

Abstract
Alzheimer’s disease (AD) is the most common form of dementia in the elderly. Current treatments for AD are not as effective as needed, nor is there any definitive antemortem diagnostic. Understanding the biological processes that occur during AD onset and/or progression will improve disease diagnosis and treatment. Recent applications of microarray technologies for analysis of messenger (m) RNA expression profiles have elucidated distinct changes in the brain as a function of AD dementia initiation and progression. However, mRNA analysis underestimates post-transcriptional modifications and therefore provides only a partial view of the molecular changes in the AD brain. Combining mRNA studies with protein expression analysis may provide a more global picture of the biological processes associated with AD dementia. Information gathered could lead to the development of select biological indices (biomarkers) for guiding AD diagnosis and therapy. We will provide a brief background on AD, followed by a review on the applications of microarray, proteomics, as well as microRNA expression profile analysis to develop novel diagnostic strategies that may be useful for the diagnosis AD and for monitoring disease progression. The availability of biomarkers that promote early disease diagnosis, particularly among asymptomatic patients, will lead to the application of personalized medicine in AD.
Keywords: Alzheimer’s disease (AD), Cognitive impairment, Dementia, Genetic risk factors, Biomarkers, Complementary DNA (cDNA) microarray analysis, Protein array analysis, Proteomics, MicroRNA (miRNA)
1. Alzheimer’s disease
Alzheimer’s disease (AD) is a chronic neurodegenerative disease of the brain that is currently afflicting roughly 5.3 million individuals in the United States (Alzheimer’s Association, 2006a). At the current rate, there are approximately 500,000 new cases reported each year, with the total number of Alzheimer’s sufferers in the US expected to grow to 13.2 million by 2050 (Hebert et al., 2009). In fact, AD moved up to 7th from 8th place among the leading causes of death in 2005, passing influenza and pneumonia, as recently reported by The Center for Disease Control (Alzheimer’s Association, 2006b). Those affected by AD survive only about half as long as those of similar age who are unaffected. Medicare/Medicaid spending in 2005 totaled $112 billion for treatment for individuals with AD and other dementias, thereby demonstrating a profound health and financial burden that this disease has placed on our society. With the aging of the baby-boomer population in America, in fact, this spending total is expected to increase to $216 billion by 2015 (The Alzheimer’s Study Group, 2005).
Early AD diagnosis in combination with new classes of neuro-protective or disease-modifying drug treatments may delay or prevent the neurodegenerative effects of AD. Thus, the development of a practical, inexpensive and reliable diagnostic methodology for AD is of profound importance. Unfortunately, AD is difficult to diagnose, particularly for physicians who lack specialized training in the field. The problem is exacerbated for patients from rural communities, in addition to those coming from lower socioeconomic backgrounds, who might not have access to, or may not be able to afford the current advanced diagnostic procedures for AD, such as imaging or other procedures available in university research settings, which increase the sensitivity and reliability of AD diagnosis. In fact, data shows that as many as two-thirds of dementia cases may go undiagnosed (Jason van Steenburgh, 2003).
Problems that interfere with our capacity to reliably diagnose AD at early stages of disease have important implications for the development of AD therapeutics. Commercial development of pharmaceuticals to treat neurological disorders such as AD is often particularly difficult, time-consuming, and expensive. The primary expenses (and development risks) for drug development are typically associated with the late stage clinical trials required for approval by the FDA, and these difficulties arise primarily from two related issues. First, it is difficult to identify and recruit patients who have a specific disease and who might derive a clinical benefit from the drug treatments designed to specifically target that disease. Second, it is difficult to measure and quantify what the precise beneficial clinical effects are of the drug being tested. Both of these problems are related to a current lack of reliable, non-invasive and cost-effective biomarkers for many neurological disorders, including AD.
2. AD from neuropathology to diagnosis
The fundamental challenge to early diagnosis of AD is that the disease is currently defined histopathologically (by the presence of neuritic plaques and neurofibrillary tangles in post-mortem dissections of brain tissue), but disease diagnosis is based on clinical symptoms. There are two issues related to this diagnostic methodology. First, the assumption is made that there is a direct causal linkage between the histopathology and the behavioral symptoms. A second assumption is that the correlation between these areas can be measured accurately and reliably using existing tests. Numerous scientific studies have questioned both of these assumptions (Bussiere et al., 2003; von et al., 2005; Braak and Braak, 1997; Haroutunian et al., 1998, 1999, 2007; Morris et al., 2001; Nelson et al., 2007). This has led researchers and clinicians alike to the same conclusion, that the current diagnostic methodology (NINCDS-ADRDA and DSM-IV criteria (McKhann et al., 1984)) is inadequate for guiding clinical care and evaluating the effects of drug therapies. The consensus is that new biomarkers must be developed and validated to measure effects directly related to the disease process, as well as effects on the brain that manifest as behavioral symptoms. Development of such biomarkers for AD remains elusive for three primary reasons: (1) an incomplete understanding of the underlying disease process, (2) the inherent complexity related to comorbidities of other types of dementia (e.g., frontotemporal dementia, vascular dementia, etc.) that have similar symptoms to AD, and (3) direct access to the diseased tissue of living subjects is limited.
2.1. Clinical implications for diagnostic biomarker discovery
According to the National Institute of Health, a biomarker is “a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.” An ideal biomarker should meet the above definition, in addition to being non-invasive and cost-effective. This kind of biomarker for AD would be useful both clinically and for the development and testing of new pharmaceutical treatments.
A rigorous application of the ideal biomarker would be to distinguish AD (as defined by an increased amyloid plaque and neurofibrillary tangle burden) from types of dementias (e.g. vascular dementia, frontotemporal dementia). This is critically important in two areas. First, treatment regimens for these diseases may be different, especially as new precisely-targeted pharmaceuticals become available. For example, AD drugs that are designed to target the amyloid cascade (Selkoe, 2001) may not be appropriate for a patient with frontotemporal dementia. Second, differential diagnosis incorporating biomarkers for an early and reliable diagnosis of AD can also facilitate the recruitment of appropriate patients into clinical trials of pharmaceutical compounds. Testing an amyloid-cascade targeting drug on patients who are believed to have AD, but in reality have frontotemporal dementia, for example, would negatively skew the results of a drug treatment study.
