

Abstract
Background
Detection of a gene using magnetic resonance imaging (MRI) is hindered by the magnetic resonance (MR) targeting gene technique. Therefore it may be advantageous to image gene-expressing cells labeled with superparamagnetic iron oxide (SPIO) nanoparticles by MRI.
Methods
The GFP-R3230Ac (GFP) cell line was incubated for 24 h using SPIO nanoparticles at a concentration of 20 μg Fe/mL. Cell samples were prepared for iron content analysis and cell function evaluation. The labeled cells were imaged using fluorescent microscopy and MRI.
Results
SPIO was used to label GFP cells effectively, with no effects on cell function and GFP expression. Iron-loaded GFP cells were successfully imaged with both fluorescent microscopy and T2*-weighted MRI. Prussian blue staining showed intracellular iron accumulation in the cells. All cells were labeled (100% labeling efficiency). The average iron content per cell was 4.75±0.11 pg Fe/cell (P<0.05 versus control).
Discussion
This study demonstrates that the GFP expression of cells is not altered by the SPIO labeling process. SPIO-labeled GFP cells can be visualized by MRI; therefore, GFP, a gene marker, was tracked indirectly with the SPIO-loaded cells using MRI. The technique holds promise for monitoring the temporal and spatial migration of cells with a gene marker and enhancing the understanding of cell- and gene-based therapeutic strategies.
Keywords: GFP-R3230Ac (GFP) cell line, magnetic resonance imaging (MRI), optical imaging, superparamagnetic iron oxide (SPIO) labeling
Introduction
To ensure the success of gene and cell therapies, it is of prime importance to develop technologies for non-invasive monitoring of the gene and cell in vivo. Cell and gene imaging have primarily been accomplished using light and fluorescence microscopy. The activity of the gene can be monitored by the magnitude of the fluorescent signal [1]. However, the method is mainly used for an end-point assessment in animal studies.
More recently, it has become possible to monitor biologic processes at cellular and molecular levels using magnetic resonance imaging (MRI), positron emission tomography (PET), near-infrared optical imaging, scintigraphy and autoradiography. Of primary interest is the development of imaging methods to monitor the progress of targeted therapies non-invasively [2]. MRI is particularly useful as a method to image the cell because it has the advantage of very high spatial resolution and the ability to measure more than one physiologic parameter using different radiofrequency pulse sequences. For further enhancement of the tissue, superparamagnetic iron oxide (SPIO; Feridex, Advanced Magnetics Inc, Cambridge, MA, USA) nanoparticle labeling can be used as compartmental, targeted or smart probes within MRI. The SPIO MRI agent sequestered within cells can generate a large magnetic moment that creates substantial disturbances in the local magnetic field. This leads to rapid dephasing of protons, including those surrounding the targeted cell [3,4]. MRI techniques such as gradient echo (GRE) sequences can provide substantial sensitivity for detecting the presence of iron oxide magnetic nanoparticles as apparent signal voids [4–6].
Cell tracking using MRI can provide the means to visualize targeted cells and monitor cell therapy directly [7–10]. The ability to track cell migration, cell homing and cellular fate non-invasively in vivo is of pivotal importance for understanding the complex roles of transplanted cells at pre-clinical trials [7,11]. Cells can be labeled directly with SPIO nanoparticles before their application for subsequent in vivo visualization of their distribution using MRI. The ability to follow cell trafficking and survival dynamically over longer periods of time is contributing to our understanding of the potential mechanism of the targeted cells, which is not possible by histologic study [8,10,12]. On the other hand, necropsy is mainly used as an end-point assessment in animal studies.
The ability to map gene markers non-invasively has tremendous implications for biomedical research [13]. However, dynamic monitoring of therapeutic genes in the tissues is challenging and the distribution of the genes is uncertain [13]. Real-time and direct MRI monitoring of genes within tissues has been hindered by the target techniques [12]. We hypothesized that SPIO labeling is a suitable technique for MRI tracking of gene markers. The purpose of the study was to validate that the gene expression of cells is not influenced by the SPIO-labeling process. We attempted to verify that MRI of labeled GFP-R3230Ac (GFP) cells can potentially track the GFP marker in vitro.
Methods
Magnetic resonance contrast agent
The iron oxide magnetic resonance (MR) contrast agent SPIO is commercially available (Feridex). The core diameter of the SPIO nanoparticle is about 6.5 nm and the total particle diameter about 70–140 nm.
The basic structure of Feridex is an iron oxide core coated with a dextran polymer. The core is a single-domain iron oxide crystal and thus has superparamagnetic properties, meaning that the magnetic moment of the particle strongly aligns with an external magnetic field. Coating of the core is necessary to prevent aggregation of the nano-scale particles and improve biocompatibility. The mechanism of intracellular contrast enhancement with SPIO is based on the difference in susceptibility between regions of labeled cells and background tissue. The change in susceptibility results in a field inhomogeneity in the vicinity of the labeled cells, causing dephasing in that region, which can be detected with MRI. The sensitivity for labeled cells is related to the degree of intracellular iron loading.
