

Abstract
Smokers have a significantly higher risk for developing coronary and cerebrovascular disease than nonsmokers. Advanced glycation end products (AGEs) are reactive, cross-linking moieties that form from the reaction of reducing sugars and the amino groups of proteins, lipids, and nucleic acids. AGEs circulate in high concentrations in the plasma of patients with diabetes or renal insufficiency and have been linked to the accelerated vasculopathy seen in patients with these diseases. Because the curing of tobacco takes place under conditions that could lead to the formation of glycation products, we examined whether tobacco and tobacco smoke could generate these reactive species that would increase AGE formation in vivo. Our findings show that reactive glycation products are present in aqueous extracts of tobacco and in tobacco smoke in a form that can rapidly react with proteins to form AGEs. This reaction can be inhibited by aminoguanidine, a known inhibitor of AGE formation. We have named these glycation products “glycotoxins.” Like other known reducing sugars and reactive glycation products, glycotoxins form smoke, react with protein, exhibit a specific fluorescence when cross-linked to proteins, and are mutagenic. Glycotoxins are transferred to the serum proteins of human smokers. AGE-apolipoprotein B and serum AGE levels in cigarette smokers were significantly higher than those in nonsmokers. These results suggest that increased glycotoxin exposure may contribute to the increased incidence of atherosclerosis and high prevalence of cancer in smokers.
Smoking has been identified to be the single greatest preventable cause of morbidity and mortality in the United States (1). In 1985 alone there were approximately 400,000 deaths in the United States that could be directly attributable to cigarette smoking (2). The number one cause for death related to smoking is myocardial infarction, but smoking also increases the risk of death from peripheral vascular disease, cerebrovascular disease, chronic obstructive pulmonary disease (chronic bronchitis and emphysema), and lung cancer and other malignancies (3, 4).
The mechanism by which cigarette smoking contributes to atherosclerosis is not precisely known; however, smokers have been reported to have increased serum levels of cholesterol, triglycerides, very low density lipoproteins, and low density lipoproteins (LDL) and a decreased serum level of high density lipoproteins and apolipoprotein A1 (5). These lipid and lipoprotein abnormalities have been presumed to act in concert with smoking-induced changes in vascular wall homeostasis and blood coagulation to adversely affect cardiovascular integrity (6–8).
Cigarette smoking is also strongly associated with lung, bladder, pancreatic, esophageal, and other types of cancer (1). Cigarette smoke contains many carcinogens. For example, benzepyrene, 2-naphthylamine, and 4-aminobiphenyl are metabolized by the body’s P450 system to form reactive substances that can chemically interact with DNA nucleotides and cause mutations (9, 10). It recently has been shown that a reactive metabolite of benzepyrene can mutagenize hot spots in the p53 gene, which have been associated with lung cancer (11).
In previous work, we and others (12–18) have reported that reducing sugars, such as glucose, and other reactive glycation products can react nonenzymatically with proteins, lipids, and nucleic acids to produce a structurally diverse class of molecules known as advanced glycation end products (AGEs). In experimental models, the formation of AGEs is associated with atherosclerotic vascular disease (19, 20) and DNA mutations (21–26). AGEs arise by a succession of chemical steps that begin with the spontaneous, nonenzymatic addition of reducing sugars to amino groups to form reversible Schiff bases. Initially, these adducts rearrange to form Amadori products, but over time further rearrangements occur to produce irreversibly bound moieties that exhibit fluorescence, cross-linking properties, and a yellow-brown color (12, 27). Although the chemical structures of only a few AGEs that form in vivo have been determined (28–31), the chemical pathway, the Maillard reaction, leading to the formation of these products is well known to food chemists who have determined the structures of a number of small molecules that form during the cooking and processing of food (8, 31). Food chemists continue to study Maillard reaction products intensively, as they are responsible for much of the flavor and aroma of cooked foods.
Because the curing of tobacco also involves conditions conducive to Maillard chemistry, we reasoned that tobacco products might be a significant source of reactants capable of promoting AGE formation and thereby provide a possible mechanism to explain the high prevalence of atherosclerotic vascular disease and cancer in smokers. We describe the identification of tobacco-derived reactive glycation products in tobacco smoke that can readily promote AGE formation in vitro and in vivo.
MATERIALS AND METHODS
Preparation of Aqueous Tobacco Extracts.
Tobacco (360 mg/ml) from cigarettes was extracted in PBS for 1 h at 25°C with agitation and filter sterilized through a 0.45-μm Millex-HA filter unit (Millipore).
Preparation of Cigarette Smoke Condensate.