As mentioned above, an accurate biomarker for AD is a significant missing component in the drug development efforts of many pharmaceutical companies. Not being able to recruit the target patients and not being able to measure the direct effects of therapies have forced these companies to perform large population studies. This approach dramatically increases development risks as well as risks to patients, in addition to driving up the associated costs and complicating the necessary data analysis and reporting requirements for FDA approval. These complications have led to the failure of several high-profile drug trials, even though clear benefits were demonstrated in some of the patient cohorts (Pepeu and Giovannini, 2009; Rafii and Aisen, 2009; Ghosh et al., 2008; Wolfe, 2008; Relkin, 2008).
2.2. Limitations of current diagnostic biomarkers
As discussed previously, the diagnosis of AD is confirmed only with histopathological evidence of amyloid plaque accumulations in the medial temporal lobe (MTL) during autopsy (Pearson et al., 1985; McKhann et al., 1984). However, the diagnosis of AD in living patients is complicated by the fact that such pathological evidence is not available. Moreover, even if it is available during life, such pathological evidence may not always manifest as behavioral symptoms. The ideal diagnostic criteria for AD, then, should include evidence of abnormal pathology as well as cognitive decline. Since publication of the AD diagnostic criteria in 1984, AD research in various neurological domains has led to a deeper understanding of the underlying disease process. These domains include: (i) risk factors, (ii) molecular biology, (iii) neuroanatomy, (iv) brain metabolism, (v) cortical processing, and (vi) behavior. To develop effective AD biomarkers, it is important to understand the relationship between these neurological domains and how they relate to AD.
As a genetic risk factor, Apolipoprotein E (ApoE) has shown some promising results. In-vitro and animal studies have shown that ApoE helps in the degradation and clearing of the toxic β-amyloid (Aβ) plaques observed in AD. Animals with the ApoE ε4 allele showed an impaired ability of degrading the Aβ plaques, making the ε4 allele a genetic risk factor for late-onset AD (Corder et al., 1993). However, ApoE ε4 has not been directly implicated in the development of AD.
The molecular biology domain has demonstrated some of the most promising results towards the development of AD biomarkers. Concentrations of proteins such as Aβ1–42 and phosphorylated Tau in the cerebrospinal fluid (CSF) have shown a high degree of correlation with AD clinical symptoms (Shoji et al., 2002). Similarly, positron emission tomography using Pittsburgh compound B (PIB-PET) can be used to directly image the Aβ plaque burden in the brain. Most importantly, increasing cortical PIB binding correlates well with declining cognitive function in patients with mild AD (Rabinovici et al., 2007). However, the diagnosis of AD based on CSF or PIB-PET biomarkers is complicated by the fact that a significant percentage of healthy subjects exhibit high cortical amyloid plaque burden. Moreover, CSF analysis requires invasive lumbar punctures while the short half-life of radioactive PIB (~20 min) makes PIB-PET imaging cumbersome, expensive, and therefore, impractical in primary healthcare settings.
In the neuroanatomy domain, magnetic resonance imaging (MRI) and computer aided tomography (CAT) scans can be used to image large-scale brain structures. In AD, the MTL, particularly the hippocampal formation, is the earliest region where toxic Aβ plaques are observed. The presence of Aβ plaques has been implicated in neuronal cell death. Based on these observations, researchers have proposed that atrophy in the hippocampal or the MTL region, measured using brain imaging, could be a useful biomarker for early-stage AD (Verhoeff et al., 2004; Suemoto et al., 2004; Kung et al., 2004). However, there are concerns that this approach may lack the specificity needed in a biomarker to indicate AD. In particular, biological processes other than formation of Aβ plaques can lead to cellular atrophy in these regions, and in fact, brain atrophy has been observed even in healthy aging. To discount the effects of healthy-aging-related atrophy, researchers have suggested using the rate of hippocampal or MTL atrophy as a biomarker of AD. There are two potential issues related to this strategy. First, the difference in the rate of brain atrophy between healthy aging subjects and AD subjects has only been demonstrated at group levels, complicating the diagnosis at an individual level. Secondly, evaluating the rate of atrophy entails collection of multiple scans at regular time intervals, making the approach time-consuming and prohibitively expensive.
As a result of brain atrophy and/or other disease processes, patients in early stages of AD demonstrate reduced levels of brain metabolism, another neurological domain that can be investigated using various biomarker modalities (Chen et al., 2005; Salmon et al., 2006). Functional magnetic resonance imaging (fMRI) and PET scans with flurodeoxy glucose (FDG-PET) are routinely used as secondary measures of metabolic activity in various parts of the brain (Small et al., 2008; Wermke et al., 2008; Wrase et al., 2008; Nestle et al., 2009). fMRI measures changes in oxygen concentrations related to regional cortical blood flow, while FDG-PET measures glucose metabolism in neuronal populations. Since these measures are intimately coupled with brain atrophy, they share all the confounding factors of brain atrophy measures (described above). Additionally, the relationship between the underlying neural activity and the emergence of these signals is poorly understood, raising questions about the utility of fMRI and FDG-PET in the diagnosis of AD.
Taken together, measurements in the risk factors, molecular biology, neuroanatomy, and brain metabolism domains provide useful information regarding the etiology of AD. However, these tests only perform a supportive role. A positive indication of mild AD based on these measurements, in and of itself, is insufficient with regards to assessing the cognitive impairment that is the hallmark of dementia. Measurements in the cortical processing and behavioral domains are generally used to assess cognitive function. For accurate assessment of cognitive impairment, it is important to understand cortical processing, behavior, and their relationship to the other neurological domains. Traditionally, measurements in the behavioral domain are used for the assessment of cognitive function. However, neuropsychological testing is expensive, time-consuming, and is only available at specialty dementia clinics. In addition to the difficulties associated with neuropsychological tests, behavior is essentially an emergent property of cortical processing. Even the simplest of behavioral responses involve cortical processing at multiple stages, such as allocation of attention, sensory information processing and classification, storage in immediate memory, and response selection (Opris and Bruce, 2005). As a consequence of cortical plasticity, it can take a long time before cortical processing impairments are manifested as detectable behavioral impairments. The resulting delay in diagnosis, especially in the case of AD, can lead to loss of valuable time for disease-modifying treatments.
