

Abstract
A series of lavendamycin analogues with two, three or four substituents at the C-6, C-7 N, C-2′, C-3′ and C-11′ positions were synthesized via short and efficient methods and evaluated as potential NAD(P)H:quinone oxidoreductase (NQO1)-directed antitumor agents. The compounds were prepared through Pictet-Spengler condensation of the desired 2-formylquinoline-5,8-diones with the required tryptophans followed by further needed transformations. Metabolism and toxicity studies demonstrated that the best substrates for NQO1 were also the most selectively toxic to NQO1-rich tumor cells compared to NQO1-deficient tumor cells.
Keywords: Lavendamycin analogues; Quinoline-5,8-diones; Antitumor; NQO1; Cytotoxicity
1. Introduction
Lavendamycin (1), a naturally occurring 7-aminoquinoline-5,8-dione antitumor antibiotic, was first isolated from the fermentation broth of Streptomyces lavendulae by Balitz et al.1 and its structure was determined by Doyle et al.2 The latter study determined that lavendamycin is a pentacyclic structure with two moieties including quinoline-5,8-dione and indolopyridine (β-carboline) (Chart).2 Lavendamycin is similar to another potent antibiotic antitumor agent streptonigrin (2).1–5 The clinical use of both of these antibiotics has been precluded because of their toxicity toward human cells.2,6–8 Initial structure-activity relationship (SAR) studies have demonstrated that the essential moiety for the cytotoxic activity of lavendamycin and streptonigrin is the 7-aminoquinoline-5,8-dione moiety.7

As it was thought that some analogues of the naturally occurring antibiotic 1 might show activity toward human tumors, efficient methods were needed to prepare these derivatives. There were two previous reports on the synthesis of lavendamycin methyl ester (3) (above and Table 1) by Kende9 and Boger10 but both were impractical, since they produced compound 3 in an overall yield of less than 2% and in 9- and 20- steps, respectively. Our group was able to develop two synthetic methods11,12 that produced 3 in an overall yield of about 40% in only 5-steps. These efficient methods, particularly those reported in 1996,12 made it possible to prepare a large number of substituted lavendamycins needed for various in vitro and in vivo screening tests.
Table 1.
Structures of lavendamycins discussed in the Introduction and Discussion sections.
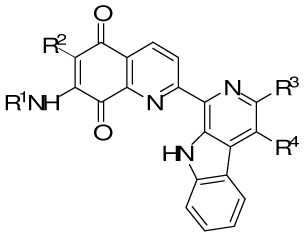 | ||||
|---|---|---|---|---|
| No. | R1 | R2 | R3 | R4 |
| 3 | H | H | CO2CH3 | CH3 |
| 4 | CH3CO | H | CONH2 | H |
| 5 | H | H | CONH2 | H |
| 6 | CH3CO | H | CO2C8H17-n | H |
| 7 | CH3CO | H | CO2(CH2)2CH(CH3)2 | H |
| 8 | CH3CO | H | CO2CH3 | CH3 |
| 9 | 2-furyl-CO | H | CO2CH3 | H |
| 10 | CH3CO | H | CONHC4H9-n | H |
| 11 | H | H | CH2OH | H |
| 12 | H | H | CO2C8H17-n | H |
| 13 | CH3CO | H | CO2CH3 | H |
| 14 | H | Cl | CO2CH3 | CH3 |
| 15 | H | OCH3 | CO2CH3 | CH3 |
| 16 | H | H | CO2(CH2)2OH | H |
We previously reported that a significant number of these lavendamycin analogues have potent biological activity including anti-HIV reverse transcriptase13 and antitumor activity.14–16 More importantly, several screening tests on a number of lavendamycin analogues have shown that a significant number of these compounds possess low animal toxicity.13,17 The NCI in vivo studies have shown that the maximum tolerated dose of three derivatives 3, 4 and 5 (Table 1) in mice is 400 mg/kg which is 31 and 1000 times higher than that for lavendamycin and streptonigrin, respectively.13,17 The lavendamycin analogue 6 reduced tumor volume (up to 80%) in mice bearing tumor xenografts at a daily dose of 300 mg/kg for 10 days without exhibiting drug-related weight loss or lethality.17 A recent study also demonstrated that the normal rat kidney epithelial cell line (NRK-52E) exhibited much less sensitivity to several lavendamycin analogues compared to the tumor cells with the same origin.17 When lavendamycin analogues 7 and 6 at a daily dose of 300 mg/kg and 8 at a daily dose of 100 mg/kg were administered to nude mice for eight and seven days, respectively, no drug-related deaths or toxicity were observed.17 Analogues 8, 7 and 6 (Table 1) inhibited tumor growth in nude mice by 69, 88 and 78%, respectively, when administered at 100, 150 and 300 mg/kg for 7, 8 and 8 days, respectively.17
NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase) is a two-electron reductase, characterized by its capacity for utilizing either NADH or NADPH as reducing cofactors and by its inhibition by dicoumarol.18 It is primarily located in the cytosol (>80%), but a recent study has reported a substantial pool of NQO1 in the nuclei of cancer cells.19 NQO1 is generally categorized as a detoxification enzyme, and it can protect the cell from a broad range of chemically reactive metabolites including electrophiles and reactive oxygen species.20 NQO1 can also function as an activating enzyme, specifically for the reductive activation of antitumor quinones and other bioreductive anticancer agents.21
NQO1 can be induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and polycyclic aromatic hydrocarbons.21 Induction by procarcinogens and the capacity of NQO1 for detoxifying reactive metabolites suggest that changes in expression of NQO1 may occur during carcinogenesis.22 Marked elevations in NQO1 activity and mRNA content have been documented in both preneoplastic tissues and established tumors. Tumors or cancer cell lines where increased NQO1 expression has been observed include those from the lung, liver, colon, and breast.23 In addition, advanced, metastatic tumors appear to express higher levels of NQO1 than non-metastatic tumors.24
Correlations between NQO1 activity in cancer cells and cytotoxicity of antitumor quinones to those cells have been reported.25–27 In our previous studies with several series of indole- and quinolinequinone antitumor agents, including analogues of lavendamycin, those compounds that were the best substrates for NQO1 were also found to be selectively toxic to cell lines with high levels of NQO1 compared to cell lines that were deficient in NQO1.15,16,28–30 In this report, we describe the synthesis, metabolism by recombinant human NQO1 and anticancer activity of novel analogues of lavendamycin with functional group substitutions on both the quinoline-5,8-dione and indolopyridine moieties.
2. Results and Discussion
2.1. Synthetic Chemistry
Table 4 presents the structures of a total of 25 lavendamycins that were the subject of our biological studies in this report. As shown in Scheme 1, Pictet-Spengler (P-S) condensation of the quinolinedione aldehydes (Table 2) with tryptophans (Table 3) yielded the target lavendamycins 40 – 58 listed in Table 4. The amino lavendamycins 59 – 64 were prepared respectively by the acid hydrolysis of the corresponding 7-acylamino lavendamycins 40 – 45.
Table 4.
Structures of lavendamycin analogues
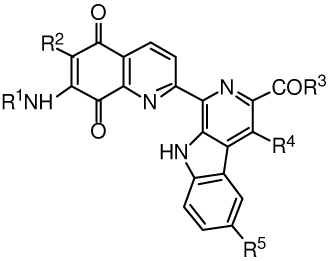 | ||||||
|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | R4 | R5 | |
| 40 | CH3CO | H | N[(CH2CH2)2]O | H | H | see ref 15 |
| 41 | CH3CO | H | O(CH2)2CH(CH3)2 | H | OH | |
| 42 | CH3CO | H | OCH3 | CH3 | OH | |
| 43 | CH3CO | H | N[(CH2)4] | H | H | see ref 15 |
| 44 | CH3CO | H | NH(CH2)2OH | H | H | |
| 45 | CH3(CH2)2CO | H | OCH3 | H | H | see ref 17 |
| 46 | CH3CO | H | OCH(CH3)CH2CH3 | H | H | |
| 47 | CH3CO | H | NHCH2CH(OH)CH2OH | H | H | |
| 48 | CH3CO | H | O(CH2)2CH3 | H | H | |
| 49 | CH3CO | H | N[(CH2CH2)2]NCH2Ph | H | H | |
| 50 | CH3CO | H | OCH2CH3 | H | H | see ref 17 |
| 51 | CH3CO | H | OCH2CH3 | H | OH | |
| 52 | (CH3)2CHCO | H | OCH3 | H | H | see ref 17 |
| 53 | HCO | H | OCH3 | H | H | |
| 54 | ClCH2CO | H | NH2 | H | H | |
| 55 | ClCH2CO | H | N[(CH2)4] | H | H | |
| 56 | 2-furyl-CO | H | OCH3 | CH3 | H | |
| 57 | HO2C(CH2)2CO | H | OC4H9-n | H | H | |
| 58 | H | Cl | OCH2CH3 | H | H | |
| 59 | H | H | N[(CH2CH2)2]O | H | H | |
| 60 | H | H | O(CH2)2CH(CH3)2 | H | OH | |
| 61 | H | H | OCH3 | CH3 | OH | |
| 62 | H | H | N[(CH2)4] | H | H | |
| 63 | H | H | NH(CH2)2OH | H | H | |
| 64 | H | H | OCH3 | H | H | |
Scheme 1.

Table 2.
Structures of aldehydes for Scheme 1.
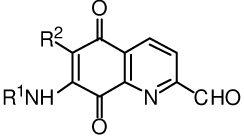
Table 3.
Structures of tryptophans for Scheme 1.