A “water-pipe” smoking device was designed and operated to allow a stream of smoke to come into contact with an aqueous solution. An aqueous solution (3 ml of PBS) was placed in a 25-ml glass Erhlenmeyer flask with a sidearm, and cigarettes were mounted in a 500-μl pipette tip that penetrated the hole in the stopper of the flask. The tip of the 500-μl pipette tip extended down past the opening of the sidearm of the flask but did not penetrate the aqueous solution. Once the device was assembled, a vacuum (approximately 20 mmHg; 1 mmHg = 133 Pa) was applied to the sidearm of the flask and the cigarette was lit. In some experiments, the pipette tip was modified: A 2 × 2-mm piece of commercial cigarette filter was used to plug the tip of the pipette tip, and then 500 mg of nothing, aminoguanidine hydrochloride, or sodium sulfate was placed between the small piece of filter and the cigarette. The preattached commercially prepared filters were not removed from the cigarettes in any of the experiments. Five cigarettes were smoked into each 3-ml aliquot of PBS. The resulting aqueous cigarette smoke condensate was filtered through a 0.45-μm Millex-HA filter unit (Millipore) before use.
In Vitro AGE Formation Assay.
Microtiter plates containing immobilized rat tail tendon collagen (Collaborative Research) were blocked with 50 μl of TPBS (PBS/0.05% Tween 20) for 1 h at 25°C, incubated with serial dilutions of either the tobacco extract or smoke-PBS solution for 18 h at 37°C, washed five times with TPBS, incubated with a rabbit polyclonal anti-AGE sera (pAb RU 9.9.93) for 1 h, washed five times with TPBS, and finally incubated with goat anti-rabbit IgG conjugated to alkaline phosphatase (Sigma). Bound glycotoxins were revealed by adding 250 μg/ml p-nitrophenyl phosphate in 100 mM diethanolamine, pH 9.5, for 45 min. The optical density was read at 405 nm on an ELISA plate reader (Dynatech, MR 5000). The software program biolynx 2.0 was used for generation of a standard curve and data extrapolation.
Glycotoxin Cross-Linking Activity.
A solution of PBS containing 2 mM EDTA was exposed to cigarette smoke as described above to obtain a smoke condensate. Forty milligrams of RNase A dissolved in PBS/EDTA containing 0, 5, or 50 mM aminoguanidine was exposed to the condensate, which contained the equivalent of eight cigarettes, and was incubated at 37°C in the dark for 0, 5, 8, 24, 48, and 72 h. Unbound and low molecular weight materials were removed using the ultrafiltration filter, Centricon 10 (Amicon). To assess the formation of RNase dimers, the samples were subjected to discontinuous SDS/PAGE under reducing conditions by using a 5% stacking gel and a 12% resolving gel and transfer to nitrocellulose paper. The blot was blocked with blocking solution (PBS/5% nonfat milk/1% BSA/0.2% Tween 20) for 1 h at 20°C, incubated with rabbit anti-ribonuclease A antibody conjugated to horseradish peroxidase (Biodesign International, Kennebunkport, ME) diluted in blocking solution for 45 min, washed extensively with PBS/0.2% Tween 20, and developed by enhanced chemiluminescence (Amersham) according to the manufacturer’s directions. The resulting fluorograph was processed using adobe photoshop and densitometric measurements of images (smoothed, sharpened, and contrast enhanced) determined by using the nih image program.
Fluorescence Determinations.
To assess RNase A-AGE-specific fluorescence, the emission at 440 nm upon excitation at 370 nm by using an LS 50B fluorescence spectrometer (Perkin–Elmer) was measured in the samples of RNase exposed to cigarette smoke condensate (preparation described above). All fluorescence values were measured at a protein concentration of 0.5 mg/ml.
Mutagenicity Studies.
The mutagenicity assay was performed as described by Maron and Ames (32). Briefly, Salmonella strains TA98, TA100, TA1535, and TA1537 (gift from B. N. Ames, Univ. of California, Berkeley) were cultured overnight at 37°C in Oxoid nutrient broth no. 2, incubated with serial dilutions of cigarette smoke condensate in 0.1 M sodium phosphate buffer, pH 7.4, in triplicate for 1 h at 25°C, and then plated on minimal glucose plates at a concentration of 0.1 ml per plate. The plates were then incubated for 48 h at 37°C, and the number of revertant mutants on each plate was counted.
Serum AGE-Apolipoprotein B (ApoB) Measurement.
The amount of AGE-modified ApoB (AGE-ApoB) was measured in human serum by sandwich ELISA employing a monoclonal anti-AGE capture antibody and an anti-ApoB-horseradish peroxidase conjugate by using a modification of the previously described assay (33). The samples were assigned a value from the standard curve and expressed in ApoB-AGE units/ml of serum.
AGE Measurement.
AGEs in human sera were measured by a competitive ELISA employing an AGE-specific monoclonal antibody (IgG1 subclass) raised to a glucose-derived AGE epitope as described (34). AGE immunoreactivity was determined in triplicate wells for samples that were serially diluted to fall within the linear range of the assay. AGE units were calculated relative to a synthetic AGE-BSA standard (33). The assay sensitivity was 5 units of AGE/ml, and the intra- and interassay coefficient of variation was 5 and 8%, respectively. Human subjects used in this study had no history of diabetes, hyperlipidemia, or renal or vascular disease.