2.3. From clinical diagnostics to cDNA microarray and proteomics in the discovery of novel surrogate indices of clinical onset and progression
One important area of AD research focuses on identifying genes that increase a person’s risk of developing AD, or that may play a role in the onset or progression of AD. At any given time and in any given tissue or cell, only a subset of genes are expressed. For active genes, the genetic information encoded in these genes is copied into an intermediary molecule called messenger RNA (mRNA) in a process known as transcription. The mRNA then is used as a template to produce the protein products encoded by the genes that are ultimately responsible for biological activities. Thus, the cDNA generated from a certain tissue type (e.g., brain tissue from certain brain regions obtained at autopsy) represents genes that were actively expressed in that brain region. By comparing cDNA patterns in the brains of people with a disease (e.g., dementia) with those of people without the disease (e.g., normal cognitive functions), researchers can identify genes whose expression varies between the two groups and, therefore, may be related to the disease.
We have examined gene expression patterns in patients with known cognitive status – no dementia, questionable dementia, mild dementia, or moderate dementia (which corresponds to early or mild AD) – for whom brain samples are available after death. We used high-throughput microarray methodologies and examined mRNA expression patterns in post-mortem brain specimens of cases characterized by normal cognitive status compared to cases characterized by moderate dementia; cognitive status was assessed by clinical dementia rating (CDR) in which normal cognitive status and moderate dementia cases are characterized by, respectively, CDR 0 and CDR 2 (Ho et al., 2005). Using a cDNA microarray platform (Incyte VI human microchip), we screened 6794 human genes and found 32 genes (25 known genes and 7 unknown genes that are identified based on expressed sequence tag or EST) that are aberrantly expressed in >1.8-fold higher or lower levels in the superior temporal gyrus of moderate dementia cases compared to cognitively normal control cases (Ho et al., 2005). Among others, we found down-regulation of the synaptic vesicle protein synapsin IIa, which normally plays an important role in synaptic vesicle turnover and neurotransmitter release (Ho et al., 2005). Based on this, we used independent Western blot analysis in further studies to explore the regulation of synapsin IIa and other known splice variants I, II, and III of the a- and b-type synapsin isoforms (Ho et al., 2001) at the protein level in the brain as a function of cognitive deterioration. We examined brain post-mortem specimens from mild cognitive impairment (MCI), AD dementia and normal neurological control cases, targeting brain regions that are at high risk (entorhinal cortex, EC) or relatively unaffected (visual cortex, VC) during AD. Interestingly, we found significantly, >2-fold reduced protein contents for a-type synapsin splice variants I, II, and III in the EC of MCI cases (characterized by CDR 0.5) compared to age-matched control cases with normal cognitive status (characterized by CDR 0). In contrast, no detectable change in the expression of b-type synapsin splice variant II protein nor synaptophysin protein was found in the same EC region of MCI cases. The selective altered expressions of synapsin a-type isoforms in the EC of MCI cases was specific to brain regions that are at high risk for AD, since no detectable decreases were found in VC from the same cases.
These results provide novel molecular evidence for the importance of selective changes in gene expression profiles during the earliest detectable stage of AD dementia. Since synapsin family members are important in the regulation of neurotransmitter release, our cDNA microarray studies suggest that the altered regulation of selected synapsin-mediated neurotransmitter release may be involved in the early phase of AD cognitive decline. While previous evidence suggested that MCI is characterized by neuronal loss (e.g., in the EC) and qualified neuropathologically for diagnosis of frank AD (Masliah et al., 2001), we note that the MCI cases used in our cDNA microarray studies did not meet neuropathological AD criteria (Ho et al., 2001). Most importantly, we also found that the expression of the pre-synaptic plasma membrane protein synaptophysin was unaffected in the EC of MCI cases, suggesting that the decreased expression of the a-type synapsin occurred in absence of structural loss of synapses. Thus, as summarized in Scheme 1, we proposed that initial cognitive decline leading to conversion from normal cognitive status to MCI and eventually to AD dementia may involve, in part, selective synaptic functional impairments (e.g., altered synapsin-mediated neurotransmitter release, such that the reduction of a-type synapsin precedes that of snaptophysin) among synapses that are otherwise morphologically intact. However, we also note that a more recent study indicated a minor decline in the expression of synaptophysin proteins in the frontal cortex of MCI as well as mild dementia (CDR 1) cases in comparison to cognitively normal individuals (Ho et al., 2005).
Scheme 1.
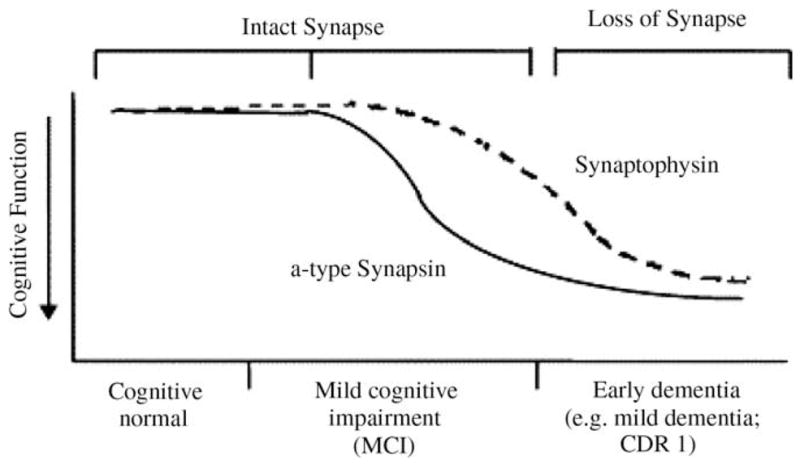
Selective alteration of functional synaptic proteins, e.g., a-type synapsin in synapses that are morphologically present may be an underlying mechanism in MCI cognitive impairment.
While the mechanisms leading to the selective reduced expression of a-type synapsin in the EC of MCI cases is unknown (Ho et al., 2001), independent studies in our lab found that excitotoxcity, but not Aβ (Aβ1–42)-mediated toxicity, recapitulated selective reduction of a-type synapsin expression in cortico-hippcampal neuron cultures (Ho and Pasinetti, unpublished observations). Finally, because recent evidence suggests that MCI clinically progresses to increasingly severe dementia at rates dependent on the level of cognitive impairment, and that MCI cases often have the neuropathologic features of AD (Ho et al., 2001), our studies provide a novel biological basis for a better understanding of the mechanisms underlying the conversion from MCI to frank AD dementia. Collectively, our cDNA microarray studies strengthen the utility of a systematic approach to explore molecular events in the brain of MCI cases and the subsequent development of independent studies to further clarify underlying mechanisms of clinical dementia progression from normal cognitive status to MCI and frank AD dementia.