GFP-R3230Ac cell line
A stable expression of GFP in the R3230Ac cell line (GFP-R3230Ac) was used in this study. The GFP cell line was kindly provided by M. W. Dewhirst’s group (Department of Radiation Oncology, Duke University Medical Center, Durham, NC, USA). The biologic background of the GFP-expressing cell line has been described previously [14,15].
Cell cultures and labeling
The GFP cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Grand Island, NY, USA) with 10% fetal bovine serum (FBS; Biosource, Logen, UT, USA) and penicillin/streptomycin at 37°C in air with a 5% CO2 atmosphere in a 75-cm2 culture flask. Media were replaced every 3–4 days. The cells were then incubated for 24 h in the culture medium with an SPIO suspension. The iron concentration of SPIO was 20 μg/mL for cell labeling and 10 mL of the SPIO culture medium was used to label 4.5×106 cells. A control group of cells was incubated with culture medium without SPIO. Unincorporated iron particles were removed by repeated washing with phosphate-buffered saline (PBS). Samples of the labeled cells were set aside for evaluation of cell-labeling efficiency (n=6, 1×104 cells/experiment), iron particle uptake (n=3, 2×106 cells/experiment), viability (n=6, 1×104 cells/experiment) and GFP expression (n=6, 2×104 cells/experiment). The remaining cells (c. 7×106/experiment) were used as MRI sample preparations (n=6).
Histologic analysis
After 24 h incubation within the SPIO suspension (cells without SPIO were treated as a control group), cells were washed three times with PBS to remove unbound SPIO. Samples of both labeled and control cells were set aside for evaluation.
Cell-labeling efficiency by light microscopy
Microscope coverslips (circle, diameter 15 mm, sterilized) were prepared in six-well plates and cleaned with ethanol, washed with PBS and stored at 37°C until use. The SPIO-labeled and control cells were placed in six-well plates and grown on the glass coverslips. After 5 h the labeled cells adhered to the coverslips; then the cells were generally fixed for about 10 min with 4% glutaraldehyde. These fixed cells were washed three times with PBS and stained with Prussian blue. Cells were incubated with a 1:1 mixture of 6% HCl (Sigma, Cream Ridge, NJ, USA) and 2% potassium ferrocyanide (Sigma) for another 30 min. The cells were then washed in PBS and observed using light microscopy. Cells were considered Prussian blue positive if intra-cytoplasmic blue shading could be detected. Labeling efficiency was determined by manual counting of Prussian blue-stained and unstained cells using a Zeiss microscope at × 40 magnification using Axiovision 4 software (Carl Zeiss Microscopy, Gottingen, Germany). The percentage of labeled cells was determined from the average of 10 high-powered fields. Prussian blue staining indicated the distribution of SPIO among individual cells. The SPIO distribution within each cell was subjected to quantitative image analysis with MetaMorph™ software (Universal Imaging, Downingtown, PA, USA). In brief, each cell was identified manually on light microscope images (TIFF images) and the MetaMorph software was used to calculate the number of pixels within each cell automatically below an empirically determined intensity threshold. Using the MetaMorph software, iron-loading histograms were calculated by counting dark pixels per cell regions of interests (ROI) in Prussian blue images.
Inductively coupled plasma atomic emission spectrometry for quantifying the iron concentration within GFP cells
The iron concentrations within labeled and control cells were quantified by inductively coupled plasma atomic emission spectrometry (ICP-AES) using a Varian model spectrometer. After labeling, washing and harvesting, cell pellets were dissolved in 1% sodium dodecyl sulfate (SDS). For iron concentration measurements, the spectrometer was set to 238.204 nm and calibrated. Iron content per cell was determined by dividing the iron concentration (pg Fe/mL) by cell concentration (cells/mL).
Confocal microscopy for GFP expression quantification within SPIO-labeled cells
The labeled and control cells were grown on alcohol-cleaned glass coverslips in six-well plates in culture medium until 50–60% confluence. The medium was then removed and the cells fixed using a fresh solution of 4% (w/v) paraformaldehyde for 15 min and rinsed thoroughly with PBS before mounting. All of the GFP cells, which included SPIO-labeled and control groups, were observed using a Zeiss confocal laser microscope (LSM-510; Carl Zeiss Microscopy) with a ×40 lens. GFP was excited using the 488 nm line of a krypton/argon laser and the emitted fluorescence detected with a 515–540 nm band pass filter. During imaging of different groups of cells, the same system configuration was used, keeping all parameters fixed, including laser power, laser line, dichroic beam splitter for separating excitation and emission, pinhole size, scanning speed, etc. All images were acquired as multi-channel TIFF. Offline image analysis was performed with MetaMorph software. We used color separation within the TIFF images to produce green-colored images. The signal intensity (SI) of each cell and background noise was measured by operator-defined ROI. The size of the ROI depended on the cell size. Average signal intensity, the mean of the measurement of high values of signal intensities, was measured using MetaMorph software for GFP, with 40 cells analyzed each group. The GFP signal intensity was normalized by background-integrated signal intensity for each cell (signal to noise ratio; SNR). SNR=SIcell/SIbackground, where SIcell is the signal intensity of the cell and SIbackground is the average signal intensity of the background.