RESULTS
Aqueous Extracts of Tobacco and Cigarette Smoke Contain Glycotoxins, Which Promote AGE Formation in Vitro.
Initially, we extracted the water-soluble reactive glycation products from tobacco leaves by soaking them in PBS. To test whether these tobacco products were capable of inducing AGE formation as assessed by specific immunoassay, we added the aqueous extract to collagen-coated plates for 18 h. The presence of newly formed AGEs on the collagen molecules was detected by using a specific anti-AGE polyclonal antibody. Fig. 1A shows a comparison of the AGE formation induced by tobacco extracts of eight brands of American cigarettes. All brands tested promoted the formation of AGE moieties. It is interesting to note that brand E, the “light” equivalent of cigarette brand D, contained more activity than brand E. This finding, which was consistently reproduced when other “regular” and “light” brands were compared, probably reflects a difference in the processing of the tobacco and also suggests that “tar” content is independent of glycotoxin content.
Figure 1.
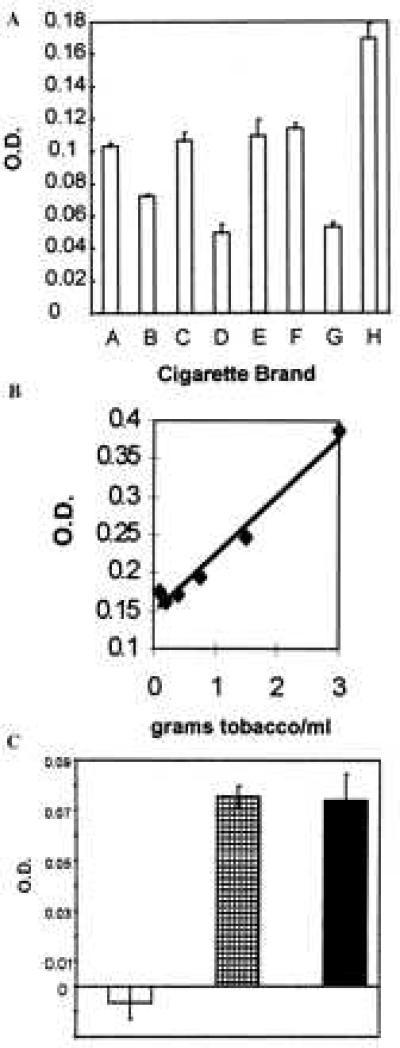
Glycotoxins in tobacco leaf extract and cigarette smoke condensate promote AGE formation in vitro. The in vitro AGE formation assay was performed on aqueous extracts of tobacco leaves from eight different brands of American cigarettes at a concentration equivalent to 90 mg of tobacco/ml (A) and on mainstream tobacco smoke at concentrations ranging from 3 to 0.375 g of tobacco/ml (B). Each bar represents a different brand of American cigarettes. Brand E is the light brand equivalent of brand D. AGE formation was revealed by incubation with a rabbit anti-AGE-specific polyclonal antibody. (C) AGE formation was inhibited by passing the mainstream cigarette smoke through a standard commercial filter + 500 mg of aminoguanidine (open bar) but not by passing it through a standard commercial filter (solid bar) or a standard commercial filter + 500 mg of sodium sulfate (striped bar).
To determine whether the reactive AGE precursors could be volatilized, we tested cigarette smoke condensate in the AGE formation assay. To collect any water-soluble volatile reactive AGE precursors, we set up a smoking apparatus in which smoke from a burning cigarette was drawn into a flask containing a solution of PBS. In an attempt to simulate the way in which lung tissue is exposed to cigarette smoke, smoke was permitted to come into contact with PBS in the bottom of the flask but was not bubbled through the solution. Immediately after preparation, the smoke-exposed PBS (cigarette smoke condensate) was added to microtiter wells coated with collagen and allowed to react overnight. Like the aqueous tobacco leaf extracts, cigarette smoke condensate was able to rapidly induce the formation of AGEs that were recognized by the rabbit anti-AGE polyclonal antisera. The production of AGEs by the cigarette smoke was concentration (Fig. 1B) and time (data not shown) dependent and could be inhibited in a dose-dependent manner by adding a soluble AGE inhibitor, aminoguanidine, directly to the wells (data not shown) or by passing the smoke through a solid column of aminoguanidine (Fig. 1C). Passing the smoke through a column of sodium sulfate had no effect on the ability of glycotoxins to form AGEs (Fig. 1C). Sodium sulfate was used as a control for the surface area of the aminoguanidine salt. The cigarette smoke condensate is approximately 10-fold less active at promoting AGE formation than the aqueous tobacco extracts, which suggests that either some of the reducing sugars are destroyed by the burning process or that the system for capturing the volatile AGEs is inefficient. The AGE-forming activity found in both the tobacco leaf extract and the cigarette smoke condensate is extremely labile: it is undetectable in this assay if the solutions are frozen at −20°C or stored at room temperature or 0°C for more than 5 h.