Consistent with our observation, Loring and colleagues (Loring et al., 2001) used cDNA microarray analyses to compare affected and unaffected brain tissue obtained after death from six patients with AD and nine control subjects. Again, these researchers found that genes related to synaptic vesicle synthesis and function were downregulated in the affected AD brain tissue, as were genes related to signal transduction, energy metabolism, stress response, calcium binding, and the molecules that help cells maintain their shape (i.e., the cytoskeleton). At the same time, some genes related to chronic inflammation, adhesion of cells to one another, cell proliferation, and protein synthesis were upregulated.
Thus, cDNA microarray analyses have led to the identification of multiple genes that appear to be important in the early stages of AD. Additional studies will be necessary to determine the exact role(s) of these genes in the development and/or progression of AD.
However, while evidence from cDNA microarray studies demonstrated that the earliest detectable stages of AD dementia is associated with selective changes in gene expressions, we are aware that since cDNA microarray methodologies are designed to assess steady-state mRNA levels, cDNA microarray studies provide only a “snapshot” of gene expression in a given tissue at a specific time and under specific conditions. It is possible that processes occurring after the transcription of DNA into mRNA – that is, during the translation process when proteins are generated from mRNA templates, as well as during subsequent protein processings – may differ between people with and without AD, and these differences may contribute to AD development and progression. Therefore, cDNA microarray technology approaches alone likely provide only a partial view of the biological processes leading to AD. Consequently, the combination of mRNA and protein expression profile analyses using, respectively, microarray and proteomic methodologies now has become the gold standard for research aiming at a more global understanding of complex biological processes that are involved in the initiation and progression of AD pathogenesis and AD dementia. The following section discusses some of the findings obtained with proteomic studies of AD using protein array technologies.
2.3.1. Protein array studies identify candidate proteins involved in AD
To identify differences in protein expression between people with MCI (who are at high risk of developing AD) and people with normal cognitive functioning, Ho and colleagues (Ho et al., 2005) used commercial protein arrays (i.e., PowerBlot arrays) that allowed for the screening of 750 independent proteins in a single assay. These analyses found 50 candidate proteins whose levels differed consistently and reproducibly between patients with high risk for developing AD and normal control subjects. The investigators found that these differentially regulated proteins can be segregated into five functional groups:
proteins related to neurotransmitters and synaptic function;
proteins related to the cytoskeleton and adhesion of cells to one another;
proteins related to the cell cycle (i.e., involved in growth and multiplication of cells as well as in cell death);
proteins related to apoptosis, or programmed cell death; and
proteins related to transcription and translation.
Most of these differentially regulated protein contents were found to be reduced by at least 2-fold in cognitively impaired compared to control normal cognitive cases, although a few these differentially regulated proteins were found in higher contents in the cognitively impaired cases. These findings are consistent with observations from microarray studies in that gene expression analysis at mRNA and protein levels point to central roles for changes in selective neurotransmitter and synaptic functions, cytoskeleton and cell adhesion, and cell proliferation in the development or manifestation of AD dementia.
Using traditional Western blot techniques we continued to evaluate the levels of these proteins in a larger number of samples from AD and MCI patients. We found that a protein called tomosyn consistently showed a decrease in content levels by at least 3-fold in the cognitively impaired patients (Ho et al., 2005). Tomosyn is important in the metabolism and refilling of the synaptic vesicles, further supporting the hypothesis that reduced synaptic functions may be associated with the cognitive decline in patients with moderate cognitive impairment. The synapsin IIa protein, whose cDNA levels were found to be reduced in the cDNA microarray studies, was not among the 750 proteins studied here; therefore, its protein levels could not be evaluated. Thus, it appears that cDNA microarray and protein array studies complemented each other in our search for molecular markers of cognitive impairment and early AD.
2.3.2. Proteomic approaches help identify biomarkers of AD
One important aspect of current AD research is the search for proteins whose presence or expression levels differ consistently between people with and without cognitive impairment or AD and that can easily be measured so that they can be used by physicians to diagnose these conditions and monitor their progression (i.e., biomarkers of AD). Thus, biomarker discovery studies focus on identifying protein profiles that can ultimately be used to develop rapid, sensitive, and specific high-throughput diagnostic tests. To achieve this goal, such biomarkers ideally should be present in biological fluids (e.g., blood or cerebrospinal fluid [CSF]) (Shaw et al., 2009, 2007; Frank et al., 2003; Sunderland et al., 2004, 2003) and not only in brain tissue, because these fluids can be attained much more easily than brain tissue samples. However, the identification of such biomarkers is challenging because they must meet at least two requirements. First, their levels must be high enough so that they can be detected among all the other proteins found in these biological fluids. Second, they must be specific to the condition in the brain being studied, as many of the proteins circulating in the blood or CSF can be produced by multiple cell types and different organs and therefore lack this specificity.
In their efforts to identify suitable biomarkers of the progression from normal cognitive functioning to MCI and AD, researchers have begun to use protein chip arrays whose surfaces have been treated chemically or biochemically so that the arrays can bind only proteins with certain structural or functional characteristics for subsequent protein expression profile analysis. For example, using this approach, a surface-enhanced laser desportion/ionization time-of-flight (SELDI-TOF) has been developed for high-throughput protein expression profile analysis (Pasinetti et al., 2006). Using SELDI-TOF technology, crude protein extracts from the tissue or fluid of interest are applied to the arrays, and the selected subsets of proteins bound by the arrays are analyzed using mass spectrometry approaches. If samples from patients with the disease and control subjects are run in parallel, differences in protein expression profiles can be identified using computerized analyses. This strategy allows for highly sensitive, quantitative, and reproducible analysis of protein expression patterns and may offer a novel means for identifying and eventually monitoring the onset and progression of AD and the associated dementia (Ho et al., 2005). Using this approach, investigators are analyzing several potential biomarkers.