Cell viability and proliferation
Cell viability was determined using trypan blue assays. The cells labeled with SPIO were washed three times with PBS and harvested by trypsinization. These cells were resuspended with PBS. The cells were transferred into Eppendorf tubes with 0.5 mL 0.4% trypan blue solution (Gibco, Gland Island, NY, USA) and 0.3 mL PBS, in 0.2 mL cell suspension in PBS (=dilution 1:5), which was allowed to stand for 5–15 min. Ten microliters of this mix were dropped into cover-slipped chambers of a hemocytometer (avoiding cell clusters by pipetting up and down) and the cell density adjusted to 20–50 cells/square. The fraction of dead cells was determined by counting blue cells; unlabeled cells served as a control group.
The proliferative capacity of cells was determined by plating 5×103 cells in standard culture dishes, followed by counting the number of cells on days 0, 3 and 5.
Sample preparation and MRI protocol
After labeling, the cells were washed three times with PBS, harvested by trypsinization and suspended in culture media. These cells were transferred to 1.5-mL test tubes and then centrifuged and fixed with 0.8 mL 10% formalin. Groups contained 2×106, 1×106 and 0.5×106 cell samples, respectively, with 2×106 unlabeled cells serving as a control sample.
MRI was performed with a clinical 3.0-T whole-body MR system (Siemens Magnetom Trio, Erlangen, Germany) using an eight-channel head coil for excitation and signal reception. To avoid susceptibility artifacts from the surrounding air in the scans, all samples were placed in a water-filled plastic container and this container was positioned within the coil. After suitable image-localization scans, the test tubes were imaged in the coronal and siggital orientations with a two-dimensional (2-D) T2*-weighted (T2*w) pulse sequence, a common negative contrast imaging method. Scanning was performed with the frequency-encoding gradient perpendicular to the Bo field.
Parameters for the 2-D T2*w MRI were repetition time (TR)/echo time (TE) = 60/10 ms, flip angle = 20°, field of view (FOV) = 200×200 mm2, band width (BW) = 620 kHz, thickness = 3 mm. These parameters yielded a resolution size of roughly 0.5 × 0.5 × 3 mm3.
For MR quantitative analysis, the average signal intensity within cell pellets and background noise were measured. The size of the ROI depended on the cell pellet diameter, with a minimum of 10 voxels per region. The measured signal intensity data were normalized by the background noise, expressed as SNR = SIcell pellet/SIbackground, where SIcell pellet is the signal intensity of the cell pellet and SIbackground is the average signal intensity of a region of the water bath, and contrast to noise ratio (CNR), expressed as SNR/σN, where σN is the standard deviation of water bath. All the data were collected from coronal orientation images.
Polymerase chain reaction quantification of GFP expression within MRI samples
After the MRI experiments, genomic DNA of SPIO-labeled and control R3230Ac cells were extracted by using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). Primers were designed for GFP (forward ATGGTGAGCAAGGGCGAGG) and reverse TTTACTTGTACAGCTCCAT) based on the coding region of the GFP gene (Invitrogen, Carlsbad, CA, USA), which amplify a 674-bp band. The polymerase chain reaction (PCR) was carried out as follows using a GeneAmp PCR system 2700 (Applied Biosystem, Palo Alto, CA, USA). Initial denaturation was performed at 94°C for 5 min, followed by 35 cycles at 94°C for 1 min (denaturation), 60°C for 30 s (annealing) and 72°C for 1 min (extension). The final extension was carried out at 72°C for 10 min. Amplified DNA fragments were separated by electrophoresis on a 1.5% agarose gel and stained with ethidium bromide. The area of DNA fragments was measured and analyzed for each group and then compared with the size of the cell pellet on MRI.
Statistics
Quantitative data are presented as mean ± SD. Comparisons between unlabeled (n = 6) and labeled (n = 6) GFP cell preparations were made using a two-tailed unpaired Student’s t-test. Significance was set at P < 0.05 for all statistical tests.
Results
Cell-labeling efficiency
Light microscopy of the Prussian blue staining within labeled cells revealed abundant intracellular uptake of SPIO into the cytoplasm. Thus the uptake of SPIO by cells exposed to SPIO was verified in this study for the given incubation time and iron concentration. No stainable iron was detected in the unlabeled cells (Figure 1A). Labeling efficiency was reproducible in approximately 100% (Figure 1B). Figure 1C shows a group of three cells from Figure 1B at a higher magnification by microscopy; most iron particles are located around the cell nucleus. No intranuclear or extracellular staining could be detected. Light micrographs of Prussian blue-stained cells quantified with MetaMorph software demonstrated a heterogeneous distribution of SPIO among individual cells, as shown in the histogram (Figure 1D).