The ability of cigarette smoke glycotoxins to induce AGE formation was also assessed by measuring the characteristic fluorescence pattern of AGEs on RNase A exposed to cigarette smoke condensate. As shown in Fig. 2A, RNase A exposed to cigarette smoke condensate for increasing amounts of time exhibited a time-dependent increase in the AGE-type fluorescence (emission at 410 nm after excitation at 370 nm). Fluorescence emission reached saturation after 24 h and was inhibited by aminoguanidine.
Figure 2.
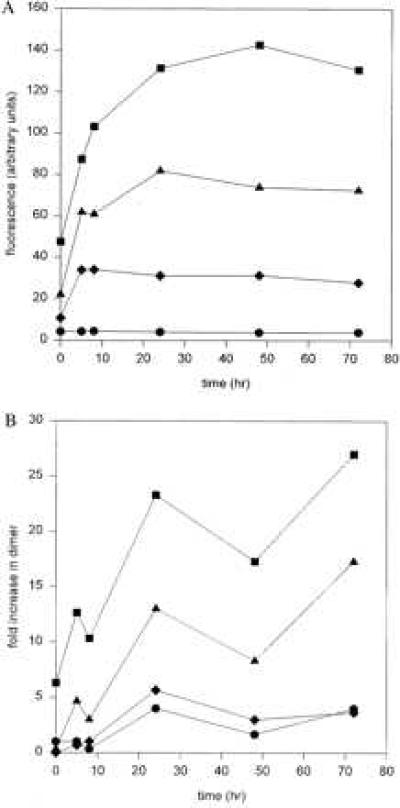
Glycotoxins in cigarette smoke condensate induce fluorescence and covalently cross-link proteins. (A) RNase A was exposed to mainstream cigarette smoke for 0, 5, 8, 24, 48, and 72 h and then assayed for RNase A-AGE-specific fluorescence by measuring emission at 440 nm upon excitation at 370 nm in the presence and absence of aminoguanidine. Circles, RNase A alone; squares, RNase A after incubation with cigarette smoke; triangles, RNase A after incubation with cigarette smoke and 5 mM aminoguanidine; diamonds, RNase A after incubation with cigarette smoke and 50 mM aminoguanidine. (B) RNase A was exposed to mainstream cigarette smoke for 0, 5, 8, 24, 48, and 72 h. To assess the formation of dimers, the samples were subjected to SDS/PAGE under reducing conditions, followed by transfer to nitrocellulose and Western blotting with a rabbit anti-RNase A antibody conjugated to horseradish peroxidase. Circles, RNase A alone; squares, RNase A after incubation with cigarette smoke; triangles, RNase A after incubation with cigarette smoke and 5 mM aminoguanidine; diamonds, RNase A after incubation with cigarette smoke and 50 mM aminoguanidine.
Glycotoxins Can Cross-Link Proteins.
One of the hallmarks of reactive glycation products is that they can also form intermolecular cross-links between the functional groups of two proteins. To show that glycotoxins, the reactive glycation products in tobacco, also had this property, we assayed glycotoxins for their ability to cross-link RNase A, a convenient model protein. RNase A was incubated with cigarette smoke condensate for increasing amounts of time, separated by molecular weight on an SDS/polyacrylamide gel, and then visualized with an anti-RNase A antisera. Using this technique, we were able to determine that RNase A-RNase A dimer formation was time dependent and reached saturation within 24 h. Dimer formation could be inhibited by aminoguanidine in a dose-dependent manner, indicating that the process depended on the presence of a carbonyl containing reactive glycation products (Fig. 2B).
Glycotoxins Are Mutagenic.
Previous studies have shown that reducing sugars can react in vitro with DNA and induce mutations in both bacterial and mammalian cells. Moreover, the presence of high concentrations of the reducing sugar, glucose-6-phosphate, in Escherichia coli leads to an increased mutation rate (23). Similarly, murine fetuses exposed to high blood sugar concentrations caused by maternal diabetes have been shown to have a 2-fold increase in their mutation rate compared with fetuses obtained from normoglycemic mothers (26).
In the present experiments, we exposed a number of strains of Salmonella typhimurium (TA98, TA100, TA1535, and TA1537) to cigarette smoke condensate in a typical Ames test. A brisk mutation activity was noted in strain TA98 but not in the other strains (Fig. 3). The exposure of TA98 to increasing amounts of freshly prepared cigarette smoke condensate results in a dose-dependent increase in mutations. When TA98 is exposed to a mutagenic agent that is able to cause a frameshift mutation, it will grow on histidine-deficient media. The passage of the smoke through a dry column of aminoguanidine before the contact with PBS removed the carbonyl containing molecules and decreased the number of revertant mutants. This activity may have been overlooked in the past as previous studies generally used smoke extracts prepared in organic solvents and analyzed the mutagenic activity after a considerable period of time. The lability of the glycotoxins in the aqueous extracts requires the rapid analysis of the mutagenic activity. Aqueous cigarette smoke condensates that were allowed to incubate either at room temperature or on ice for more than 5 h were no longer mutagenic.