One direction of investigation using the SELDI-TOF technology has been focused on simultaneous analysis of all Aβ peptide species in biological fluids. As mentioned earlier, one of the characteristic features of AD is the formation of Aβ plaques in the brain that consist mainly of Aβ peptides. Several variants of the Aβ peptide exist, and precise determination of the patterns of these various Aβ peptide species in tissue extracts or body fluids could conceivably provide a more concise prognostic biomarker for AD and would provide a better understanding of AD pathogenesis. Therefore, in addition to traditional molecular biological techniques, researchers have used the protein chip array/SELDI-TOF technique to simultaneously detect the various Aβ peptide variants in order to gain a better understanding of the processes involved in AD development and progression. To date, this strategy has been used to study AD pathogenesis (Vehmas et al., 2001; Xiang et al., 2002; Goldstein et al., 2003; Davies et al., 1999) and the processes occurring during the breakdown of APP into the Aβ peptide species (Beher et al., 2002; Frears et al., 1999; Austen et al., 2000). Other studies using this strategy were aimed at exploring new drugs for AD treatment (Davies et al., 1999; Beher et al., 2002) or potential development of a vaccine therapy for AD (Vehmas et al., 2001).
Other efforts in the search for AD biomarkers have been focused on exploring the diagnostic value of using a combination of several proteins to distinguish patients with AD dementia from cognitively normal patients, or from patients with other forms of dementia unrelated to Alzheimer’s disease. One group of researchers reported that, using protein chip arrays and SELDI-TOF, they identified a panel of five proteins whose expression was significantly altered in AD patients compared to healthy control subjects (Carrette et al., 2003). Other researchers proposed using the ratio of different variants of the Aβ peptide in the CSF as a diagnostic tool for distinguishing patients with AD from healthy people and patients with other forms of dementia (Shoji, 2002; Shoji et al., 2002); however, this approach appears to be less specific and sensitive than the one using the panel of five proteins.
2.3.3. S100A7: a novel biomarker of AD involved in the attenuation of β-amyloid neuropathology in AD
Using SELDI-TOF mass spectrometry we have explored the identification of potential novel biomarkers associated with the onset and eventual progression of the cognitive decline associated with early AD. Using protein microchips that had been treated so that they selectively interacted with negatively charged or copper-binding proteins, Ho and colleagues (Ho et al., 2002) detected a novel protein biomarker whose function is still unknown, but whose levels were found to be increased in the CSF of patients with early MCI in comparison to control patients. We continued to explore the potential biological role of this novel candidate AD biomarker protein species. Following purification, we sequence-identified this protein species as S100A7, a protein known to be involved in immune responses (Qin et al., 2009). Using an adenoviral-S100A7 expression system, we continued to explore the potential role of S100A7 in AD amyloid neuropathology in an in-vitro model of AD. We found that the expression of exogenous S100A7 in primary cortico-hippocampal neuron cultures derived from Tg2576 transgenic mouse models of AD inhibits the generation of β-amyloid peptides, coincidental with a selective promotion of “non-amyloidogenic” α-secretase activity via induction of ADAM (a disintegrin and metalloproteinase)-10. Finally, a selective expression of human S100A7 in the brains of transgenic mice resulted in a significant promotion of α-secretase activity. This most recent studies in our lab suggests that S100A7 may be a novel biomarker of AD dementia and supports the hypothesis that promotion of S100A7 expression in the brain may selectively promote α-secretase activity in the brain of AD, thereby precluding the generation of amyloidogenic β-amyloid peptides.
3. From microarray and proteomics to hidden molecular biomarker signatures in the development of personalized medicine in AD
Biomarker discovery studies, such as those described above, offer clinicians and scientists a novel approach for treating and understanding diseases such as AD. In modern medicine, physicians and scientists alike generally view and interpret disease at the ‘visual’ level, namely the level of the organism, the organ, and, more recently, the tissue. With the advent of genomic and proteomic technologies, personalized medicine offers the promise and potential of uncovering the largely ‘unseen’ details of disease causality, onset, and progression. A broad aim of personalized medicine is to use a molecular characterization approach to create a better system for disease classification. This work is anticipated to lead to earlier interventions and more specific treatments predicated on the individual’s specific biochemical fingerprint. This is in stark contrast to current medical practice. Appreciating the differences in these approaches discussed here might benefit from likening them to viewing an iceberg: up to now, scientists and physicians alike have been limited to viewing and interpreting the easily visible aspects of disease – that is, at the level of the organism, organ and tissue; by contrast, personalized medicine promises to reveal a far deeper and more comprehensive view of the largely ‘unseen’ details of disease causality, onset and progression by enabling them to view a disease from its onset and to monitor its progression at the molecular and cellular levels. As an example, the usual clinical appearance of AD (e.g., deterioration of memory functions, among other neuropsychological indexes) is conceptually analogous to the “tip of the iceberg.” However, and most importantly, our understanding of the disease is enhanced by being able to view the “hidden, submerged unseen mass” of the disease from its onset, i.e., being able to monitor its progression at the molecular level through the use of novel genomic technologies (e.g., miRNA and protein biomarkers at cellular-molecular levels, as depicted in the scheme of the working hypothesis shown below in Scheme 2). To date, some specific examples of personalized medicine approaches have been utilized to benefit diagnosis and treatment of diseases in patients, as discussed below (see Scheme 2).
Scheme 2.

Schematic subdivision of AD as clinical and biochemical entity at various functional levels within an organism. The top bracket embraces those levels of disease for which traditional medicine has been successful in the last two centuries (tip of the iceberg), such that only observable pathology is addressed. The lower bracket embraces the extended region accessible by personalized medicine attempting to define the disease at cellular/biochemical levels.
3.1. Evidence supporting the feasibility of using personalized medicine in preventative strategy
Prognostic molecular signatures found in biomarker discovery and validation studies can be used to monitor and better understand disease onset, progression and treatment. Examples of this currently being used in clinical practice includes the C-reactive protein as an index for risk of cardiovascular disease, and LDL and HDL cholesterol as indices for risk of atherosclerosis (Nissen et al., 2005; Nissen, 2005). Detecting abnormal levels of markers such as these may trigger interventions aimed at preventing future disease. There are two genetic tests currently on the market which can identify disease susceptibility and guide preventive care. First, a recent molecular predictive indicator of disease which has received wide attention is the test for BRCA1 and BRCA2, genetic variants that indicate a hereditary propensity for breast and ovarian cancers (Nelson et al., 2005). Women with BRCA1 or BRCA2 genetic risk factors have a 36–85% lifetime chance of developing breast cancer, compared with a 13% chance among the general female population, and these genetic tests can be used to guide preventive measures, such as increased frequency of mammography, prophylactic surgery, and chemoprevention. Second, the treatment of early-stage breast cancer in women may be transformed, for example, by several assays in development that scan a panel of genes correlated with risk of disease recurrence and response to therapy (Cronin et al., 2004; Paik et al., 2004). One such assay now being used in clinical settings is Oncotype DX™, which analyzes the expression of 21 genes (Hornberger et al., 2005; Paik, 2006).