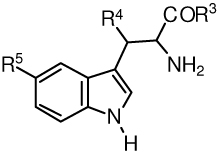 | ||||
|---|---|---|---|---|
| No. | R3 | R4 | R5 | |
| 25 | OCH3 | H | H | see ref 15 |
| 26 | N[(CH2CH2)2]O | H | H | see ref 15 |
| 27 | O(CH2)2CH(CH3)2 | H | OH | |
| 28 | OCH3 | CH3 | OH | |
| 29 | N[(CH2)4] | H | H | see ref 15 |
| 30 | NHCH2CH2OH | H | H | |
| 31 | NHCH2CH(OH)CH2OH | H | H | |
| 32 | N[(CH2CH2)2]NCH2Ph | H | H | |
| 33 | O(CH2)2CH3 | H | H | |
| 34 | OCH(CH3)CH2CH3 | H | H | |
| 35 | OCH2CH3 | H | H | see ref 17 |
| 36 | OCH2CH3 | H | OH | |
| 37 | OCH3 | CH3 | H | see ref 31 |
| 38 | OC4H9-n | H | H | see ref 13 |
| 39 | NH2 | H | H | see ref 13 |
High Resolution Mass Spectroscopy of 67 (Scheme 2) did not produce a signal for the molecular ion M+, but rather gave one at 354.109. This corresponds to a molecular formula of C18H16N3O5 (M − 2H2O + H)+, that may be due to the fragment shown in Scheme 2, produced by the dehydration of 67 under HRMS conditions.
Scheme 2a.

aReagents and conditions: (a) see ref 16; (b) succinic anhydride, NaOAc, Na2SO3, DMF, Ar, 18 h, rt, 82%; (c) K2Cr2O7, CH3COOH, 20 h, rt, 44%; (d) SeO2, 1,4-dioxane, H2O, 11 h, reflux, 57%.
Similar to our reported synthesis of β-methyltryptophan methyl ester,31 the nitrobutanoate 71 (Scheme 3) and the precursor to the ester 28, existed in two stereoisomers A and B in a ratio of 1/1 as shown by NMR. However, upon recrystallization of the mixture, stereoisomer B was completely converted and only the less soluble isomer A was obtained in pure form. When 71A was reduced with H2 in the presence of Ra-Ni the product was 5-benzyloxy β-methyltryptophan methyl ester (28, 73%).
Scheme 3a.

aReagents and conditions: (a) CH3CH=N(CH3)2, CH3COOH, toluene, 4 days, 5 °C, 38%; (b) NaCH2CH3CO2CH3, Et3N, toluene, Ar, 1 h, rt, then 9 h at 103–105 °C, 97%; (c) H2, Pd-C 10%, CF3COOH, 21 h, rt, 93%.
Alkyl esters 27, 33, 34 and 36 (Scheme 5) were prepared by the Fischer esterification of the corresponding tryptophan or hydroxytryptophan with the required alcohol. The preparation of 7-foramido-2-formylquinoline-5,8-dione (20), necessary for the synthesis of lavendamycin 53 is described in the Experimental Section.
Scheme 5.

2.2. Biological Studies
2.2.1. Metabolism
Metabolism of the lavendamycin analogues by recombinant human NQO1 was examined. Reduction rates by NQO1 were measured using a spectrophotometic assay that employs cytochrome c as the terminal electron acceptor15 and gives initial rates of lavendamycin analogue reduction (Table 5). The initial reduction rates (µmol cytochrome c reduced/min/mg NQO1) were calculated from the linear portion (0–30 s) of the reaction graphs.
Table 5.
Metabolism of lavendamycin analogues by recombinant human NQO1 monitored by spectrophotometric cytochrome c assay.
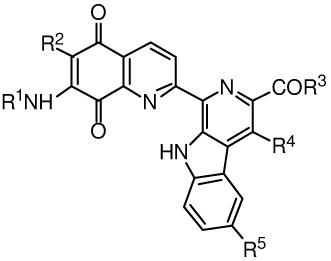 | ||||||
|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | R4 | R5 | Metabolism by NQO1 |
| (µmol/min/mg) | ||||||
| 43 | CH3CO | H | N[(CH2)4] | H | H | 43 ± 7 |
| 44 | CH3CO | H | NH(CH2)2OH | H | H | 2.2 ± 0.7 |
| 46 | CH3CO | H | OCH(CH3)CH2CH3 | H | H | 3.9 ± 2.0 |
| 47 | CH3CO | H | NHCH2CH(OH)CH2OH | H | H | 36 ± 6 |
| 48 | CH3CO | H | O(CH2)2CH3 | H | H | 6.8 ± 3.5 |
| 49 | CH3CO | H | N[(CH2CH2)2]NCH2Ph | H | H | 8.7 ± 1.5 |
| 50 | CH3CO | H | OCH2CH3 | H | H | 3.6 ± 0.3 |
| 51 | CH3CO | H | OCH2CH3 | H | OH | 21 ± 6 |
| 52 | (CH3)2CHCO | H | OCH3 | H | H | 0.04 ± 0.08 |
| 53 | HCO | H | OCH3 | H | H | 20 ± 8 |
| 54 | ClCH2CO | H | NH2 | H | H | 13 ± 2 |
| 55 | ClCH2CO | H | N[(CH2)4] | H | H | 34 ± 5 |
| 56 | 2-furyl-CO | H | OCH3 | CH3 | H | 5.8 ± 1.7 |
| 57 | HO2C(CH2)2CO | H | OC4H9-n | H | H | 5.2 ± 2.6 |
| 58 | H | Cl | OCH2CH3 | H | H | 6.9 ± 1.1 |
| 59 | H | H | N[(CH2CH2)2]O | H | H | 103 ± 8 |
| 60 | H | H | O(CH2)2CH(CH3)2 | H | OH | 2.9 ± 1.4 |
| 61 | H | H | OCH3 | CH3 | OH | 31 ± 7 |
| 62 | H | H | N[(CH2)4] | H | H | 35 ± 7 |
| 63 | H | H | NH(CH2)2OH | H | H | 26 ± 4 |
| 64 | H | H | OCH3 | H | H | 28 ± 2 |
Compound 59 with NH2 and CON[(CH2CH2)2]O groups at R1 and R3 positions, respectively, displayed the highest metabolism rate by NQO1 among the lavendamycin analogues (Table 5). This could be due to the fact that the NH2 group, a small substituent, did not produce steric hindrance with the internal wall of the NQO1 active site resulting in favorable positioning of 59 for hydride ion reception from FAD and quinone reduction. Also, the morpholino group at R3 could potentially form hydrogen bonding or van der Waals interactions with the residues of the active site to increase 59 binding affinity in the active site. Our recent molecular modeling studies have demonstrated that lavendamycin analogues with a small to medium size substituent at the R1 position and substituents at R3 that are capable of hydrogen bonding or van der Waals interactions with the key residues of the active site or FAD are good NQO1 substrates.15,16 Compounds 43, 47, 55, 61 and 64 displayed good metabolism rates in the range of 28 to 43 µmol/min/mg NQO1 (Table 5).
Compound 52, however, with (CH3)2CHCONH and CO2CH3 groups at the R1 and R3 positions, respectively, exhibited the lowest metabolism rate by NQO1 and ranked as the poorest substrate (Table 5). The decreased reduction rate of 52 can be explained by steric hindrance between the quinolinedione moiety of 52 and the NQO1 active site due in part to the large and branched isobutyramide group at R1. This steric interaction could result in unfavorable positioning of 52 in the active site with subsequent poor hydride ion reception and quinone reduction capability. Lavendamycin analogue 56 also displayed a low metabolism rate by NQO1 (Table 5) possibly due to the failure of the 2-furyl-CONH group at the R1 position to intercalate between and form van der Waals interactions with internal wall residues such as Trp-105 and Phe-106.16 A similar 2-furyl-CONH compound 9 (Table 1) with H instead of CH3 at R4 was also ranked as a poor NQO1 substrate by our recent molecular docking study.16 We previously demonstrated that the presence of substituents at R2 also increases steric interactions of lavendamycin ligands with the internal wall of the active site.15,16 Accordingly, compound 58 with a Cl group at this position was ranked as a poor substrate for NQO1 (Table 5).
2.2.2. In Vitro Cytotoxicity
Cytotoxicity studies were also performed on the lavendamycin analogues with cell survival being determined by the MTT colorimetric assay. We demonstrated an excellent positive linear correlation between the IC50 values for clonogenic and MTT assays in a previous study with lavendamycin analogues.15 We utilized the BE human colon adenocarcinoma cells stably transfected with human NQO1 cDNA.32 The BE cells had no measurable NQO1 activity whereas activity in the transfected cells (BE-NQ) was 337 nmol/min/mg total cell protein using dichlorophenolindophenol (DCPIP) as the standard electron acceptor. In the present study, the cytotoxicity of the lavendamycin analogues (Table 6) was compared using these cell lines.
Table 6.
Cytotoxicity of lavendamycin analogues toward BE (NQO1-deficient) and BE-NQ (NQO1-rich) human colon adenocarcinoma cell lines.