Figure 3.
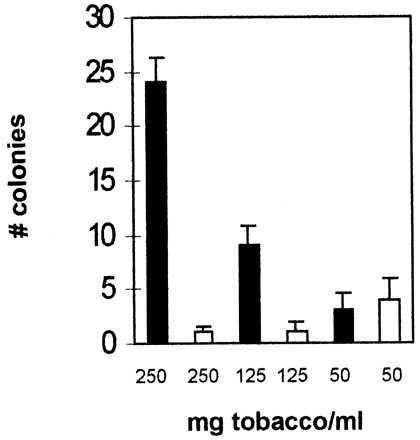
Glycotoxins are mutagenic. Salmonella strain TA98 was incubated with serial dilutions of cigarette smoke (solid bars) and cigarette smoke passed through aminoguanidine (open bars) at concentrations ranging from 250 mg of tobacco/ml to 50 mg of tobacco/ml for 1 h and then plated. Forty-eight hours later, the number of colonies on each plate was counted. Each bar represents the mean of three plates ± SD.
Glycotoxins Promote the Formation of AGEs on Serum Proteins in Vivo.
We next investigated the possibility that the glycotoxins could be absorbed in vivo after inhalation of cigarette smoke. Thus, we examined the amount of AGEs that were formed on serum proteins as a surrogate marker for total body exposure. We examined blood samples obtained from human smokers and nonsmokers. In this study, AGE serum protein levels and the more specific short-lived protein AGE-ApoB were assayed (Fig. 4). All individuals in the smoking group smoked more than one pack per day, but otherwise their smoking history was not controlled. The AGE-ApoB levels in smokers (units/ml ± standard deviation) (247 ± 76 ApoB-AGE units/ml, n = 17) were significantly higher (P = 0.018, unpaired Student t test) than nonsmokers (190 ± 54 ApoB-AGE units/ml, n = 17), and the serum AGE levels in smokers (210 ± 51 AGE units/ml, n = 17) were significantly higher (P = 0.01) than the serum AGE levels in nonsmokers (161 ± 53 AGE units/ml). Future studies may be able to correlate the amount of AGE attached to ApoB or other serum proteins present in the blood with the degree of exposure to cigarette smoke. This integral of exposure will allow analysis of cumulative exposure to first and second hand smoke. In effect, this test would be similar to the hemoglobin A1C test, which gives an integral of the blood glucose level for a 30-day period. It is interesting to note that the levels of AGE-serum proteins are nearly the same as that previously seen in patients with diabetes (AGE-serum proteins 200–400 units/ml).
Figure 4.
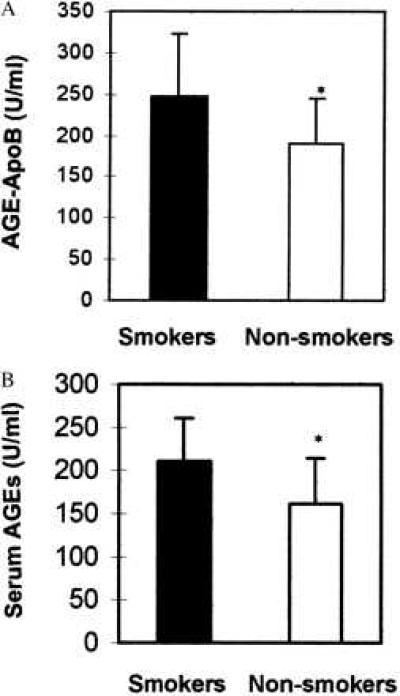
Serum ApoB-AGE (A) and total serum protein-AGE levels (B) were measured in healthy nondiabetic smokers and healthy, nondiabetic nonsmokers. AGE-ApoB levels in smokers (mean ± SD, 247 ± 76 units/ml, n = 17) were significantly higher (P = 0.018, unpaired Student t test) than nonsmokers (mean ± SD, 190 ± 54, n = 17). Serum AGE levels in smokers (210 ± 51 AGE units/ml, n = 17) were significantly higher (P = 0.01) than the serum-AGE levels in nonsmokers (161 ± 53 AGE units/ml, n = 17). AGE units are expressed relative to an ApoB-AGE standard (1 unit = 1 ng of ApoB, ICN Pharmaceuticals, Biochemical Division, Aurora, OH) and a BSA-AGE standard in the respective assays. Subjects were not controlled for age, sex, or number of pack-years smoked.
DISCUSSION
The observation that many of the vascular complications associated with cigarette smoking are also seen in diabetic patients who have high circulating and tissue-bound AGE levels prompted us to investigate the possibility that cigarette smoking promotes AGE formation. We show here that both aqueous extracts of tobacco and cigarette smoke contain glycotoxins, highly reactive glycation products that can rapidly induce AGE formation on proteins in vitro and in vivo and cause DNA mutations in vitro. Both of these activities can be eliminated by passing the samples through a dry packed column of aminoguanidine, a potent and specific inhibitor of AGE formation.