Figure 1.

Optical microscope view stained with Prussian blue to demonstrate the uptake of SPIO particles. (A) No stainable iron was detected in the unlabeled cells. SPIO particles are visible within all cells in (B) as blue iron stain. All of the iron is around the nucleus (N); no intranuclear or extracellular SPIO could be detected (C). (D) Quantification of SPIO particle distribution inside each cell by light microscopy. Note individual cells take up different amounts of SPIO particles (n = 339).
The mean iron load per cell was determined by ICP-AES. After 24 h of SPIO labeling, labeled cells contained 4.75 ± 0.11 pg Fe/cell and the control group 0.19 ± 0.01 pg Fe/cell (mean ± SD). The iron content per cell in the labeled group was significantly higher compared with that in the control group (P < 0.05).
Cell viability and proliferation following iron loading
Cell viability was observed using trypan blue staining with a light microscope, after the labeling procedure described above. There was no significant difference between the control and SPIO-labeled groups (control versus labeled, mean ± SD, 5.86 ± 0.52 versus 5.93 ± 0.46; P > 0.05). No significant difference in death rate between SPIO-labeled cells and control cells was observed at day 5 following labeling (control versus labeled, 6.05 ± 0.30 versus 5.80 ± 0.49; P > 0.05).
GFP cell proliferation was not affected by SPIO labeling compared with unlabeled control cells. The doubling time of the cells was approximately 23 h, as calculated from the growth curve. After 5 days, total cell numbers were (8.47 ± 2.0) × 104 in the labeled group versus (8.39 ± 3.0) × 104 in the control group. The mean doubling time for labeled and unlabeled cells was not significantly different (P > 0.05).
GFP expression of the cells
Confocal fluorescence microscopy (Figure 2A) revealed the GFP distribution within the cells. A phase microscopy image of the same view showed that iron magnetic endosomes, dark spots, were only present within the cytoplasm (Figure 2B). Fluorescence was detected and contained within the cellular membrane (Figure 2A). The GFP signal intensity within each cell was subjected to quantitative image analysis with MetaMorph software. The GFP SNR measurement demonstrated that the SPIO-labeling process did not affect the GFP expression of the cells (Figure 2C; control versus labeled, 1.63 ± 0.05 versus 1.65 ± 0.04; P > 0.05). There was no significant difference in GFP signal intensity between labeled and control cells.
Figure 2.
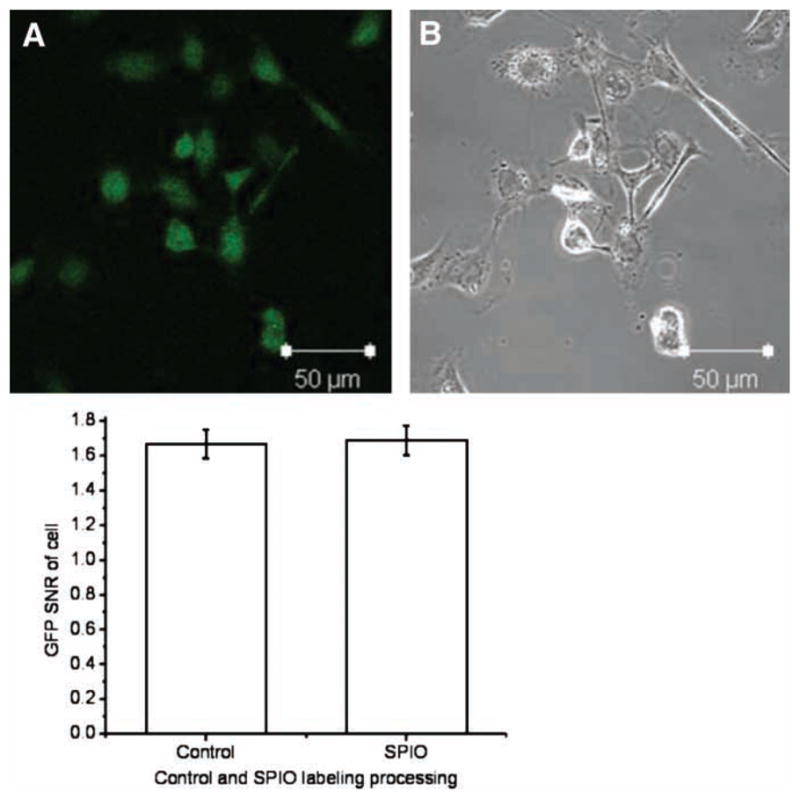
Confocal microscopic view of labeled cells with Prussian blue staining. (A) GFP expression of whole labeled cells; (B) a light microscope image of the same view demonstrating the uptake of SPIO particles (dark particles in cytoplasm). (C) Quantitative SNR analysis with MetaMorph™ software; no difference was observed in GFP expression between labeled and unlabeled groups, P > 0.05.