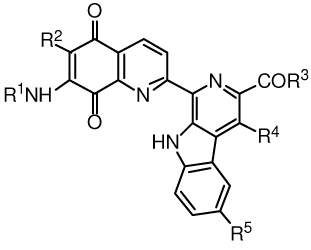 | ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 (µM) | Selectivity Ratio | |||||||
| No. | R1 | R2 | R3 | R4 | R5 | BE-NQ | BE | BE/BE-NQ |
| 43 | CH3CO | H | N[(CH2)4] | H | H | 6.2 ± 0.2 | 13 ± 1 | 2.1 |
| 44 | CH3CO | H | NH(CH2)2OH | H | H | >50 | >50 | - |
| 46 | CH3CO | H | OCH(CH3)CH2CH3 | H | H | 40 ± 10 | 33 ± 8 | 0.8 |
| 47 | CH3CO | H | NHCH2CH(OH)CH2OH | H | H | 18 ± 2 | >50 | >2.8 |
| 48 | CH3CO | H | O(CH2)2CH3 | H | H | 2.4 ± 0.1 | 3.2 ± 0.2 | 1.3 |
| 49 | CH3CO | H | N[(CH2CH2)2]NCH2Ph | H | H | 9.8 ± 0.8 | 6.5 ± 0.5 | 0.7 |
| 50 | CH3CO | H | OCH2CH3 | H | H | 10 ± 1 | 13 ± 1 | 1.3 |
| 51 | CH3CO | H | OCH2CH3 | H | OH | 4.8 ± 0.2 | 4.0 ± 0.4 | 0.8 |
| 52 | (CH3)2CHCO | H | OCH3 | H | H | >50 | >50 | - |
| 53 | HCO | H | OCH3 | H | H | 10 ± 1 | 14 ± 1 | 1.4 |
| 54 | ClCH2CO | H | NH2 | H | H | 5.8 ± 0.6 | 12 ± 1 | 2.1 |
| 55 | ClCH2CO | H | N[(CH2)4] | H | H | 3.0 ± 0.5 | 8.0 ± 1.6 | 2.7 |
| 56 | 2-furyl-CO | H | OCH3 | CH3 | H | >50 | >50 | - |
| 57 | HO2C(CH2)2CO | H | OC4H9-n | H | H | 7.6 ± 0.2 | 12 ± 1 | 1.6 |
| 58 | H | Cl | OCH2CH3 | H | H | >50 | >50 | - |
| 59 | H | H | N[(CH2CH2)2]O | H | H | 2.0 ± 0.1 | 18 ± 1 | 9.0 |
| 60 | H | H | O(CH2)2CH(CH3)2 | H | OH | >50 | >50 | - |
| 61 | H | H | OCH3 | CH3 | OH | 5.7 ± 0.6 | 14 ± 1 | 2.5 |
| 62 | H | H | N[(CH2)4] | H | H | 45 ± 3 | >50 | >1.1 |
| 63 | H | H | NH(CH2)2OH | H | H | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.0 |
| 64 | H | H | OCH3 | H | H | 22 ± 3 | 45 ± 3 | 2.0 |
Lavendamycin analogues such as 47, 55, 59 and 61 that were good substrates for NQO1 (Table 5) were also more toxic to the NQO1-rich BE-NQ cell line than the NQO1-deficient BE cell line (Table 6). Compound 59, the best substrate for NQO1 (Table 1), had the greatest differential toxicity with a selectivity ratio [IC50 (BE) / IC50 (BE-NQ)] of 9 (Table 6). Our previous studies also determined that good lavendamycin substrates for NQO1 were selectively toxic towards BE-NQ versus BE cells.15,16 Recently studied lavendamycin analogues 10, 11, 12, 5, 3 and 16 (Table 1) exhibited high selective toxicity toward BE-NQ cells (selectivity ratios = 30, 11, 10, 9, 9 and 7, respectively).15,16 Four analogues, 8, 13, 14 and 15 (Table 1), suppressed the clonogenic survival of A549 human lung carcinoma cells at concentrations less than 100 nM with 14 being the most potent analogue that inhibited A549 colony growth by 70% at a concentration of 100 nM.14 Also, the colony outgrowth of the cells was reduced by 70% at 10 nM and by 100% at 100 nM concentration of the lavendamycin analogue 5.14 Although the PC-3 human prostate cancer cell line is rather resistant to antitumor effects of many antitcancer agents, these cells displayed sensitivity toward the cytotoxic properties of 5 at a concentration as low as 10 nM.14 Compound 5 also exhibited promising antiproliferative activities against cancer cell lines of the National Cancer Institute (NCI) 60-cell line panel including colon, ovarian and renal cells, and cancer cells in a hollow fiber tumorigenesis assay.14
Lavendamycin analogues 44, 46, 48, 49, 50, 52, 56, 57 and 58 that were poor substrates for NQO1 (Table 5) demonstrated no selective toxicity toward BE-NQ cells or had no measurable cytotoxicity (IC50 > 50 µM) (Table 6). Overall, our results suggest that the best lavendamycin substrates for NQO1 were also the most selectively toxic to the high-NQO1 BE-NQ cell line compared to NQO1-deficient BE cells, consistent with our previous studies.15,16
3. Conclusions
Twenty-five novel lavendamycin analogues were synthesized, characterized and evaluated as NQO1-directed antitumor agents. In line with our earlier computational and structure-activity studies,15,16 analogues with small substituents at the R1 position and groups capable of hydrogen bonding/van der Waals interactions at the R3 position generally made the best substrates for NQO1. Consequently, many of these compounds were also selectively toxic to the cancer cells with elevated NQO1 activity. The best substrate for NQO1 was 59, the 2'- CON[(CH2CH2)2]O - 7-NH2 derivative that also was nine times more toxic to the high-NQO1 BE-NQ cells than the NQO1-deficient BE cells. These data advance our understanding of the structure-activity relationships for NQO1-directed antitumor quinones.
4. Experimental
4.1. Chemistry
4.1.1. General Methods
For general methods, see reference 16 (J. Med. Chem., 2008, 51, 3104). In nearly all of the experimental work-ups, solvents were evaporated under reduced pressure and heat using a rotaevaporator. In each experiment, the progress of the reaction was monitored by TLC and the reaction stopped when TLC showed its completion. In some instances, no NMR signals were observed for active H’s, especially when DMSO-d6 was the solvent of use.
4.1.2. 7-N-Formamido-2-formylquinoline-5,8-dione (20)
In a 25 mL two-necked round-bottomed flask, equipped with a stirring bar, a condenser and an argon filled balloon, 7-formamido-2-methylquinoline-5,8-dione33 (7, 0.246 g, 1.14 mmol), selenium dioxide (0.202 g, 1.82 mmol) were added to 18 mL of dried distilled 1,4-dioxane and 0.2 mL water and refluxed for 24 h. Solid black selenium metal was allowed to settle and the supernatant was filtered. The filter paper containing selenium was placed in a beaker, 10 mL of dioxane was added, and heated for a few minutes on a steam bath to boiling. The mixture was filtered and the solid was again washed with 15 mL of dichloromethane. The combined filtrates were evaporated and tothe resulting solid, 50 mL of dichloromethane was added and placed in a separatory funnel. The solution was washed with sodium bicarbonate (2 × 50 mL), the organic layer was dried (MgSO4) and evaporated. The brown yellow solid was dissolved in dichloromethane, and then concentrated to a small volume. Addition of enough n-hexane gave the yellow solid of pure 5 (78.6 mg, 30%): mp 192–195 °C; 1H NMR (CDCl3) δ 8.07 (s, 1H), 8.34 (d, 1H, J = 7.0), 8.60 (s, 1H), 8.64 (d, 1H, J = 6.6), 8.80 (s, 1H), 10.31 (s, 1H); HRMS m/e calcd for C11H6N2O4, 230.032; found, 230.032.
4.1.3. 7-N-(3-Carboxypropionyl)amino-2-formylquinoline-5,8-dione (23)
Dione 68 (225.1 mg, 0.781 mmol) was added to 4 mL of dioxane and 0.1 mL of water. To the mixture 167.9 mg (1.5 mmol) of selenium dioxide was added and refluxed with stirring under argon for 11 h. The reaction mixture was filtered hot and the filter cake was returned to the flask and refluxed for 5 min with 5 mL of dioxane and then filtered. This process was repeated once more and the combined filtrates were evaporated to produce 130 mg (55%) of the yellow orange solid 23: mp 225.5 °C (dec); 1H NMR (DMSO-d6) δ 2.50 (m, 2H), 2.88 (m, 2H), 7.8 (s, 1H), 8.28 (d, 1H, J = 8.0), 8.54 (d, 1H, J = 8.0), 10.16 (s, 1H), 10.23 (s, 1H), 12.17 (br s, 1H); HRMS m/e C14H10N2O6, 303.061; found, 303.062.
4.1.4. 7-N-(3-Carboxypropionyl)amino-2-methylquinoline-5,8-dione (68)
To a solution of potassium dichromate (1.10 g) in 29 mL of water/glacial acetic acid mixture (13.4 /15.6), 0.51 g (1.3 mmol) of amide 67 was added and the reaction mixture was stirred at rt for 20 h and then extracted with dichloromethane (5 × 50 mL). The combined extracts were dried (MgSO4) and evaporated to give 0.26 g (69%) of the yellow solid 53: mp 144 °C (dec); 1H NMR (DMSO-d6) δ 2.73 (m, 7H), 7.71 (s, 1H), 7.73 (d, 1H, J = 8.3), 8.24 (d, 1H, J = 7.9), 9.98 (s, 1H); HRMS m/e calcd for C14H12N2O5, 288.075; found, 288.077.
4.1.5. 5,7-Bis(3-carboxypropionylamino)-8-hydroxy-2-methylquinoline (67)
5,7-Diamino-8-hydroxy-2-methylquinoline dihydrogen chloride salt16 (66, 1 g, 3.81 mmol) was added to stirred solution of sodium acetate (2 g), sodium sulfite (2 g) in 80 mL DMF under argon. A solution of succinic anhydride (3.05 g) in 30 mL DMF was dropwise added and stirred for 18 h at rt. The reaction mixture was filtered, water (3 mL) added to the filtrate and after1 h, 500 mL of dichloromethane was added and kept in the refrigerator for 36 h. The orange red solid was filtered and dried (1.2034 g, 81%): mp 180–180.6 °C; 1H NMR (DMSO-d6) δ 2.54 (m, 4H), 2.64 (m, 4H), 2.70 (s, 3H), 7.35 (d, 1H, J = 8.8), 8.06 (s, 1H), 8.18 (d, 1H, J = 8.8), 9.65 (br s, 1H), 9.98 (br s, 1H); HRMS m/e calcd for C18H16N3O5 (M − 2 H2O + H), 354.109; found, 354.108.