Glycotoxins from cigarette smoke have many unique characteristics; foremost is their high reactivity. In marked contrast to glucose or glucose 6-phosphate, both of which induce AGE formation over a period of days to weeks (34, 35), glycotoxins can induce AGE formation in hours. Furthermore, glycotoxins seem to readily cross cell membranes of bacterial and mammalian cells. Glycotoxins in smoke also seem to be readily absorbed through the lungs from cigarette smoke and in turn react with serum proteins, such as apoB. Although we have not yet determined the chemical structure(s) of glycotoxins, we can surmise that they have carbonyl groups and probably dicarbonyls because they are removed by aminoguanidine. The glycotoxins are probably a product of the rearrangement of products formed during the Maillard reaction, which is initiated during the tobacco curing process. The instability of the glycotoxins upon isolation implies that they and other Maillard products, which are responsible for the aroma and flavor of smoke, may be constantly formed and degraded as the tobacco is cured and aged.
The formation of AGEs in vivo by tobacco glycotoxins may be in part responsible for the increased incidence of atherosclerosis in cigarette smokers. There is abundant in vitro and in vivo evidence that AGEs play a role in the pathogenesis of atherosclerosis. (i) AGEs have been shown to cross-link connective tissue collagen, which serves to increase connective tissue rigidity (36). (ii) Collagen-linked AGEs serve as reactive “foci” to covalently trap circulating serum proteins, such as lipoproteins (37). (iii) Binding of endothelial cell AGE receptors will increase vascular permeability, decrease synthesis of the anti-coagulant thrombomodulin, and increase synthesis of the procoagulant tissue factor (38). (iv) Tissue-bound AGEs can chemically quench cell-derived nitric oxide activity, thereby inhibiting the nitric oxide-dependent vascular relaxation (39). (v) AGE modification of ApoB prevents the normal uptake of LDL by tissue LDL receptors, thereby increasing the circulating LDL cholesterol levels (40, 41). (vi) Markedly elevated vascular tissue and circulating AGEs have been linked to the accelerated vasculopathy of end-stage diabetic renal disease (20, 42–44). (vii) Exogenous AGE-modified albumin injected intravascularly into normal young rats and rabbits causes vascular alterations similar to those seen in animals with age- and diabetes-related vascular disease. Animals injected with exogenous AGE albumin show increases in vascular wall permeability, increased mononuclear cell migratory activity in subendothelial and periarteriolar spaces, and defective blood pressure response after challenge with acetylcholine and nitrogylcerin (19). It is reasonable to expect that the AGEs formed by the reaction of glycotoxins with serum and tissue proteins would have the same activities as the previously described AGEs.
Glycotoxins may also contribute to the increased incidence of cancer in cigarette smokers. In addition to reacting with amino groups on proteins and lipids, reactive glycation products can also react with the amino groups of nucleic acids. Incubation of either DNA or single nucleotides with reducing sugars in vitro produced absorbance and fluorescence changes similar to those observed for the AGE compounds bound to proteins and lipids. The incubation of bacterial plasmid DNA or mammalian shuttle vector DNA incubated with glucose or glucose-6-phosphate displayed considerable increase in mutation rates (21, 22, 24).
The most compelling evidence for the mutagenicity of reducing sugars in mammals comes from work done by Lee et al. (26) on the effect of a maternal hyperglycemic environment on developing embryos in mice. Embryonic cells, like the lens of the eye and red blood cells, do not require insulin for glucose transport across their membranes. Transgenic mouse embryos that contained integrated copies of the bacterial gene lacI, a well characterized mutagenesis marker, were transplanted into the uteri of streptozotocin-induced diabetic and control surrogate mothers. The lacI mutation frequency was significantly higher in the DNA from the embryos transplanted into the diabetic mothers than in those transplanted into control mice.
These experimental data are consistent with retrospective clinical studies that have shown that birth defects are three times more common in surviving infants born to diabetic mothers than to nondiabetic mothers. The incidence and severity of these defects correlates with the level of hyperglycemia immediately before conception or in the first trimester (45–49).
Although diabetic individuals do not have a demonstrable increase in any type of cancer, it is important to note that the intracellular glucose concentration in most cells is independent of the serum glucose level. However, tobacco-derived glycotoxins are far more potent than glucose. Like glucose and glucose 6-phosphate, they can cause insertion and/or deletion mutations in an aminoguanidine-dependent manner, but unlike these reducing sugars, glycotoxins can readily cross cell membranes. Further studies are needed to determine if glycotoxins can cause mutations in cultured mammalian cells or in vivo.
In summary, our findings argue that glycotoxins, found in cigarette smoke, are responsible in part for the increased rates of atherosclerotic vascular disease and cancer seen among cigarette smokers. During the process of smoking, high concentrations of glycotoxins are inhaled into the alveoli, where they may be both absorbed into the blood stream and taken up into the lung parenchymal cells. Once in the blood stream, glycotoxins may induce the formation of AGE moieties on both serum and vascular wall proteins and thereby accelerate the development of atherosclerosis. Glycotoxins may also react with DNA in the nuclei of the lung parenchymal cells to induce mutations and possibly cancer.