We hypothesize that similar approaches will soon be available for treating neurodegenerative disorders such as AD. The information provided will be fundamental for guiding selection of treatment strategies and developing monitoring decisions based on the foreknowledge of disease progression, time to event, and likelihood of treatment benefit (Habel et al., 2006; Paik, 2006). While the AD phenotype varies from patient to patient, monitoring progression at the molecular and cellular level promises to provide invaluable, novel information on pathophysiological changes associated with the course of the disease. The degree of success of individualizing medicine in AD and in other age-related neurodegenerative disorders will, however, depend on the degree to which the molecular aspects of the disease can be elucidated and measured. Aside from messenger RNA-directed studies, recent developments suggest that cellular microRNA may provide a promising target for biomarker studies.
MicroRNA (miRNA) species are naturally present 21- to 22- nucleotide base RNAs, functioning to silence their target genes’ messenger RNAs either by binding to the coding region and promoting cleavage and degradation, or by inhibiting their translation by binding at the 3′-untranslated region (UTR). MiRNAs are recognized for their roles in regulating many cellular processes (Giannakakis et al., 2007), and deregulation of miRNA function has been implicated in human diseases, including cancer (Fabbri et al., 2007), heart disease (Chien, 2007) and Alzheimer’s disease (Lukiw, 2007). Thus, specific changes in miRNA might be relevant to AD pathogenesis. Ongoing studies in our laboratory suggest that the combination of miRNA and proteomic technologies will provide a novel tool for preventative medicine strategies in AD (Pasinetti and Ho, personal communication). Consistent with our hypothesis, recent evidence for example identified a causal relationship between miRNA-29a/b-1 expression and β-amyloid generation in-vitro supporting the hypothesis that specific miRNAs can contribute to increased amyloid neuropathology in sporadic AD (Hebert et al., 2009). Thus, the application of miRNA profiling, DNA microarrays, new bioinformatics predictions, and eventually, in situ hybridization as well as biochemical validation will help to clarify the important role of miRNA as recently demonstrated for miRNA-107, possibly involved in accelerated disease progression through regulation of the APP secretase, BACE1 (Wang et al., 2008).
4. Future perspective
It is clear that the combination of high-throughput cDNA, protein microarrays and miRNA will have a significant effect on neurobiological research, in particular on efforts to discover the mechanisms involved in AD and monitoring the progression from MCI to full-blown AD. Although these approaches are powerful, we note that they should be used to complement other traditional methods, such as those used in molecular biology and genetics. Most importantly, they should be used with great care and rigor to avoid misidentification of false positives. The relatively youthful stage of proteomic techniques has left the field without a “gold standard” to guide investigators and enhance comparability of proteomics and biomarker discovery studies in the field of AD. A recent consensus panel suggested that an ideal biomarker for AD should detect a fundamental feature of AD neuropathology that needs to be validated in neuropathologically confirmed cases. This biomarker should have a diagnostic sensitivity >80% for detecting AD and a specificity of >80% for distinguishing other forms of dementia. Recommended steps to establish a biomarker include (1) confirmation by at least two independent studies conducted by qualified investigators with the results published in peer-reviewed journals, and (2) evidence supporting the utility of the biomarker in assessing a beneficial effect in AD disease-modifying therapy. This line of work has only just begun.
Acknowledgments
Supported by UO1-AG0293310 by the NIA, and the Veteran’s Affairs Medical Center (Bronx, NY) Geriatrics Research, Education and Clinical Center (GRECC) to G.M.P.
Footnotes
Financial disclosure: The authors declare that they have no competing financial interests.
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- Alzheimer’s Association. Early Onset Dementia: A National Challenge, A Future Crisis. 2006a. [Google Scholar]
- Alzheimer’s Association. Fact Sheet: Alzheimer’s Disease. 2006b. [Google Scholar]
- Austen BM, Frears ER, Davies H. The use of seldi proteinchip arrays to monitor production of Alzheimer’s betaamyloid in transfected cells. J Pept Sci. 2000;6:459–469. doi: 10.1002/1099-1387(200009)6:9<459::AID-PSC286>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Beher D, Owens AP, Wrigley JD, Shearman MS. Generation of C-terminally truncated amyloid-beta peptides is dependent on gamma-secretase activity. J Neurochem. 2002;82:563–575. doi: 10.1046/j.1471-4159.2002.00985.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Gold G, Kovari E, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, Hof PR. Stereologic analysis of neurofibrillary tangle formation in prefrontal cortex area 9 in aging and Alzheimer’s disease. Neuroscience. 2003;117:577–592. doi: 10.1016/s0306-4522(02)00942-9. [DOI] [PubMed] [Google Scholar]
- Carrette O, Demalte I, Scherl A, Yalkinoglu O, Corthals G, Burkhard P, Hochstrasser DF, Sanchez JC. A panel of cerebrospinal fluid potential biomarkers for the diagnosis of Alzheimer’s disease. Proteomics. 2003;3:1486–1494. doi: 10.1002/pmic.200300470. [DOI] [PubMed] [Google Scholar]
- Chen WP, Matsunari I, Noda A, Yanase D, Yajima K, Takeda N, Yamada M, Minoshima S, Nishimura S. Rapid scanning protocol for brain 18F-FDG PET: a validation study. J Nucl Med. 2005;46:1633–1641. [PubMed] [Google Scholar]