4.1.6. 5-Hydroxytryptophan isoamyl ester (27)
5-Hydroxytryptophan (1.101g, 5 mmol) in 60 mL anhydrous isoamyl alcohol with 10 mL of hydrogen chloride ether solution (1.0 M) was refluxed for 22 h. The reaction mixture was evaporated, the solid salt was mixed with 50 mL of dichloromethane and neutralized with a solution of 14% ammonium hydroxide. The aqueous layer was extracted with EtOAc (3 × 75 mL) and the combined organic extracts were dried (MgSO4) and then evaporated to give 1.28 g (91%) of product 27 as a gel: 1H NMR (DMSO-d6) δ 0.89 (d, 6H, J = 6.2), 1.30 (m, 2H), 1.60 (m, 1H), 2.8 (m, 1H), 2.9 (m, 1H), 4.14 (t, 2H, J = 7.0), 6.55 (s, 1H), 6.75 (m, 1H), 6.95 (m, 1H), 7.10 (d, 1H, J = 8.8), 8.56 (m, 1H), 10.51 (s, 1H); HRMS m/e calcd for C16H22N2O3, 290.163; found, 290.162
4.1.7. Methyl (2RS,3SR)-2-amino-3-[3-(5-hydroxyindolyl)]butanoate (28)
In a 500 mL heavy-walled hydrogenation flask equipped with a magnetic stirring bar, a cool solution of trifluroacetic acid (0.96 g, 8.44 mmol) in 145 mL absolute ethanol was placed. To this, powdered benzyloxy nitro ester 71A (1.03 g, 2.81 mmol) was added and the mixture was stirred for 1 h for most of the solid to be dissolved. The magnetic bar was removed and 1.65 g of 10% Pd-C was added. Using a Parr hydrogenator, the mixture was hydrogenated at 40 psi at rt for 21 h, then filtered through a layer of Celite. The filter cake was washed with absolute ethanol (3 × 15 mL) and the filtrate was evaporated to dryness. To the residue, 60 mL ether, 2 mL water, 3 mL 14% ammonium hydroxide were added and the yellow ether layer was separated. The aqueous layer was extracted with ether (5 × 50 mL), then the combined ether extracts were washed with 10% sodium chloride solution (2 × 10 mL), dried (MgSO4) and evaporated to give a solid. The product was redissolved in some ether and evaporated. This process was repeated nine more times and the off-white crystals were dried on a vcuum pump, producing 0.65 g (93%) of 28: mp 88–95 °C (unable to recrystallize 28 further): 1H NMR (CDCl3) δ 1.28 (d, 3H, J = 7.1), 2.15 (br s, 2H), 3.57 (m, 1H), 3.65 (s, 3H), 3.89 ( d, 1H, J = 3.8), 6.74 (m, 1H), 6.98 (d, 1H, J = 2.2), 7.03 (d, 1H, J = 2.5), 7.04 (s, 1H), 7.17 (d, 1H, J = 3.7), 7.95 (br s, 1H); HRMS m/e calcd for C13H16N2O3; 248.115; found, 248.115.
4.1.8. Methyl 3-[3-(5-benzyloxyindolyl)]nitrobutanoate (71)
In a 500 mL three-necked round-bottomed flask equipped with a magnetic stirring bar, thermometer, condenser under argon flow was placed indole 70 (1.72 g, 5.54 mmol) in 19.5 mL anhydrous toluene. To this, 0.74 g (7.29 mmol) of freshly distilled triethylamine and 1.0 g (8.42 mmol) of methyl nitroacetate were added. The solution was stirred for 1 h at rt, then heated at 103–105 °C for 9 h and allowed to cool to rt. The solution was extracted with a10% hydrochloric acid solution (3 × 24 mL) followed by water (2 × 13 mL). The organic layer was dried (MgSO4), evaporated and the solid was dried under a vacuum pump at 60 °C overnight (1.98 g, 97%). NMR showed the product to be a mixture of the two stereoisomers 71A and 71B. Careful recrystallization of this product with a mixture of chloroform: hexane (60:40) produced only 71A. An analytical sample was obtained by the recrystallization of the product with 95% ethanol giving 71A as off-white crystals: mp 110–111 °C; 1H NMR (CDCl3) δ 1.53 (d, 3H, J = 3.5), 3.53 (s, 3H), 4.10 (m, 1H), 5.11 (s, 2H), 5.60 (d, 1H, J = 9.0), 6.94 (dd, 1H, J = 9.0, 2.3), 7.05 (dd, 1H, J = 9.0, 2.5), 7.10 (m, 1H), 7.23 (s, 1H), 7.38 (m, 5H), 9.01 (br s, 1H); anal calcd for C20H20N2O5; C, 65.21; H, 5.47; N, 7.60, found; C, 65.04; H, 5.54; N, 7.57; HRMS m/e calcd, 368.141; found, 368.137.
4.1.9. 3-(Isoporopyylaminoethylidene)-5-benzyloxyindole (70).31,34
A 250 mL three-necked round-bottomed flask equipped with a magnetic stirring bar, thermometer, dropping funnel, Claisen head, a condenser with flowing ice-water (using an ice-water circulator) and a calcium chloride drying tube, was immersed in an ice-salt bath. A solution of 5-benzyloxyindole 69 (3.06 g, 0.014 mol) in 50 mL of glacial acetic acid was placed in the flask and then freshly distilled ethylideneisopropylamine34 (1.31 g, 0.015 mol) in 23 mL anhydrous toluene was dropwise added over an h, maintaining the mixture temperature below 15 °C. The reaction mixture was stirred at this temperature for 30 more min and then kept in the refrigerator for 4 days. With vigorous stirring, the mixture was added to 115 mL of ice-water, 38 mL of ether and then the dark upper ether layer was separated from the aqueous layer. The ether solution was extracted with 1 M potassium hydrogen sulfate (3 × 23 mL) and all the aqueous solutions were combined and basified with 10 M NaOH to pH = 11 or higher, while the solution temperature was kept below 25 °C. Enough base was added to produce an oily layer at the top of the solution. Solidification of the oil was facilitated by the addition of 5 mL methylcyclohexane and scratching the side of the beaker inside the oil. The mixture was allowed to stand in the refrigerator overnight, the resulting solid filtered off, and washed thoroughly with cold water to remove any remaining NaOH. The crystals of 70 were vacuum dried (1.61 g, 38%): mp 132–132.5 °C; 1H NMR (CDCl3) δ 1.01 (d, 3H, J = 4.0), 1.04 (d, 3H, J = 4.0), 1.40 (br s, 1H), 1.46 (d, 3H, J = 6.6), 2.82 (m, 1H), 4.17 (q, 1H, J = 6.6), 5.10 (s, 2H), 6.92 (m, 1H), 7.06 (d, 1H, J = 2.3), 7.22 (s, 2H), 7.37 (m, 5H), 7.89 (br s, 1H); anal calcd for C20H24N2O; C, 77.89; H, 7.84; N, 9.08; found, C, 77.55; H, 7.74; N, 8.86; HRMS m/e calcd, 308.193; found, 308.189.
4.1.10. Tryptophan 2-hydroxyethanamide (30)
In a 100 mL round-bottomed flask equipped with a magnetic stirring bar and an argon filled balloon, a mixture of amide 73 (1.45 g, 3.82 mmol), isopropanol (66 mL), 10 % Pd-C (1.785 g) and dry ammonium formate (1.204 g, 19 mmol) was stirred at rt for 5 h. The reaction mixture was filtered and the filtrate was evaporated to give 30 as a gel (774 mg, 82%); 1H NMR (CDCl3) δ 3.02 (m, 2H), 3.39 (m, 2H), 3.65 (m, 2H), 3.67 (m, 1H) 7.09 (s, 1H), 7.15 (m, 1H), 7.21 (m, 1H), 7.37 (d, 1H, J = 8.0), 7.62 (br s, 1H), 7.83 (d, 1H, J = 7.3), 8.07 (br s, 1H); HRMS m/e calcd for C13H17N3O2, 248.139; found, 248.139.
4.1.11. N-Carbobenzyloxytryptophan 2-hydroxyethanamide (73)
In a 25 mL round-bottomed flask equipped with a magnetic stirring bar and an argon filled balloon, 544 mg (1.25 mmol) of succinimide ester 72, and 76.4 mg (1.25 mmol) of 2-hydroxyethanol were added to a mixture of isopropyl alcohol (5.2 mL), chloroform (5.2 mL) and triethylamine (0.8 mL). The reaction mixture was stirred at rt for 23 h and then mixed with 60 mL EtOAc. The organic layer was washed with 10 mL portions of brine, 10% citric acid and water, dried (MgSO4) and then evaporated to give tryptophan hydroxyamide 73 as a gel (464.6 mg, 98%): 1H NMR (CDCl3) δ 2.65 (br s, 2H), 3.40 (m, 1H), 3.48 (m, 1H), 3.67 (s, 2H), 5.10 (m, 5H), 5.43 (m, 1H), 7.08 (m, 1H), 7.15 (m, 1H), 7.29 (m, 7H), 7.55 (m, 1H), 8.41 (br s, 1H); HRMS m/e calcd for C21H24N3O4 (MH+), 382.176; found, 382.176.
4.1.12. Tryptophan 2,3-dihydroxypropanamide (31)
In a set up similar to that for 30 and in a 50 mL flask, amide 74 (411 mg, 1 mmol), anhydrous methanol (18 mL), 10% Pd-C (315.3 mg) and anhydrous ammonium formate were mixed and stirred at rt for 30 min. The mixture was filtered and the filtrate evaporated to give 31 as a colorless gel (236 mg, 85%); 1H NMR (DMSO-d6) δ 2.80 (m, 1H), 2.97 (m, 2H), 3.11 (m, 1H), 3.25 (m, 3H), 3.46 (m, 2H), 3.56 (m, 1H), 6.98 (t, 1H, J = 8.1), 7.07 (t, 1H, J = 8.1), 7.17 (s, 1H), 7.34 (d. 1H, J = 8.4), 7.58 (d, 1H, J = 8.4), 8.06 (br s, 1H), 8.29 (s, 1H), 10.89 (br s, 1H); HRMS m/e calcd for C14H20N3O3 (MH+), 278.150; found, 278.150.