MRI of GFP cells
A representative 2-D T2*w MRI image of labeled cell pellets is shown in Figure 3. The control GFP cells appeared hype-intense on the image. A strong signal decline was observed on the image of labeled cells. Signal loss within voxels containing labeled cells was observed with the labeled cells. Figure 4A shows the measurement of the susceptibility-induced signal void area (SVA; cm2). The different numbers of cell samples (SVA) of the labeled samples was significantly different (two million versus one million, 0.59 ± 0.02 versus 0.49 ± 0.03; two-million versus half-million, 0.59 ± 0.02 versus 0.36 ± 0.02; one million versus half-million, 0.49 ± 0.03 versus 0.36 ± 0.02; all P < 0.05). The SVA of the control group with two million unlabeled cells was 0.08±0.01, which was significantly smaller than that of the labeled groups (all P < 0.01).
Figure 3.
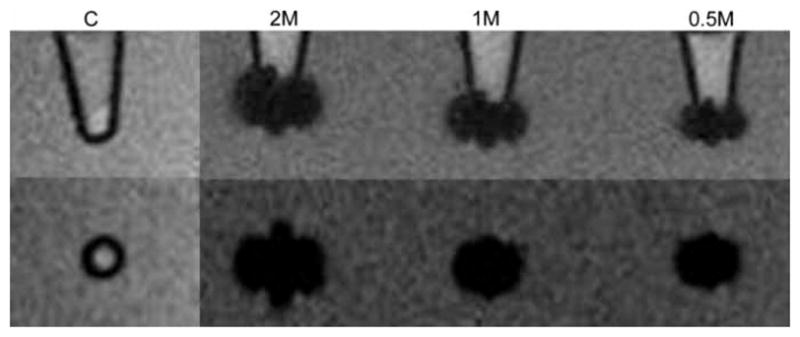
A representative T2*w MR image of GFP cell pellets: LABELED two, one and half-million cells and two million unlabeled cells as a control group. All labeled cell pellets show a marked susceptibility effect compared with the control group. The images on the top row are siggitally orientated, and the bottom row is coronally orientated. C, two million unlabeled cells as a control; 2M, two million labeled cells; 1M, one-million labeled cells; 0.5M, half-million labeled cells.
Figure 4.

(A) Measurement of the susceptibility-induced signal void area for each sample on T2*w MRI. (B) Measured signal intensity (expressed as CNR) of samples on MR images.
Figure 4B shows that the CNR of two million, one million and half-million labeled cells was similar with no significant difference (two million versus one million, − 255.36 ± 4.01 versus − 257.37 ± 8.05; two million versus half-million, − 255.36 ± 4.01 versus − 255 ± 5.71; one million versus half-million, − 257.37 ± 8.05 versus − 255 ± 5.71; all P > 0.05). The CNR of the control group with two million unlabeled cells was 59.94 ± 5.26, which was significantly higher than those of the labeled groups (all P < 0.01).
PCR of GFP expression
Following MRI, we performed PCR experiments for identification of GFP. GFP expression was observed within each of the samples (Figure 5A). The total area of DNA fragments was measured for each sample. Figure 5B shows that the size of the DNA fragments corresponded to the total number of cells in the sample. There were significant differences with different numbers of labeled cells (two million versus one million, 2.12 ± 0.03 versus 0.98 ± 0.04 cm2; two million versus half-million, 2.12 ± 0.03 versus 0.51 ± 0.04 cm2; one million versus half million, 0.98 ± 0.04 versus 0.51 ± 0.04 cm2; all P < 0.05). However, the area of DNA fragment of the control group with two million unlabeled cells was 2.13 ± 0.02, which was not significant compared with the two million labeled cell group (P > 0.05).
Figure 5.

A representative PCR analysis for detecting the size of GFP DNA fragments. The size of the amplified fragment was 436 bp. (A) Lane M is a ladder marker (100 bp to 1.5 kbp). Lanes 1–4 are from our MR samples: lane 1 is from the two million unlabeled cells; lane 2, from the labeled two million cells; lane 3, from the one million labeled cells; lane 4, from the half-million labeled cells. (B) Measurements of GFP DNA fragment area size on PCR, showing that samples with more cells contain more GFP fragments.
Discussion
The study has validated that the gene marker of cells is not influenced by the SPIO contrast agent-labeling process. The results have shown that SPIO labeling is a suitable technique for GFP tracking in vitro.
The ability to image cellular migration in vivo could be very useful for studying the effects of cell therapy, inflammation, tumors and the immune response. A number of investigators have shown that cells labeled with a commercially available SPIO can be imaged for weeks with MRI, and that the labeling does not damage the cells in vitro [16,17]. In vivo cell tracking by MRI can provide a way to observe biologic processes and directly monitor cell therapy [17,18]. We have demonstrated that SPIO do not affect GFP expression, cell viability and function. We have developed and characterized an assay that combines MRI and optical microscopy to evaluate living GFP cells. Furthermore, SPIO-labeled GFP cells can be monitored using both a clinical 3T MR scanner and fluorescence microscopy. This means that SPIO-labeled GFP cells are compatible for both MRI and intravital microscopy imaging studies.