- Chien KR. Molecular medicine: microRNAs and the tell-tale heart. Nature. 2007;447:389–390. doi: 10.1038/447389a. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cronin M, Ghosh K, Sistare F, Quackenbush J, Vilker V, O’Connell C. Universal RNA reference materials for gene expression. Clin Chem. 2004;50:1464–1471. doi: 10.1373/clinchem.2004.035675. [DOI] [PubMed] [Google Scholar]
- Davies H, Lomas L, Austen B. Profiling of amyloid beta peptide variants using SELDI protein chip arrays. Biotechniques. 1999;27:1258–1261. [PubMed] [Google Scholar]
- Fabbri M, Ivan M, Cimmino A, Negrini M, Calin GA. Regulatory mechanisms of microRNAs involvement in cancer. Expert Opin Biol Ther. 2007;7:1009–1019. doi: 10.1517/14712598.7.7.1009. [DOI] [PubMed] [Google Scholar]
- Frank RA, Galasko D, Hampel H, Hardy J, De Leon MJ, Mehta PD, Rogers J, Siemers E, Trojanowski JQ. Biological markers for therapeutic trials in Alzheimer’s disease. Proceedings of the biological markers working group; NIA initiative on neuroimaging in Alzheimer’s disease. Neurobiol Aging. 2003;24:521–536. doi: 10.1016/s0197-4580(03)00002-2. [DOI] [PubMed] [Google Scholar]
- Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of beta-amyloid. Neuroreport. 1999;10:1699–1705. doi: 10.1097/00001756-199906030-00014. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Gemma S, Tang J. Beta-secretase as a therapeutic target for Alzheimer’s disease. Neurotherapeutics. 2008;5:399–408. doi: 10.1016/j.nurt.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakakis A, Coukos G, Hatzigeorgiou A, Sandaltzopoulos R, Zhang L. miRNA genetic alterations in human cancers. Expert Opin Biol Ther. 2007;7:1375–1386. doi: 10.1517/14712598.7.9.1375. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Muffat JA, Cherny RA, Moir RD, Ericsson MH, Huang X, Mavros C, Coccia JA, Faget KY, Fitch KA, Masters CL, Tanzi RE, Chylack LT, Jr, Bush AI. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet. 2003;361:1258–1265. doi: 10.1016/S0140-6736(03)12981-9. [DOI] [PubMed] [Google Scholar]
- Habel LA, Shak S, Jacobs MK, Capra A, Alexander C, Pho M, Baker J, Walker M, Watson D, Hackett J, Blick NT, Greenberg D, Fehrenbacher L, Langholz B, Quesenberry CP. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res. 2006;8:R25. doi: 10.1186/bcr1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Purohit DP, Perl DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–718. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Davies P, Vianna C, Buxbaum JD, Purohit DP. Tau protein abnormalities associated with the progression of alzheimer disease type dementia. Neurobiol Aging. 2007;28:1–7. doi: 10.1016/j.neurobiolaging.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Hebert SS, Horrq K, Nicola L, Bergmans B, Papadopoulou AS, Delacourte A, De Strooper B. MicroRNA regulation of Alzheimer’s amyloid precursor protein expression. Neurobiol Dis. 2009;33:422–428. doi: 10.1016/j.nbd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Ho L, Guo Y, Spielman L, Petrescu O, Haroutunian V, Purohit D, Czernik A, Yemul S, Aisen PS, Mohs R, Pasinetti GM. Altered expression of a-type but not b-type synapsin isoform in the brain of patients at high risk for Alzheimer’s disease assessed by DNA microarray technique. Neurosci Lett. 2001;298:191–194. doi: 10.1016/s0304-3940(00)01753-5. [DOI] [PubMed] [Google Scholar]
- Ho L, Gineste C, Pompl P, Dang A, Schall M, Pasinetti GM. Expression of psoriasin and cystatin C in the CSF of early Alzheimer’s disease dementia cases correlates with Aβ1–40 content and severity of dementia. Soc Neurosci Meet 2002 [Google Scholar]
- Ho L, Sharma N, Blackman L, Festa E, Reddy G, Pasinetti GM. From proteomics to biomarker discovery in Alzheimer’s disease. Brain Res Brain Res Rev. 2005;48:360–369. doi: 10.1016/j.brainresrev.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Hornberger J, Cosler LE, Lyman GH. Economic analysis of targeting chemotherapy using a 21-gene RT-PCR assay in lymph-node-negative, estrogen-receptor-positive, early-stage breast cancer. Am J Manag Care. 2005;11:313–324. [PubMed] [Google Scholar]
- Kung MP, Hou C, Zhuang ZP, Skovronsky D, Kung HF. Binding of two potential imaging agents targeting amyloid plaques in postmortem brain tissues of patients with Alzheimer’s disease. Brain Res. 2004;1025:98–105. doi: 10.1016/j.brainres.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer’s disease. DNA Cell Biol. 2001;20:683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, Deteresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- Nelson HD, Huffman LH, Fu R, Harris EL. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2005;143:362–379. doi: 10.7326/0003-4819-143-5-200509060-00012. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66:1136–1146. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestle U, Weber W, Hentschel M, Grosu AL. Biological imaging in radiation therapy: role of positron emission tomography. Phys Med Biol. 2009;54:R1–R25. doi: 10.1088/0031-9155/54/1/R01. [DOI] [PubMed] [Google Scholar]
- Nissen SE. Halting the progression of atherosclerosis with intensive lipid lowering: results from the Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) trial. Am J Med. 2005;118 (Suppl 12A):22–27. doi: 10.1016/j.amjmed.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Tuzcu EM, Schoenhagen P, Crowe T, Sasiela WJ, Tsai J, Orazem J, Magorien RD, O’Shaughnessy C, Ganz P. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med. 2005;352:29–38. doi: 10.1056/NEJMoa042000. [DOI] [PubMed] [Google Scholar]
- Opris I, Bruce CJ. Neural circuitry of judgment and decision mechanisms. Brain Res Rev. 2005;48:509–526. doi: 10.1016/j.brainresrev.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Paik S. Molecular profiling of breast cancer. Curr Opin Obstet Gynecol. 2006;18:59–63. doi: 10.1097/01.gco.0000192970.52320.29. [DOI] [PubMed] [Google Scholar]