4.1.13.N-Carbobenzyloxytryptophan 2,3-dihydroxypropanamide (74)
In a similar procedure to that of 73, succinimide ester 72 (1.74 g, 4 mmol), 2,3-dihydroxypropanamine (364 mg, 4 mmol) were added to a mixture of 8 mL of isopropyl alcohol, 16 mL of chloroform and 1.12 mL of triethylamine and stirred at rt for 24 h. To the mixture, 120 mL EtOAc was added and the solution was washed consecutively with 10 mL portions of brine, 10% citric acid and then water. The organic layer was dried (Mg SO4) and evaporated to give 1.66 g (71%) of 74 as a gel: 1H NMR (DMSO-d6) δ 3.10 (m, 5H), 4.25 (m, 2H), 4.50 (m, 1H), 4.72 (m, 1H), 5.17 (m, 4H), 7.00 (m, 2H), 7.13 (s, 1H), 7.35 (m, 5H), 7.60 (m, 1H), 7.98 (br s, 1H), 10.80 (br s, 1H); HRMS m/e calcd for C22H26N3O5 MH+), 412.187; found, 412.187.
4.1.14. Tryptophan N-benzylpiperazine amide (32)
In a setup similar to that for 30, and in a 50 mL flask, a mixture of amide 75 (250 mg, 0.5 mmol), ammonium formate (158 mg, 2.5 mmol) and 10% Pd-C (790 mg) were added to 20 mL anhydrous isopropyl alcohol and stirred at rt for 35 min. The reaction mixture was filtered and the filter cake was washed with 20 mL isopropanol. The solid obtained upon the evaporation of the filtrate was suspended in 3 mL of dichloromethane and introduced on top of a 4 in diameter flash chromatography column. The column was first eluted with a mixture of dichloromethane/methanol (10/1) to remove the unreacted starting materials and then the product was eluted with methanol/dichloromethane (5/1). Evaporation of the eluant gave a yellowish white solid that was dried under a vacuum pump at 70–80 °C over 48 h, yielding 95 mg (52%) of the white solid 32: mp 86–87 °C; 1H NMR (DMSO-d6) δ 1.70 (m, 2H), 2.16 (m, 2H), 2.80 (m, 1H), 2.90 (m, 1H), 3.29 (m, 8H), 3.91 (m, 1H), 6.95 (m, 1H), 7.01 (m, 1H), 7.12 (s, 1H), 7.27 (m, 5H), 7.35 (d, 1H, J = 8.0), 7.47 (d, 1H, J = 7.8), 10.85 (s, 1H); HRMS m/e calcd for C22H27N4O (MH+), 363.217; found, 363.218.
4.1.15. N-Carbobenzyloxytryptophan N-benzylpiperazine amide (75)
In a similar set up to that for 73, succinimide ester 72 (435 mg, 1 mmol), N-benzylpiperazine (176 mg, ~ 0.18 mL, 1 mmol) were added to 5 mL chloroform, absolute ethanol (3 mL), and 0.14 ml (1 mmol) of triethylamime and was stirred at rt for 48 h. The mixture was evaporated and the solid was dissolved in 90 mL dichloromethane and the solution was washed with 30 mL of water followed by 2 × 30 mL of 10% citric acid. The aqueous layer was extracted with 2 × 10 mL dichloromethane and the combined organic extracts were washed with 30 mL of 1 M sodium bicarbonate solution, followed by 5 mL of water, dried (MgSO4) and evaporated to dryness to give a gel. This material was further dried under a vacuum pump at 60 °C overnight to produce the white solid of 75 (455 mg, 92%): mp = 134–136 °C; 1H NMR (CDCl3) δ 1.32 (m, 4H), 2.01 (m, 4H), 3.16 (m, 2H), 3.30 (s, 2H), 4.96 (m, 1H), 5.11 (s, 2H), 5.76 (m, 1H), 7.25 (m, 14H) 7.64 (d, 1H, J = 7.7), 8.08 (s, 1H); HRMS m/e calcd for C30H32N4O3, 496.247; found, 496.247.
4.1.16. DL-Tryptophan n-propyl ester (33)
Using a similar procedure to that for ester 27, ester 33 was obtained in 88% yield by the reaction of n-propyl alcohol with DL-tryptophan in the presence of anhydrous HCl in ether as a thick honey colored paste. TLC showed the product to be pure: Rf = 0.58 (3/2 acetone/EtOAc); 1H NMR (CDCl3) δ 0.90 (m, 3H), 1.6 (s, 2H), 1.75 (m, 2H), 3.04 (m, 1H), 3.27 (m, 1H), 3.83 (m, 1H), 4.05 (m, 2H), 7.06 (s, 1H), 7.12 (m, 1H), 7.20 (m, 1H), 7.35 (d, 1H, J = 8.1), 7.63 (d, 1H, J = 8.1), 8.30 (br s, 1H).
4.1.17. L-Tryptophan sec-butyl ester (34)
L-Tryptophan (1 g, 4.9 mmol) and sec-butyl alcohol (60 mL) were placed in a 100 mL round-bottomed flask equipped with a magnetic stirring bar, a condenser and an oil bubbler. The flask was cooled in an ice-bath for 10 min and then 12 mL of an anhydrous HCl-ether solution was added via a syringe through a rubber septum at the top of the condenser. The flask was removed from the ice-bath, placed in an oil bath and then refluxed for 22 h. Three drops of conc. sulfuric acid was added and the solution was refluxed for an additional 2 h. When thin layer chromatography indicated the completion of the reaction, the mixture was allowed to cool to rt to provide a solid residue. Dichloromethane (50 mL) was added to the residue, then placed in a separatory funnel and treated with a 5% solution of sodium bicarbonate until pH ~ 8. The mixture was extracted with dichloromethane (4 × 40 mL), washed with 40 mL of brine, dried (MgSO4) and then evaporated to yield 0.88 g (69%) of 34 as a light brown gel. 1H NMR (CDCl3) δ 0.90 (m, 3H), 1.20 (m, 3H), 1.60 (m, 4H), 3.00 (m, 1H), 3.30 (m, 1H), 3.80 (m, 1H), 4.87 (m, 1H), 7.07 (s, 1H), 7.20 (m, 2H), 7.35 (d, 1H, J = 7.7), 7.65 (d, 1H, J = 7.7), 8.22 (br s, 1H); HRMS m/e calcd for C15H20N2O5, 260.152; found, 260.151.
4.1.18. 5-Hydroxytryptophan ethyl ester (36)
In a 100 mL round-bottomed flask, 5-hydroxtrypyophan (1.101 g, 5 mmol), ethanol (50 mL) and 10 mL of hydrogen chloride ether solution (1.0 M) was heated in an oil bath at 88 °C for 24 h. The reaction mixture was evaporated, the solid salt was mixed with 50 mL of dichloromethane and neutralized with a solution of 14% ammonium hydroxide. The aqueous layer was extracted with EtOAc (3 × 75 mL) and the combined organic extracts were dried (MgSO4) and then evaporated to give 367 mg (29%) of ester 36 as a gel: 1H NMR (CDCl3) δ 1.22 (t, 3H, J = 7.2), 1.80 (br s, 2H), 2.02 (s, 1H), 3.00 (m, 1H), 3.22 (m, 1H), 3.77 (m, 1H), 4.15 (q, 2H, J = 7.2), 6.72 (m, 1H), 7.01 (m, 2H), 7.22 (m, 1H), 7.95 (br s, 1H); HRMS m/e calcd for C13H16N2O3, 248.115; found, 248.115.
4.1.19. 7-N-Acetyl-11'-hydroxydemethyllavendamycin isoamyl ester (41)
In a 250 mL three-necked round-bottomed flask, equipped with a magnetic stirring bar and a condenser under an argon flow, isoamyl ester 27 (84 mg, 0.3 mmol) in 10 mL anhydrous DMF and 100 mL anhydrous anisole were placed. To this solution, aldehyde 17 (73.2 mg, 0.3 mmol) was added and heated to 150 °C over 3 h and then the heat was maintained for an additional 5 h. The golden yellow solution was allowed to cool to rt and then evaporated to give an orange solid that was washed with 3 mL of acetone followed by 3 mL of ether. The filtrate also gave some product as a brown solid. Total weight of the product 41 was 80.2 mg (53%): mp >290 °C; Rf = 0.23 (0.2/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 1.01 (d, 6H, J = 6), 1.70 (m, 1H), 2.30 (m, 2H), 2.42 (s, 3H), 4.38 (t, 2H, J = 6.4), 7.20 (m, 1H), 7.35 (d, 1H, J = 8.8), 7.65 (d, 1H, J = 1.4), 7.71 (s, 1H), 8.40 (d, 1H, J = 8.1), 8.72 (d, 1H, J = 8.1), 8.79 (s, 1H), 9.45 (s, 1H), 10.12 (br s, 1H), 11.48 (br s, 1H); HRMS m/e calcd for C28H24N4O6 512.170; found, 512.169.
4.1.20. 7-N-Acetyl-11'-Hydroxylavendamycin methyl ester (42)
In a set up similar to that for 41, in a 500 mL flask, aldehyde 17 (180.7 mg, 0.58 mmol), 5-hydroxy-β-methyltryptophan methyl ester (28, 198.8 mg, 0.8 mmol) in 390 mL of anhydrous anisole were slowly heated to 162 °C over a period of 3 h and then allowed to continue at this temperature for 26 h. In the last 2 h of the reaction, argon flow was replaced by an oxygen flow. The reaction mixture was evaporated to give a dark red solid (201 mg, 59%): mp 180–181 °C (dec); Rf = 0.49 (0.4/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 2.30 (s, 3H), 2.96 (s, 3H), 3.94 (s, 3H), 7.16 (d, 1H, J = 8.8), 7.38 (d, 1H, J = 8.8), 7.61 (br s, 1H), 7.71 (s, 1H), 8.36 (d, 1H, J = 8.2), 8.69 (d, 1H, J = 8.3), 9.42 (s, 1H), 9.42 (s, 1H), 10.12 (s, 1H), 11.56 (br s, 1H); HRMS m/e calcd for C25H18N4O6, 470.122; found, 470.121.