Reporter gene expression studies constitute a valuable and necessary step in furthering our understanding of gene tracking and transfer possibilities using cells. GFP has been a traditional marker gene of choice in many studies [1,2]. Its performance has been characterized extensively in many species, in part because of its high intrinsic fluorescence, which allows for easy and convenient detection of in vitro cells using in vivo optical microscopy. GFP is appealing as a marker gene because of its ease of use in different types of in vitro and in vivo studies. In vivo grafting of GFP cells has revealed the usefulness of GFP as a marker gene for macroscopic and cellular analyzes of graft performance [19,20]. For our experimental design we used SPIO-labeled GFP cells for confocal microscopy and MRI detection. We have verified that SPIO do not affect the stable marker gene on the cells.
Feridex, which has been approved by the US Food and Drug Administration (FDA), is attractive for cellular MRI because it accumulates in high concentrations within cells of the target tissue or organ. A very small amount of SPIO (0.015 mmol Fe/kg) has been shown to be sufficient to produce sufficient contrast between normal tissue and liver cancer lesions [21]. The biocompatibility of Feridex is ensured by a low molecular weight dextran coating around the iron core that stabilizes the particulate. The contrast agent has been reported previously in other cell lines for cell labeling and has proven to be non-toxic in both in vitro and in vivo studies [22,23]. Additionally, it is advantageous that the particles reside exclusively within the cells and that they are not attached to the cell exterior, as is the case with other cell-labeling approaches [24,25]. This makes it possible to follow cell proliferation and trafficking, provided that the vacuoles containing SPIO are shared during the mitotic process. We have verified that the SPIO do not affect the viability, proliferation and GFP expression of GFP cells
We used SPIO for labeling GFP cells. The results confirm that SPIO are internalized inside GFP cells. The data also show that each cell contains many vacuole compartments containing iron, by Prussian blue staining (Figure 1C). SPIO are thus suited for labeling GFP-expressing cells. Labeling efficiency was assessed by Prussian blue staining and showed that all cells were labeled, although the degree of labeling varied from cell to cell.
Confocal microscopy studies revealed the GFP distribution within the whole cell and verified that the GFP intensity of the labeled cells was not significantly different compared with unlabeled cells (P < 0.05), as quantified using MetaMorph image analysis. These results were confirmed by PCR analysis after MR imaging.
The changes in GFP cell viability after SPIO labeling were analyzed by trypan blue. The results confirmed no significant changes in GFP cell viability following the labeling procedure. They verified that the SPIO-labeling methods do not affect GFP cell viability at a concentration of 20 μg/mL (3 × 104 cells/cm2). SPIO taken up by the cells and sequestered within the endosomes may be released over time and reused in normal iron metabolic pathways. In our experiment, there was no effect on cell viability when using a 20 μg/mL iron concentration with GFP cells after labeling for 24 h. This finding indicates that the iron was not released into the cytoplasm as a free radical, which would have been toxic to the cells and increased cell death compared with unlabeled cells. Growth characteristics of labeled GFP cells were assessed using a cell proliferation assay. The growth of labeled GFP cells was not affected by incubation with SPIO.
We have demonstrated that the SPIO-labeling procedure does not affect GFP signal intensity within the cells. This suggests that SPIO-labeled cells can be used as a vehicle to monitor GFP gene markers indirectly and non-invasively with MRI. All cell pellets were visible on the T2*w images as signal voids. The size depended on the number of labeled cells contained in the samples. The susceptibility effect on the signal void area of SPIO-labeled GFP cell pellets was larger than control cell pellets for the same total amount of cells (P > 0.05). The signal intensity of cell pellets demonstrated negative enhancement on T2*w MRI. However, the degree of negative enhancement (CNR) did not depend on the total number of labeled cells and was similar on labeled groups.
A high-resolution protocol was used with a standard gradient imaging set. 3.0T and higher field scanners have demonstrated a clear advantage over 1.5T and lower fields in many applications that require T2*w protocols for contrast enhancement, such as functional MRI, tissue oxygenation and susceptibility weighted venography scans. This also applies to detection of SPIO-labeled cell populations because susceptibility artifacts increase quadratically with Bo [26]. SNR also improves at higher fields as a result of the increased polarization of the nuclear spins present in the magnetic field, making it possible to increase either the speed of the imaging protocols or the spatial resolution. For cell detection, spatial resolution can be increased by up to 40% compared with 1.5T using the same imaging time at equivalent SNR, with some additional CNR gains because of the shorter T2* relaxation times in the presence of iron at the same echo time [27].
