alter heart rate and rhythmicity in Drosophila. J Comp Physiol [B] 2006;176:253–263. doi: 10.1007/s00360-005-0046-7. [DOI] [PubMed] [Google Scholar]
- Schneider M, Khalil AA, Poulton J, Castillejo-Lopez C, Egger-Adam D, Wodarz A, Deng WM, Baumgartner S. Perlecan and dystroglycan act at the basal side of the Drosophila follicular epithelium to maintain epithelial organization. Development. 2006;133:3805–3815. doi: 10.1242/dev.02549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbata HR, Yatsenko AS, Patterson L, Sood VD, Nudel U, Yaffe D, Baker D, Ruohola-Baker H. Dissecting muscle and neuronal disorders in a Drosophila model of muscular dystrophy. EMBO J. 2007;2:481–493. doi: 10.1038/sj.emboj.7601503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;2:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JE, Korenjak M, Brehm A. Tumour suppressors – a fly's perspective. Eur J Cancer. 2003;10:1355–1362. doi: 10.1016/s0959-8049(03)00263-6. [DOI] [PubMed] [Google Scholar]
- Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS, Metzger JM. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol Ther. 2007;15:1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- Van der Plas MC, Pilgram GS, Plomp JJ, de Jong A, Fradkin LG, Noordermeer JN. Dystrophin is required for appropriate retrograde control of neurotransmitter release at the Drosophila neuromuscular junction. J Neurosci. 2006;26:333–344. doi: 10.1523/JNEUROSCI.4069-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh TV, et al. Cardiac enhancer activity of the homeobox gene tinman depends on CREB consensus binding sites in Drosophila. Genesis. 2000;1:55–66. [PubMed] [Google Scholar]
- Watchko J, O'Day T, Wang B, Zhou L, Tang Y, Li J, Xiao X. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther. 2002;12:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005;14:1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- Wessells RJ, Bodmer R. Screening assays for heart function mutants in Drosophila. Biotechniques. 2004;37:58–60. doi: 10.2144/04371ST01. [DOI] [PubMed] [Google Scholar]
- Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet. 2004;36:1275–1281. doi: 10.1038/ng1476. [DOI] [PubMed] [Google Scholar]
- Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol. 2007;292:H846–H855. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- Wolf MJ, Amrein H, Izatt JA, Choma MA, Reedy MC, Rockman HA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci USA. 2006;103:1394–1399. doi: 10.1073/pnas.0507359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. doi: 10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan DR. Microdystrophins gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffran S, Frasch M. Early signals in cardiac development. Circ Res. 2002;91:457–469. doi: 10.1161/01.res.0000034152.74523.a8. [DOI] [PubMed] [Google Scholar]
- Zaffran S, Astier M, Gratecos D, Sémériva M. The held out wings (how) Drosophila gene encodes a putative RNA-binding protein involved in the control of muscular and cardiac activity. Development. 1997;124:2087–2098. doi: 10.1242/dev.124.10.2087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Cardiac parameters in wild-type and dys mutants at 1 week, 3 weeks, 5 weeks and 7 weeks of age. (A) Mean heart period in seconds (s) [±standard error of the mean (SEM)] illustrating shorter heart period for dys mutants compared to age-matched controls yw/CantonS flies obtained from 1-min movies at the indicated ages; n = 15–20 flies per data point. Note that dys8-2/dyskx43 flies show shorter heart period at 1–7 weeks old compare to age-matched controls, and dysExel6184/dyskx43 from 3-week-old on. Significant differences were determined by t-test; P values < 0.05 were considered significant (*). (B) Standard deviation of the heart period is used as a measure of the arrhythmicity (‘arrhythmicity index’). Note all time points (except 1-week-old) dys mutants are more rhythmical than the controls. (C) Mean diastolic intervals (±SEM) for dys mutants are shorter than the controls at different ages. Significant differences were determined by sample t-test. P values < 0.05 were considered significant (*P < 0.01). (D) Percentage of total flies showing asystolic episodes (diastolic intervals > 1.3 s) in 60-s movies. Significant differences were estimated by t-test, *P < 0.05. (E) Mesodermal knockdown of the dys long-form dystrophin-like products does not exhibit elevated heart failure rates in response to electrical pacing compared with controls dysRNAi/CantonS. The hetezygous dys flies dysExel6184/cantons do not show either an increase in heart failure compared to controls.
Fig. S2 (A, B, C) Heart parameters in wild-type and dys mutants. (A, B) Dystrophin-like product mutants dysExel6184/dyskx43 and dys8-2/dyskx43, as well as the heterozygotes dysExel6184/cantonS and dyskx43/cantonS have a larger diastolic (A) and systolic diameters (B) than age-matched controls. Significant differences were determined by t-test. P values < 0.05 were considered significant (*P < 0.0001; P < 0.001 for dysExel6184/cantons at 1-week-old and 7-week-old). (C) Plot of fractional shortening for the controls and dys mutants. The dys Exel6184/dyskx43 shows lower fractional shortening from younger age (1 week) to 5-week-old, whereas the mutant dys8-2/dyskx43 illustrates lower fractional shortening from 3-week-old. P values < 0.05 were considered significant (*P < 0.001 for 1 week; P < 0.0001 for 3 weeks and 5 weeks; dys8-2/dyskx43 at 7 weeks P < 0.05). Note the dilated hearts for dysExel6184/CantonS and dyskx43/CantonS do not show impairment in systolic function. All data are expressed as mean ± standard error of the mean.
Suppl. movies: View of a beating adult cardiac tube for wild type and dys mutants flies (1 week old).