4.1.21. 7-N-Demethyllavendamycin 2-hydroxyethanamide (44)
Using a similar set up as that for 41, in a 500 mL flask, tryptophan (2-hydroxyethan)amide (30, 125.8 mg, 0.5 mmol) was dissolved in a mixture of 10 mL anhydrous DMF and 220 mL anisole, heated to 84 °C and to this, 122 mg (0.5 mmol) of 7-acetamido-2-formylquinoline-5,8-dione (17) was added. The reaction mixture was gradually heated to 150–155 °C over 3 h and kept at this temperature for another 3 h. The mixture was evaporated, the solid was washed consecutively with 5 mL portions of acetone, ether and finally petroleum ether to give 131 mg of a product. NMR showed that this product contained some of the 5,8-dihydroxy derivative that was converted to 44 by treating with 63.65 mg of DDQ in 23 mL THF and allowed to stir at rt for 20.5 h. The solid was consecutively washed with 3 mL portions of THF, acetone and petroleum ether yielding 124 mg (53%) of 44 as an orange solid: mp > 280 °C; Rf = 0.47 (0.2/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 2.32 (s, 3H), 3.53 (m, 2H), 3.62 (m, 2H), 4.86 (m, 1H), 7.42 (dd, 1H, J = 7.0, 7.0), 7.67 (m, 2H), 7.84 (s, 1H), 8.54 (m, 2H), 9.07 (s, 1H), 9.09 ( m, 1H), 9.42 (d, 1H, J = 8.4), 10.32 (br s, 1H), 11.98 (br s, 1H); HRMS m/e calcd for C25H19N5O5, 469.138; found, 469.137.
4.1.22. N-Acetyldemethyllavendamycin sec-butyl ester (46)
In a set up similar to that for 41, anhydrous and freshly distilled xylene (145 mL) was placed in the flask and to this, 7-acetamido-2-formylquinoline-5,8-dione (17, 0.2401 g, 1 mmol) was added and heated to 100 °C. To the mixture, a solution of L-Tryptophan sec-butyl ester (34, 0.2568 g, 1 mmol) in 12 mL of anhydrous and freshly distilled xylene under argon was added via a syringe and then heated for 16 h at 130 °C until the reaction was completed (TLC). The mixture was filtered off to produce a greenish solid and the filtrate was kept in the refrigerator overnight to give more product (total, 0.2785 g, 57%). An analytical sample of 46 was obtained by recrystallization with DMSO: mp = 268 °C; Rf = 0.46 (1.7/10 MeOH/CH2Cl2); 1H NMR (CDCl3) δ 1.03 (t, 3H, J = 7.7), 1.42 (d, 3H, J = 6.2), 1.8 (m, 1H), 2.30 (s, 3H), 5.2 (m, 1H), 7.34 (t, 1H, J = 8.1), 7.62 (t, 1H, J = 8.1), 7.69 (d, 1H, J = 8.4), 7.93 (m, 2H), 8.21 (d, 1H, J = 7.7), 8.35 (br s, 1H), 8.52 (d, 1H, J = 8.4), 8.88 (s, 1H), 9.17 (d, 1H, J = 8.4), 11.23 (br s, 1H); HRMS m/e cacd for C27H25N4O5 (M + 3H)+, 485.182; found, 485.183.
4.1.23. 7-N-Acetyldemethyllavendamycin 2,3-dihydroxypropanamide (47)
In a set up similar to that for 41, tryptophan 2,3-dihydroxypropanamide (31, 114.8 mg, 0.44 mmol) was dissolved in 40 mL anhydrous DMF and 240 mL anhydrous anisole and heated to 80 °C in a 500 mL flask. To this, 7-acetamido-2-formylquinoline-5,8-dione (17, 97.6 mg, 0.44 mmol) was added and the mixture was gradually heated to 150–154 °C over a period of 3 h, and then continued at this temperature for 5 additional h. The reaction mixture was concentrated to a small volume producing 47 as orange crystals. The crystals were washed with ethyl ether and dried (104.2 mg, 52% ): mp 214–215 °C (dec); Rf = 0.4 (0.3/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 2.30 (s, 3H), 3.37 (m, 1H), 3.45 (m, 2H), 3.60 (m, 1H), 4.75 (m, 1H), 5.00 (m, 1H), 7.45 (m, 1H), 7.74 (m, 2H), 7.84 (s, 1H), 8.55 (m, 2H), 9.07 (s, 1H), 9.12 (br s, 1H), 9.34 (d, 1H, J = 8.4), 11.97 (br s, 1H); HRMS m/e calcd for C26H21N5O6, 499.149; found, 499.147.
4.1.24. 7-N-Acetyldemethyllavendamycin n-propyl ester (48)
In a set up similar to that of 41, in a 500 mL flask, 7-acetamido-2-formylquinoline-5,8-dione (17, 149 mg, 0.61 mmol), tryptophan n-propyyl ester (33, 162 mg, 0.66 mmol) in 90 mL anhydrous and distilled xylene were heated to 100 °C over one h and then allowed to reflux for 3 h. While hot, some solid impurities were filtered and the filtrate was evaporated to a small volume and allowed to cool giving an orange-red solid. The product was dried on a vacuum pump, producing 80 mg (30%) of 48: mp 265 °C (dec): Rf = 0.48 (0.1/5 MeOH/CH2Cl2); 1H NMR (CDCl3) δ 1.13 (t, 3H, J = 7.3), 1.94 (m, 2H), 2.36 (s, 3H), 4.46 (t, 2H, J = 7.4), 7.40 (t, 1H, J = 8.1), 7.67 (t, 1H, J = 6.9), 7.75 (d, 1H, J = 8.0), 8.00 (s, 1H), 8.26 (d, 1H, J = 7.6), 8.44 (s, 1H), 8.58 (d, 1H, J = 8.4), 8.97 (s, 1H), 9.22 (d, 1H, J = 7.1), 11.83 (br s, 1H); HRMS m/e calcd for C26H20N4O5; 468.143; found, 468.144.
4.1.25. 7-N-Acetyldemethyllavendmycin N-benzylpiperazine amide (49)
In a set up similar to that for 41, in a 100 mL flask, aldehyde 17 (122 mg, 0.5 mmol) in 40 mL anhydrous anisole was stirred and heated to 70 °C until it was dissolved. A solution of N-benzylpiperazine amide of tryptophan (32, 180 mg, 0.5 mmol) in 5 mL dried and freshly distilled pyridine was introduced to the reaction flask using a syringe. The reaction mixture was heated to reflux (30 min) and then maintained at this temperature for 6 h. The mixture was allowed to cool to rt , the yellow lemon precipitate was filtered off and the filter cake was washed with THF (3 mL), followed by petroleum ether (3 mL). The solid was vacuum dried, suspended in 10 mL THF and then treated with DDQ (94 mg, 0.41 mmol), stirred at rt for 24 h in order to oxidize any 5,8-dihydroxy lavendamycin present in the mixture to 49. The brown reaction mixture was filtered off, the filter cake washed with THF (3 mL) followed by petroleum ether (3 mL) and dried on a vacuum pump giving 115 mg (40%): mp > 260 °C; Rf = 0.88 (1/7 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 2.32 (s, 3H), 3.30 (m, 4H), 3.57 (s, 2H), 3.76 (m, 4H), 7.32 (m, 6H), 7.70 (m, 2H), 7.81 (s, 1H), 8.47 (d, 1H, J = 7.7), 8.55 (d, 1H, J = 8.2), 8.70 (s, 1H), 8.86 (d, 1H, J = 8.2), 10.30 (br s, 1H), 11.85 (br s, 1H); HRMS m/e calcd for C34H28N6O4; 584.217; found, 584.219.
4.1.26. 7-N-Acetyl-11'-hydroxydemethyllavendamycin ethyl ester (51)
Using a similar set up to that for 41, in a 100 mL flask, 5-hydroxytryptophan ethyl ester (36, 29.76 mg, 0.12 mmol) in 4 mL of DMF was added to 40 mL anhydrous anisole and then with 29.8 mg (0.12 mmol) of 7-acetamido-2-formylquinoline-5,8-dione9,10 (17) was gradually heated to 160 °C over 3 h and kept at this temperature for 23.5 h. The orange solution was evaporated to give a dark solid. The crude product was washed with 3 mL acetone and dried under vacuum producing the dark violet 51 (27 mg, 48%): mp > 280 °C; Rf = 0.69 (0.2/5 MeOH/ CH2Cl2); 1H NMR (DMSO-d6) δ 1.44 (m, 3H), 2.30 (s, 3H), 4.40 (m, 2H), 7.17 (m, 1H), 7.40 (m, 1H), 7.67 (br s, 1H), 7.74 (s, 1H), 8.44 (d, 1H, J = 8.4), 8.78 (d, 1H, J = 8.4), 8.84 (s, 1H), 9.45 (s, 1H), 10.15 (br s, 1H), 11.53 (br s, 1H); HRMS m/e calcd for C25H18N4O6, 470.122; found, 470.121.
4.1.27. 7-N-Formyldemethyllavendamycin methyl ester (53)
In a similar set up as that for 41, 7-formamido-2-formylquinoline-5,8-dione (20, 50 mg, 0.22 mmol), and tryptophan methyl ester (46 mg, 0.21 mmol) were dissolved in 125 mL of anhydrous anisole in a 250 mL flask, heated to 130 °C over an h and continued for another 1.5 h. Anisole was evaporated and the solid residue was washed with 5 mL of acetone. The brownish solid was filtered off and washed with a few mL acetone and dried. The product 53 weighed 18.45 mg (19.6%): mp > 300 °C; Rf = 0.79 (0.2/2.3 MeOH/EtOAC/CH2Cl2); 1H NMR (CDCl3) δ 4.1 (s, 3H), 7.42 (m, 1H), 7.69 (m, 1H), 7.78 (d, 1H, J = 8.1), 8.02 (s, 1H), 8.26 (d, 1H, J = 8.1), 8.53 (br s, 1H), 8.61 (d, 1H, J = 8.4), 8.76 (br s, 1H), 9.02 (s, 1H), 9.26 (d, 1H, J = 8.4), 11.85 (br s, 1H); HRMS m/e calcd for C23H14N4O5, 426.099; found, 426.096.