Figure 1C illustrates the fact that at least some of the cells were only lightly labeled. We predict that they would only visualize for a short time in an in vivo application. However, more heavily labeled cells could contribute to the observed contrast with MRI for a longer amount of time. We conclude that interpretation of MRI images of SPIO-labeled GFP cells as a surrogate for GFP tracking will probably require additional assessment of the iron-loading distribution for future in vivo studies.
In conclusion, a commercially available FDA-approved MR contrast agent was used to label GFP-expressing cells effectively with no effects on cell viability, proliferation or GFP expression in vitro. Pre-clinical experience with use of MR contrast agents should allow translation of this method from the experimental setting in vitro to in vivo or pre-clinical trials to monitor gene markers indirectly with MRI. SPIO-labeling of cells with specific gene expression holds promise for monitoring the temporal and spatial migration of gene markers and probably enhancing our understanding of gene-based therapeutic strategies.
One of the limitations of the study was that in vitro data on cell samples did not provide an ideal replacement for an in vivo situation; thus, further studies should focus on measurements at clinical field strengths. Another limitation was that the effect on cell viability was assessed directly after incubation. There might be a possibility that SPIO uptake is toxic after a longer period of time by flow cytometry methods.
The long-term goal of this work is to establish the fields of ‘small animal MR imaging’ and ‘microscopic imaging’. MRI can be performed in vitro on tissue specimens, ex vivo on tissue preparations and in vivo. The main focus of future studies will be to bridge in vitro and in vivo studies on biologic applications of MRI.
Acknowledgments
We sincerely thank Mark W. Dewhirst’s group (Department of Radiation Oncology, Duke University Medical Center, Durham, NC 27710, USA) for kindly providing the cell line. We would also like to acknowledge Ms Mary Kay Hayes for helping to revise the manuscript.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Bryant Z, Subrahmanyan L, Tworoger M, et al. Characterization of differentially expressed genes in purified Drosophila follicle cells: toward a general strategy for cell type-specific developmental analysis. Proc Natl Acad Sci USA. 1999;96:5559–64. doi: 10.1073/pnas.96.10.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guilhon E, Voisin P, de Zwart JA, et al. Spatial and temporal control of transgene expression in vivo using a heat-sensitive promoter and MRI-guided focused ultrasound. J Gene Med. 2003;5:333–42. doi: 10.1002/jgm.345. [DOI] [PubMed] [Google Scholar]
- 3.Frank JA, Anderson SA, Kalsih H, et al. Methods for magnetically labeling stem and other cells for detection by in vivo magnetic resonance imaging. Cytotherapy. 2004;6:621–5. doi: 10.1080/14653240410005267-1. [DOI] [PubMed] [Google Scholar]
- 4.Bulte JW, Kraitchman DL. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004;17:484–99. doi: 10.1002/nbm.924. [DOI] [PubMed] [Google Scholar]
- 5.Schnorr J, Wagner S, Abramjuk C, et al. Focal liver lesions: SPIO-, gadolinium-, and ferucarbotran-enhanced dynamic T1-weighted and delayed T2-weighted MR imaging in rabbits. Radiology. 2006;240:90–100. doi: 10.1148/radiol.2393040884. [DOI] [PubMed] [Google Scholar]
- 6.Oweida AJ, Dunn EA, Karlik SJ, et al. Iron-oxide labeling of hematogenous macrophages in a model of experimental auto-immune encephalomyelitis and the contribution to signal loss in fast imaging employing steady state acquisition (FIESTA) images. J Magn Reson Imaging. 2007;26:144–51. doi: 10.1002/jmri.21005. [DOI] [PubMed] [Google Scholar]
- 7.Zhang CY, Lu J, Tsourkas A. Iron chelator-based amplification strategy for improved targeting of transferrin receptor with SPIO. Cancer Biol Ther. 2008;7:889–895. doi: 10.4161/cbt.7.6.5893. [DOI] [PubMed] [Google Scholar]
- 8.Modo M, Hoehn M, Bulte JW. Cellular MR imaging. Mol Imaging. 2005;4:143–64. doi: 10.1162/15353500200505145. [DOI] [PubMed] [Google Scholar]
- 9.Politi LS. MR-based imaging of neural stem cells. Neuroradiology. 2007;49:523–34. doi: 10.1007/s00234-007-0219-z. [DOI] [PubMed] [Google Scholar]