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Ungar LH, Lange DJ, Yemul S, Deng H, Yuan X, Brown RH, Cudkowicz ME, Newhall K, Peskind E, Marcus S, Ho L. Identification of potential CSF biomarkers in ALS. Neurology. 2006;66:1218–1222. doi: 10.1212/01.wnl.0000203129.82104.07. [DOI] [PubMed] [Google Scholar]
- Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, Powell TP. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci USA. 1985;82:4531–4534. doi: 10.1073/pnas.82.13.4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepeu G, Giovannini MG. Cholinesterase inhibitors and beyond. Curr Alzheimer Res. 2009;6:86–96. doi: 10.2174/156720509787602861. [DOI] [PubMed] [Google Scholar]
- Qin W, Ho L, Wang J, Peskind E, Pasinetti GM. S100A7, a novel Alzheimer’s disease biomarker with non-amyloidogenic alpha-secretase activity acts via selective promotion of ADAM-10. PLoS One. 2009;4:e4183. doi: 10.1371/journal.pone.0004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, Furst AJ, O’Neil JP, Racine CA, Mormino EC, Baker SL, Chetty S, Patel P, Pagliaro TA, Klunk WE, Mathis CA, Rosen HJ, Miller BL, Jagust WJ. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- Rafii MS, Aisen PS. Recent developments in Alzheimer’s disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin NR. Current state of immunotherapy for Alzheimer’s disease. CNS Spectr. 2008;13:39–41. doi: 10.1017/s1092852900027061. [DOI] [PubMed] [Google Scholar]
- Salmon E, Collette F, Magis D, Degueldre C, Bechet S, Guillaume Bcaancaad, Lekeu Fcco. IC-101-03: early FDG-pet brain metabolic impairment in questionable AD patients who convert to dementia. Alzheimer Demen. 2006;2:S649. [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Korecka M, Clark CM, Lee VM, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov. 2007;6:295–303. doi: 10.1038/nrd2176. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M. Cerebrospinal fluid Abeta40 and Abeta42: natural course and clinical usefulness. Front Biosci. 2002;7:d997–1006. doi: 10.2741/A826. [DOI] [PubMed] [Google Scholar]
- Shoji M, Matsubara E, Murakami T, Manabe Y, Abe K, Kanai M, Ikeda M, Tomidokoro Y, Shizuka M, Watanabe M, Amari M, Ishiguro K, Kawarabayashi T, Harigaya Y, Okamoto K, Nishimura T, Nakamura Y, Takeda M, Urakami K, Adachi Y, Nakashima K, Arai H, Sasaki H, Kanemaru K, Yamanouchi H, Yoshida Y, Ichise K, Tanaka K, Hamamoto M, Yamamoto H, Matsubayashi T, Yoshida H, Toji H, Nakamura S, Hirai S. Cerebrospinal fluid tau in dementia disorders: a large scale multicenter study by a Japanese study group. Neurobiol Aging. 2002;23:363–370. doi: 10.1016/s0197-4580(01)00309-8. [DOI] [PubMed] [Google Scholar]
- Small GW, Bookheimer SY, Thompson PM, Cole GM, Huang SC, Kepe V, Barrio JR. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008;7:161–172. doi: 10.1016/S1474-4422(08)70019-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suemoto T, Okamura N, Shiomitsu T, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Kudo Y, Sawada T. In vivo labeling of amyloid with BF-108. Neurosci Res. 2004;48:65–74. doi: 10.1016/j.neures.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Sunderland T, Linker G, Mirza N, Putnam KT, Friedman DL, Kimmel LH, Bergeson J, Manetti GJ, Zimmermann M, Tang B, Bartko JJ, Cohen RM. Decreased beta-amyloid1-42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA. 2003;289:2094–2103. doi: 10.1001/jama.289.16.2094. [DOI] [PubMed] [Google Scholar]
- Sunderland T, Mirza N, Putnam KT, Linker G, Bhupali D, Durham R, Soares H, Kimmel L, Friedman D, Bergeson J, Csako G, Levy JA, Bartko JJ, Cohen RM. Cerebrospinal fluid beta-amyloid1-42 and tau in control subjects at risk for Alzheimer’s disease: the effect of APOE epsilon4 allele. Biol Psychiatry. 2004;56:670–676. doi: 10.1016/j.biopsych.2004.07.021. [DOI] [PubMed] [Google Scholar]
- The Alzheimer’s Study Group. A National Alzheimer’s Strategic Plan 2005 [Google Scholar]
- van Steenburgh J. Alzheimer’s or age? Some tips to make the right diagnosis. ACP Observer. 2003 Available from: http://www.acpinternist.org/archives/2003/09/alzheimers.htm.
- Vehmas AK, Borchelt DR, Price DL, McCarthy D, Wills-Karp M, Peper MJ, Rudow G, Luyinbazi J, Siew LT, Troncoso JC. Beta-amyloid peptide vaccination results in marked changes in serum and brain Abeta levels in APPswe/PS1DeltaE9 mice, as detected by SELDI-TOF-based Protein Chip technology. DNA Cell Biol. 2001;20:713–721. doi: 10.1089/10445490152717578. [DOI] [PubMed] [Google Scholar]
- Verhoeff NP, Wilson AA, Takeshita S, Trop L, Hussey D, Singh K, Kung HF, Kung MP, Houle S. In-vivo imaging of Alzheimer disease beta-amyloid with [11C]SB-13 PET. Am J Geriatr Psychiatry. 2004;12:584–595. doi: 10.1176/appi.ajgp.12.6.584. [DOI] [PubMed] [Google Scholar]
- von GA, Kovari E, Rivara CB, Bouras C, Hof PR, Giannakopoulos P. Stereologic analysis of hippocampal Alzheimer’s disease pathology in the oldest-old: evidence for sparing of the entorhinal cortex and CA1 field. Exp Neurol. 2005;193:198–206. doi: 10.1016/j.expneurol.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of {beta}-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermke M, Sorg C, Wohlschlager AM, Drzezga A. A new integrative model of cerebral activation, deactivation and default mode function in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008;35 (Suppl 1):S12–S24. doi: 10.1007/s00259-007-0698-5. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. Inhibition and modulation of gamma-secretase for Alzheimer’s disease. Neurotherapeutics. 2008;5:391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrase J, Reimold M, Beck A, Heinz A. Multimodal imaging with PET, fMRI and genetic research. Fortschr Neurol Psychiatr. 2008;76:278–285. doi: 10.1055/s-2008-1038178. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Ho L, Yemul S, Zhao Z, Qing W, Pompl P, Kelley K, Dang A, Qing W, Teplow D, Pasinetti GM. Cyclooxygenase-2 promotes amyloid plaque deposition in a mouse model of Alzheimer’s disease neuropathology. Gene Expr. 2002;10:271–278. doi: 10.3727/000000002783992352. [DOI] [PMC free article] [PubMed] [Google Scholar]