4.1.28. 7-N-Chloroacetyldemethyllavendamycin amide (54)
In a set up similar to that of 53, 7-N-chloroacetyl-2-formylquinoline-5,8-dione15 (21, 82.7 mg, 0.3 mmol), tryptophan amide13 (39, 61 mg, 0.3 mmol), and anhydrous anisole (120 mL) were mixed, stirred and heated to 155 °C over a period of 3 h. Upon the completion of the reaction after 20 h, the mixture was allowed to cool to room temperature and the yellow solid was filtered off. The product was washed with petroleum ether and dried on a vacuum pump overnight to give 68 mg (49%) of pure 54: mp> 280 °C; Rf = 0.35 (0.25/5 MeOH/CH2Cl2). 1H NMR (DMSO-d6) δ 4.55 (s, 2H), 7.39 (t, 1H, J = 8.4), 7.70 (t, 1H, J = 8.4), 7.82 (d, 1H, J = 8.0), 8.49 (s, 1H), 8.49 (d, 1H, J = 8.1), 8.61 (d, 1H, J = 8.8), 9.02 (s, 1H), 9.12 (d, 1H, J = 9.2), 9.75 (s, 1H), 9.93 (s, 1H), 10.19 (s, 1H), 12.26 (br s, 1H); HRMS m/e calcd for C23H14ClN5O4, 462.097; found, 462.097.
4.1.29. 7-N-Chloroacetyldemethyllavendamycin pyrrolidine amide (55)
In a similar set up to that of 53, quinolinedione aldehyde 21 (42.5 mg, 0.15 mmol), tryptophan pyrrolidine amide15 (29, 39 mg, 0.15 mmol) and 55 mL anhydrous anisole were mixed in a 100 mL flask. The mixture was gradually heated to 155 °C over a period of 3 h and then heated at this temperature for an additional 18 h. The reaction mixture was filtered off hot and the filter cake was washed with 4 mL of dichloromethane followed by 4 mL of chloroform. The filtrate was evaporated, the crude product was introduced on a small column of silica gel and eluted with ethyl acetate to produce 11 mg (21%) of pure 55 as a yellow solid: mp 264 °C (dec); Rf = 0.77 (0.25/5 MeOH/CH2Cl2). 1H NMR (DMSO-d6) δ 1.96 (m, 4H), 3.64 (m, 2H), 3.98 (m, 2H), 4.66 (s, 2H), 7.41 (t, 1H, J = 8.1), 7.71 (m, 2H), 7.81 (s, 1H), 8.48 (d, 1H, J =8.1), 8.57 (d, 1H, J = 8.1), 8.88 (s, 1H), 8.94 (d, 1H, J = 8.4), 10.65 (s, 1H), 11.87 (br s, 1H); HRMS m/e calcd for C27H20ClN5O4, 513.124; found, 513.120.
4.1.30. 7-N-(2-Furyl)carbonyllavendamycin methyl ester (56)
Using a similar set up to that of 53, in a 50 mL flask, a mixture of 7-furoylamino-2-formylquinoline-5,8-dione16 (22, 29.7 mg, 0.10 mmol), β-methyltryptophan methyl ester31 (37, 23.1 mg, 0.10 mmol) and 32 mL freshly distilled anydrous xylene was gradually heated to 145 °C over a 3 h period. The heat was continued for an additional 19 h and then the mixture was evaporated to dryness. The dark orange solid was washed with a small amount of chloroform producing the light orange solid 56 (30.11 mg, 60%): mp 327–328 °C; Rf = 4.1 (0.1/5 MeOH/CH2Cl2); 1H NMR (CDCl3) δ 3.21 (s, 3H), 4.09 (s, 3H), 6.68 (m, 1H), 7.41 (m, 2H),7.68 (m, 2H), 7.82 (m, 1H), 8.06 (m, 1H), 8.36 (m, 1H), 8.53 (d, 1H, J = 8.4), 9.12 (d, 1H, J = 8.4), 9.36 (s, 1H), 11.92 (br s, 1H). HRMS m/e calcd for C28H18N4O6, 506.122; found, 506.122.
4.1.31. 7-N-(3-Carboxypropionyl)demethyllavendamycin n-butyl ester (57)
Using the same set up as that of 53, 60 mL of anhydrous anisole was placed in a 100 mL flask and then tryptophan n-butyl ester13 (38, 39.5 mg, 0.15 mmol) was added followed by 40 mg (0.13 mmol) of 7-N-(3-carboxypropionyl)-2-formylquinoline-5,8-dione (23). With stirring, this mixture was heated at 120 °C for 19.5 h. The reaction mixture was filtered to remove the solid impurity and the filtrate was evaporated. The amorphous solid was recrystallized from 10 mL ethyl acetate to give 22 mg (29%) of 57 as a red crystalline solid: mp 232.4 °C (dec); Rf = 0.83 (2.5/3.5 MeOH/CHCl3). 1H NMR (DMSO-d6) δ 1.01 (t, 3H, J = 7.3), 1.54 (m, 2H), 1.83 (m, 2H), 2.50 (m, 2H), 2.90 (m, 2H), 4.50 (m, 2H), 7.50 (m, 1H), 7.70 (m, 2H), 7.80 (s, 1H), 8.54 (d, 1H, J = 8.0), 8.59 (d, 1H, J = 8.4), 8.93 (d, 1H, J = 8.4), 9.09 (s, 1H), 10.35 (br s, 1H), 11.97 (br s, 1H); HRMS m/e calcd for C29H25N4O7 (MH+), 541.172; found, 541.172.
4.1.32. 6-Chlorodemethyllavenamycin ethyl ester (58)
In a 500 mL flask with similar set up to that of 53, a mixture of 7-amino-6-chloro-2-formylquinoline-5,8-dione (24, 47.4 mg. 0.2 mmol) and tryptophan ethyl ester 17 (35, 46.4 mg, 0.2 mmol) and 120 mL anhydrous anisole was slowly heated to 130 °C over 3 h. The reaction mixture was refluxed for 1 h and then allowed to cool to rt. The solvent was evaporated to dryness, the solid washed with acetone (20 mL), filtered and dried under vacuum giving 47 mg (49%) of 58: mp > 270 °C; Rf = 0.26 (0.1/5 MeOH/CH2Cl2); 1H NMR CDCl3) δ 1.57 (m, 3H), 4.57 (q, 2H, J = 7.2), 5.86 (br s, 2H), 7.42 (m, 1H), 7.68 (m, 1H), 7.77 (d, 1H, J = 7.3), 8.27 (d, 1H, J = 7.8), 8.67 (d, 1H, J = 8.2), 9.00 (s, 1H), 9.22 (d, 1H, J = 8.3), 11.86 (br s, 1H); HRMS m/e calcd for C23H16ClN4O4 (MH+), 447.086; found, 447.086.
4.1.33. Demethyllavendamycin morpholine amide (59)
In a 25 mL two-necked round-bottomed flask, equipped with a magnetic stirring bar, a condenser and an argon filled balloon, 7-N-acetyldemethyllavendamycin morpholine amide 15(40, 163 mg, 0.33 mmol) was placed and kept for a few min in an ice-bath. To this 15.8 mL of 70% sulfuric acid was dropwise added until the solid was dissolved. The mixture was heated at 61 °C for 6 h. To the reaction mixture, 10 mL water was added and neutralized to a pH of 8.5 with a saturated solution of sodium carbonate. The mixture was evaporated to give a solid material, that was finely ground by mortar and pestle and extracted with 160 mL of chloroform in a Soxhlet extractor overnight. The resulting solution was evaporated and dried to give 91 mg (63%) of the red orange 59: mp 275–276 °C; Rf = 0.39 (0.01/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 3.74 (m, 8H), 5.95 (s, 1H), 7.39 (t, 1H, J = 8.1), 7.48 (br s, 2H), 7.70 (m, 2H), 8.47 (d, 1H, J = 8.1), 8.49 (t, 1H, J = 8.1), 8.71 (s, 1H), 8.84 (d, 1H, J = 8.4) 11.87 (br s, 1H). HRMS m/e calcd for C25H20N5O4 (MH+), 454.154; found, 454.151.
4.1.34. 11'-Hydroxydemethyllavendamycin isoamyl ester (60)
In a procedure similar to that of 59, 7-N-acetyldemethyllavendamycin isoamyl ester (41, 25.1 mg, 0.05 mmol) and 2.4 mL of sulfuric acid (70%) was heated at 60 °C for 3.5 h. The reaction mixture was carefully neutralized with a saturated solution of sodium bicarbonate to pH = 8, and then extracted with chloroform (4 × 100 mL). The organic extracts were washed with water followed by brine and then dried (MgSO4). Evaporation of the solvent produced 16 mg (70%) of the orange product 60: mp > 280 °C; Rf = 0.23 (0.2/5 MeOH/CH2Cl2); 1H NMR (DMSOd6) δ 1.01(d, 6H, J = 6.2), 1.74 (m, 3H), 4.43 (m, 2H), 5.95 (s, 1H), 7.25 (d, 1H, J = 8.8), 7.61 (d, 1H, J = 8.8), 7.78 (br s, 1H), 8.54 (d, 1H, J = 8.4), 8.90 (d, 1H, J = 8.4), 8.97 (s, 1H), 9.46 (s, 1H), 11.82 (br s, 1H); HRMS m/e calcd for C26H22N4O5, 471.167; found, 471.169.