Acknowledgments
We thank Dr. Gregory Finch of the Lovelace Inhalation Toxicology Research Institute for advice and Drs. Michael Yamin and Thomas Donnelly for help with the editing and preparation of the manuscript. A.C. serves on the Board of Directors of Alteon Inc. A.C. and C.C. are paid consultants of Alteon Inc.
ABBREVIATIONS
- LDL
low density lipoprotein
- AGE
advanced glycation end products
- ApoB
apolipoprotein B
References
- 1.Bartecchi C E, MacKenzie T D, Schrier R W. N Engl J Med. 1994;330:907–912. doi: 10.1056/NEJM199403313301307. [DOI] [PubMed] [Google Scholar]
- 2.Robbins S L, Kumar V, Cotran R S. Pathological Basis of Disease. 5th Ed. Philadelphia: Saunders; 1994. pp. 379–430. [Google Scholar]
- 3.Shah P K, Helfant R H. Chest. 1988;94:449–452. doi: 10.1378/chest.94.3.449. [DOI] [PubMed] [Google Scholar]
- 4.Sherman C B. Clin Chest Med. 1991;12:643. [PubMed] [Google Scholar]
- 5.Muscat J E, Harris R E, Haley N J, Wynder E L. Am Heart J. 1991;121:141–147. doi: 10.1016/0002-8703(91)90967-m. [DOI] [PubMed] [Google Scholar]
- 6.Sugiishi M, Takatsu F. Circulation. 1993;87:76–79. doi: 10.1161/01.cir.87.1.76. [DOI] [PubMed] [Google Scholar]
- 7.Deanfield J E, Shea M J, Wilson R A, Horlock P, de Landsheere C M, Selwyn A P. Am J Cardiol. 1986;57:1005–1009. doi: 10.1016/0002-9149(86)90665-x. [DOI] [PubMed] [Google Scholar]
- 8.Krupski W C. Ann Vasc Surg. 1991;5:291–304. doi: 10.1007/BF02329389. [DOI] [PubMed] [Google Scholar]
- 9.Demarini D M, Shelton M L, Levine J G. Carcinogenesis. 1995;16:2535–2542. doi: 10.1093/carcin/16.10.2535. [DOI] [PubMed] [Google Scholar]
- 10.Florin I, Rutberg L, Curvall M, Enzell C R. Toxicology. 1980;8:219–232. doi: 10.1016/0300-483x(80)90055-4. [DOI] [PubMed] [Google Scholar]
- 11.Denissenko M F, Pao A, Tang M, Streck R J. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 12.Cerami A. Trends Biochem Sci. 1986;11:311–314. [Google Scholar]
- 13.Papoulis A, Al-Abed Y, Bucala R. Biochemistry. 1995;34:648–655. doi: 10.1021/bi00002a032. [DOI] [PubMed] [Google Scholar]
- 14.Al-Abed Y, Ulrich P, Kapurnitu A, Lolis E, Bucala R. Bioorg Med Chem Lett. 1995;5:2929–2930. [Google Scholar]
- 15.Al-Abed Y, Bucala R. Bioorg Med Chem Lett. 1995;5:2161–2162. [Google Scholar]
- 16.Al-Abed Y, Mitsuhashi T, Bucala R. Bioorg Med Chem Lett. 1996;6:1577–1578. [Google Scholar]
- 17.Henie T, Walter A W, Haebner R, Klostermeyer H. Z Lebensm Unters Forsch. 1994;199:55–58. doi: 10.1007/BF01193454. [DOI] [PubMed] [Google Scholar]
- 18.Konishi Y, Hayase F, Kato H. Biosci Biotechnol Biochem. 1994;58:1953–1955. doi: 10.1271/bbb.58.18. [DOI] [PubMed] [Google Scholar]
- 19.Vlassara H, Fuh H, Makita Z, Krungkrai S, Cerami A. Proc Natl Acad Sci USA. 1992;89:12043–12047. doi: 10.1073/pnas.89.24.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brownlee M, Cerami A, Vlassara H. N Engl J Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 21.Bucala R, Model P, Russel M, Cerami A. Proc Natl Acad Sci USA. 1985;82:1670–1674. doi: 10.1073/pnas.82.24.8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee A T, Cerami A. Mutat Res. 1987;179:151–158. doi: 10.1016/0027-5107(87)90305-8. [DOI] [PubMed] [Google Scholar]
- 23.Lee A T, Cerami A. Proc Natl Acad Sci USA. 1987;84:8311–8314. doi: 10.1073/pnas.84.23.8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee A T, Cerami A. Mutat Res. 1990;238:185–191. doi: 10.1016/0165-1110(90)90010-9. [DOI] [PubMed] [Google Scholar]
- 25.Lee A T, Cerami A. Mutat Res. 1991;249:125–133. doi: 10.1016/0027-5107(91)90139-f. [DOI] [PubMed] [Google Scholar]