- 10.Daldrup-Link HE, Rudelius M, Piontek G, et al. Migration of iron oxide-labeled human hematopoietic progenitor cells in a mouse model: in vivo monitoring with 1.5-T MR imaging equipment. Radiology. 2005;234:197–205. doi: 10.1148/radiol.2341031236. [DOI] [PubMed] [Google Scholar]
- 11.Frank JA, Anderson SA, Kalsih H, et al. Methods for magnetically labeling stem and other cells for detection by in vivo magnetic resonance imaging. Cytotherapy. 2004;6:621–5. doi: 10.1080/14653240410005267-1. [DOI] [PubMed] [Google Scholar]
- 12.Bowen CV, Zhang X, Saab G, et al. Application of the static dephasing regime theory to superparamagnetic iron-oxide loaded cells. Magn Reson Med. 2002;48:52–61. doi: 10.1002/mrm.10192. [DOI] [PubMed] [Google Scholar]
- 13.Arbab AS, Frank JA. Cellular MRI and its role in stem cell therapy. Regen Med. 2008;3:199–215. doi: 10.2217/17460751.3.2.199. [DOI] [PubMed] [Google Scholar]
- 14.Shan S, Sorg B, Dewhirst MW. A novel rodent mammary window of orthotopic breast cancer for intravital microscopy. Microvasc Res. 2003;65:109–17. doi: 10.1016/s0026-2862(02)00017-1. [DOI] [PubMed] [Google Scholar]
- 15.Li CY, Shan S, Huang Q, et al. Initial stages of tumor cell-induced angiogenesis: evaluation via skin window chambers in rodent models. J Natl Cancer Inst. 2000;92:143–7. doi: 10.1093/jnci/92.2.143. [DOI] [PubMed] [Google Scholar]
- 16.Guzman R, Uchida N, Bliss TM, et al. Long-term monitoring of transplanted human neural stem cells in developmental and pathological contexts with MRI. Proc Natl Acad Sci USA. 2007;104:10211–6. doi: 10.1073/pnas.0608519104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers WJ, Meyer CH, Kramer CM. Technology insight: in vivo cell tracking by use of MRI. Nat Clin Pract Cardiovasc Med. 2006;3:554–62. doi: 10.1038/ncpcardio0659. [DOI] [PubMed] [Google Scholar]
- 18.Wu YJ, Muldoon LL, Varallyay C, et al. In vivo leukocyte labeling with intravenous ferumoxides/protamine sulfate complex and in vitro characterization for cellular magnetic resonance imaging. Am J Physiol Cell Physiol. 2007;293:1698–708. doi: 10.1152/ajpcell.00215.2007. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Kon T, Li C, et al. High intensity focused ultrasound-induced gene activation in sublethally injured tumor cells in vitro. J Acoust Soc Am. 2005;118:3328–36. doi: 10.1121/1.2041247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott G, McGrath J, Crockett-Torabi E. Green fluorescent protein: a novel viability assay for cryobiological applications. Cryobiology. 2000;40:360–9. doi: 10.1006/cryo.2000.2258. [DOI] [PubMed] [Google Scholar]
- 21.Zech CJ, Herrmann KA, Dietrich O, et al. Black-blood diffusion-weighted EPI acquisition of the liver with parallel imaging: comparison with a standard T2-weighted sequence for detection of focal liver lesions. Invest Radiol. 2008;43:261–6. doi: 10.1097/RLI.0b013e31816200b5. [DOI] [PubMed] [Google Scholar]
- 22.Terrovitis JV, Bulte JW, Sarvananthan S, et al. Magnetic resonance imaging of ferumoxide-labeled mesenchymal stem cells seeded on collagen scaffolds: relevance to tissue engineering. Tissue Eng. 2006;12:2765–75. doi: 10.1089/ten.2006.12.2765. [DOI] [PubMed] [Google Scholar]
- 23.Pawelczyk E, Arbab AS, Pandit S, et al. Expression of transferrin receptor and ferritin following ferumoxides-protamine sulfate labeling of cells: implications for cellular magnetic resonance imaging. NMR Biomed. 2006;19:581–92. doi: 10.1002/nbm.1038. [DOI] [PubMed] [Google Scholar]
- 24.Kamaly N, Kalber T, Ahmad A, et al. Imodal paramagnetic and fluorescent liposomes for cellular and tumor magnetic resonance imaging. Bioconjug Chem. 2008;19:118–29. doi: 10.1021/bc7001715. [DOI] [PubMed] [Google Scholar]
- 25.Sitharaman B, Tran LA, Pham QP, et al. Gadofullerenes as nanoscale magnetic labels for cellular MRI. Contrast Media Mol Imaging. 2007;2:139–46. doi: 10.1002/cmmi.140. [DOI] [PubMed] [Google Scholar]
- 26.Bi X, Deshpande V, Simonetti O, et al. Three-dimensional breathhold SSFP coronary MRA: a comparison between 1.5T and 3.0T. J Magn Reson Imaging. 2005;22:206–12. doi: 10.1002/jmri.20374. [DOI] [PubMed] [Google Scholar]
- 27.Fischbach F, Bruhn H, Unterhauser F, et al. Magnetic resonance imaging of hyaline cartilage defects at 1.5T and 3.0T: comparison of medium T2-weighted fast spin echo, T1-weighted two-dimensional and three-dimensional gradient echo pulse sequences. Acta Radiol. 2005;46:67–73. doi: 10.1080/02841850510012625. [DOI] [PubMed] [Google Scholar]