- 26.Lee A T, Plump A, DeSimone C, Cerami A, Bucala R. Diabetes. 1995;44:20–24. doi: 10.2337/diab.44.1.20. [DOI] [PubMed] [Google Scholar]
- 27.Cerami A. In: Proceedings of the 5th International Symposium on the Maillard Reaction—Maillard Reactions in Chemistry, Food and Health. Labuza T, Reineccius G, Monnier V, O’Brien J, Baynes J, editors. Cambridge, U.K.: R. Soc. Chem.; 1994. pp. 1–10. [Google Scholar]
- 28.Njoroge F G, Sayre L M, Monnier V. Carbohydr Res. 1987;167:211–220. doi: 10.1016/0008-6215(87)80280-x. [DOI] [PubMed] [Google Scholar]
- 29.Sell D R, Monnier V. J Biol Chem. 1989;264:21597–21602. [PubMed] [Google Scholar]
- 30.Odetti P, Fogarty J, Sell D R, Monnier V. Diabetes. 1992;41:153–159. doi: 10.2337/diab.41.2.153. [DOI] [PubMed] [Google Scholar]
- 31.Pongor S, Ulrich P C, Bencsath F A, Cerami A. Proc Natl Acad Sci USA. 1984;81:2684–2688. doi: 10.1073/pnas.81.9.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maron D M, Ames B N. Mutat Res. 1983;113:173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 33.Makita Z, Vlassara H, Cerami A, Bucala R. J Biol Chem. 1992;267:5133–5138. [PubMed] [Google Scholar]
- 34.Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Proc Natl Acad Sci USA. 1993;90:6434–6438. doi: 10.1073/pnas.90.14.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bucala R, Cerami A. In: The Maillard Reaction: Consequences for the Chemical and Life Sciences. Ikan R, editor. Philadelphia: Wiley; 1996. pp. 161–181. [Google Scholar]
- 36.Kohn R R, Cerami A, Monnier V. Diabetes. 1984;33:57–59. doi: 10.2337/diab.33.1.57. [DOI] [PubMed] [Google Scholar]
- 37.Monnier V, Kohn R R, Cerami A. Proc Natl Acad Sci USA. 1984;81:583–587. doi: 10.1073/pnas.81.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esposito C, Gerlach H, Brett J, Stern D, Vlassara H. J Exp Med. 1989;179:1387–1407. doi: 10.1084/jem.170.4.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bucala R, Tracey K J, Cerami A. J Clin Invest. 1991;87:432–438. doi: 10.1172/JCI115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bucala R, Makita Z, Vega G, Grundy S, Koschinsky T, Cerami A, Vlassara H. Proc Natl Acad Sci USA. 1994;91:9441–9445. doi: 10.1073/pnas.91.20.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bucala R, Mitchell R, Arnold K, Innerarity T, Vlassara H, Cerami A. J Biol Chem. 1995;270:10828–10832. doi: 10.1074/jbc.270.18.10828. [DOI] [PubMed] [Google Scholar]
- 42.Makita Z, Bucala R, Rayfield E J, Friedman E A, Kaufman A M, Korbet S M, Barth R H, Winston J A, Fuh H, Manogue K R, Cerami A, Vlassara H. Lancet. 1994;343:1519–1522. doi: 10.1016/s0140-6736(94)92935-1. [DOI] [PubMed] [Google Scholar]
- 43.Makita Z, Radoff S, Rayfield E J, Yang Z, Skolnik E, Delaney V, Friedman E. N Engl J Med. 1991;325:836–842. doi: 10.1056/NEJM199109193251202. [DOI] [PubMed] [Google Scholar]
- 44.Sell D R, Monnier V. J Clin Invest. 1990;85:380–384. doi: 10.1172/JCI114449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mills J L. Teratology. 1982;25:385–394. doi: 10.1002/tera.1420250316. [DOI] [PubMed] [Google Scholar]
- 46.Cousins L. Am J Obstet Gynecol. 1983;147:333–338. doi: 10.1016/0002-9378(83)91122-5. [DOI] [PubMed] [Google Scholar]
- 47.Mills J L, Baker L, Goldman A S. Diabetes. 1979;28:292–293. doi: 10.2337/diab.28.4.292. [DOI] [PubMed] [Google Scholar]
- 48.Greene M F, Hare J W, Cloherty J P, Benacerraf B R, Soeldner J S. Teratology. 1989;39:225–231. doi: 10.1002/tera.1420390303. [DOI] [PubMed] [Google Scholar]
- 49.Fuhrmann K, Reiher H, Semmler K, Fischer F, Fischer M, Glockner E. Diabetes Care. 1983;6:219–223. doi: 10.2337/diacare.6.3.219. [DOI] [PubMed] [Google Scholar]