4.1.35. 11´-Hydroxylavendamycin methyl ester (61)
In a similar set up and procedure to that of 59, acetyllavendamycin ester 42 (98.4 mg, 0.21 mmol) was slowly added to 15 mL of a 70% solution of sulfuric acid at 0 °C and then heated at 60 °C for 3 h. The reaction mixture was treated with a saturated solution of sodium carbonate to pH = 8 and extracted with 8 × 75 mL of ethyl acetate, dried and evaporated to give 61 as red crystals. The aqueous layer was evaporated to dryness, the solid was ground and extracted with 150 mL ethyl acetate for 24 h in a Soxhelet extractor. Evaporation of the solution afforded more of 61 (total weight 33.7 mg, 38%): mp > 280 °C; Rf = 0.72 (0.4/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 2.85 (s, 3H), 3.90 (s, 3H), 5.85 (s, 1H), 7.06 (d, 1H, J = 8.8), 7.30 (d, 1H, J = 8.8), 7.41 (br s, 2H), 7.49 (s, 1H), 8.23 (d, 1H, J = 8.1), 8.50 (d, 1H, J = 8.1), 9.30 (s, 1H), 11.36 (s, 1H); HRMS m/e calcd for C23H16N4O5, 428.112; found, 428.113.
4.1.36. Demethyllavendamycin pyrrolidine amide (62)
In a similar procedure to that for 59, 51 mg (0.11 mmol) of 7-N-acetyldemethyllavendamycin pyrrolidine amide15 (43), was hydrolyzed with 5.2 mL of 70% sulfuric acid to give 24 mg (50%) of the orange red product 62: mp 300 °C; Rf = 0.22 (0.1/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 1.92 (m, 4H), 3.63 (m, 2H), 3.98 (m, 2H), 5.95 (s, 1H), 7.39 (m, 2H), 7.77 (m, 3H), 8.45 (m, 2H), 8.84 (m, 2H), 11.89 (br s, 1H); HRMS m/e calcd for C25H20N5O3 (MH+) 438.156; found 438.156.
4.1.37. Demethyllavendamycin 2-hydroxyethanamide (63)
Lavendamycin 44 (70.5 mg, 0.15 mmol) was added slowly to 7.2 mL of 70% sulfuric acid and heated at 60 °C for 3.5 h. The mixture was treated with a saturated solution of sodium carbonate to pH = 8 and evaporated to dryness. The residue was extracted with 70 mL of methanol/dichloromethane (10/60) and then with 35 mL of the same mixture. The extracts were concentrated to a small volume and the red crystals of 63 were filtered off (18.9 mg, 30%): mp > 280 °C; Rf = 0.31 (0.3/5 MeOH/CH2Cl2); 1H NMR (DMSO-d6) δ 3.83 (m, 2H), 4.40 (m, 2H), 5.02 (br s, 2H), 5.96 (s, 1H), 7.40 (m, 1H), 7.73 (m, 2H), 7.78 (d, 1H, J = 8.0), 8.5 (d, 1H, J = 8.0), 8.85 (d, 1H, J = 8.1), 8.92 (d, 1H, J = 8.1), 9.12 (s, 1H), 12.01 (br s, 1H); HRMS m/e calcd for C23H17N5O4, 427.128; found, 427.130.
4.1.38. Demethyllavendamycin methyl ester (64)
Using a similar set up as that for 59, 7-N-butyryldemethyllavendamycin methyl ester17 (45, 22 mg, 0.05 mmol) was slowly added to 2 mL of 70% sulfuric acid in a 5 mL flask with stirring. The mixture was heated at 60 °C for 4 h, then carefully neutralized with a saturated solution of sodium carbonate to pH = 8 and extracted with chloroform (2 × 100 mL and 50 mL). The extracts were evaporated to give 64 as a red solid (17 mg, 85%): mp 220 °C (dec); Rf = 0.35 (0.1/5 MeOH/CH2Cl2); 1H NMR (CDCl3) δ 4.00 (s, 3H), 5.35 (br s, 2H), 6.15 (s, 1H), 7.41 (m, 1H), 7.70 (m, 2H), 7.81 (d, 1H, J = 8.0), 8.27 (d, 1H, J = 8.0), 8.60 (d, 1H, J = 8.3), 9.20 (d, 1H, J = 8.3), 12.00 (br s, 1H); HRMS m/e calcd for C22H14N4O4, 398.101; found, 398.099.
4.2. Biology
4.2.1. Cell culture
BE human colon adenocarcinoma cells and stably NQO1-transfected BE-NQ cells32 were a gift from Dr. David Ross (University of Colorado-Denver, Denver, CO). Cells were grown in a minimum essential medium (MEM) with Earle’s salts, non-essential amino acids, L-glutamine and penicillin/streptomycin, and supplemented with 10% fetal bovine serum (FBS), sodium bicarbonate and HEPES. Cell culture medium and supplements were obtained from Gibco, Invitrogen Co., Grand Island, NY. The cells were incubated at 37 °C under a humidified atmosphere containing 5% CO2.
4.2.2. Spectrophotometric cytochrome c assay
Lavendamycins analogue reduction was monitored using a spectrophotometric assay in which the rate of reduction of cytochrome c was quantified at 550 nm. Briefly, the assay mixture contained cytochrome c (70 µM), NADH (1 mM), recombinant human NQO1 (0.1–3 µg) (gift from Dr. David Ross, University of Colorado-Denver, Denver, CO) and lavendamycins (25 µM) in a final volume of 1 mL Tris-HCl (25 mM, pH 7.4) containing 0.7 mg/mL BSA and 0.1% Tween-20. Reactions were carried out at room temperature and started by the addition of NADH. Rates of reduction were calculated from the initial linear part of the reaction curve (0–30 s) and results were expressed in terms of µmol of cytochrome c reduced/min/mg of NQO1 using a molar extinction coefficient of 21.1 mM−1 cm−1 for cytochrome c. All reactions were carried out at least in triplicate.
4.2.3. Cell viability assay
Growth inhibition was determined using the MTT colorimetric assay. Cells were plated in 96-well plates at a density of 10,000 cells/mL and allowed to attach overnight (16 h). Lavendamycin analogue solutions were applied in medium for 2 hours. Lavendamycin analogues solutions were removed and replaced with fresh medium, and the plates were incubated at 37 °C under a humidified atmosphere containing 5% CO2 for 3–5 days. MTT (50 µg) was added and the cells were incubated for another 4 hours. Medium/MTT solutions were removed carefully by aspiration, the MTT formazan crystals were dissolved in 100 µL DMSO, and absorbance was determined on a plate reader at 560 nm. IC50 values (concentration at which cell survival equals 50% of control) were determined from semi-log plots of percent of control vs. concentration. Selectivity ratios were defined as the IC50 value for the BE cell line divided by the IC50 value for the BE-NQ cell line.
Scheme 4a.

aReagents and conditions: (a) H2N(CH2)2OH for 73, H2NCH2CHOHCH2OH for 74, N(CH2CH2)2NBn for 75, CHCl3, Et3N, alcohol, Ar, 29 h for 73, 24 h for 74, and 48 h for 75, rt; (b) HCOONH4, Pd-C 10%, isopropanol for 30, 32 and methanol for 31, Ar, 4.5 h for 30, 30 min for 31 and 32, rt.
Acknowledgements
The financial support by the National Institutes of Health is greatly appreciated (NIH grants: R15CA74245, MB; R15CA78232 and P20RR017670, HDB). We also thank professor David Williams and the staff at the mass spectroscopy laboratory at Indiana University for their help in obtaining mass spectral data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balitz DM, Bush JA, Bradner WT, Doyle TW, O'Herron FA, Nettleton DE. J. Antibiot. 1982;35:259. doi: 10.7164/antibiotics.35.259. [DOI] [PubMed] [Google Scholar]
- 2.Doyle TW, Balitz DM, Grulich RE, Nettleton DE, Gould SJ, Tann C-H, Meows AE. Tetrahedron Lett. 1981;22:4595. [Google Scholar]
- 3.Erickson WR, Gould SJ. J. Am. Chem. Soc. 1985;107:5831. [Google Scholar]
- 4.Erickson WR, Gould SJ. J. Am. Chem. Soc. 1987;109:620. [Google Scholar]
- 5.Rao KV, Biemann K, Woodward RB. J. Am. Chem. Soc. 1963;85:2532. [Google Scholar]
- 6.Hackethal CA, Golbey RB, Tan CTC, Karnofsky DA, Burchenal JH. Antibiot. Chemother. 1961;11:178. [PubMed] [Google Scholar]
- 7.Boger DL, Mitscher YM, Drake SD, Kitos PA, Thompson SC. J. Med. Chem. 1987;30:1918. doi: 10.1021/jm00393a040. [DOI] [PubMed] [Google Scholar]
- 8.Wilson WL, Labra C, Barrist E. Antibiot. Chemother. 1961;11:147. [PubMed] [Google Scholar]
- 9.Kende AS, Ebetino FH. Tetrahedron Lett. 1984;25:923. [Google Scholar]
- 10.Boger DL, Duff SR, Panek JS, Yasuda M. J. Org. Chem. 1985;50:5790. [Google Scholar]
- 11.Behforouz M, Gu Z, Cai W, Horn MA, Ahmadian M. J. Org. Chem. 1993;58:7089. [Google Scholar]
- 12.Behforouz M, Haddad J, Cai W, Arnold MB, Mohammadi F, Sousa AC, Horn MA. J. Org. Chem. 1996;61:6552. doi: 10.1021/jo960794t. [DOI] [PubMed] [Google Scholar]
- 13.Behforouz M, Cai W, Stocksdale MG, Lucas JS, Jung JY, Briere D, Wang A, Katen KS, Behforouz NC. J. Med. Chem. 2003;46:5773. doi: 10.1021/jm0304414. [DOI] [PubMed] [Google Scholar]
- 14.Fang Y, Linardic CM, Richardson DA, Cai W, Behforouz M, Abraham RT. Mol. Cancer Ther. 2003;2:517. [PubMed] [Google Scholar]
- 15.Hassani M, Cai W, Holley DC, Lineswala JP, Maharjan BR, Ebrahimian GR, Seradj H, Stocksdale MG, Mohammadi F, Marvin CC, Gerdes JM, Beall HD, Behforouz M. J. Med. Chem. 2005;48:7733. doi: 10.1021/jm050758z. [DOI] [PubMed] [Google Scholar]
- 16.Hassani M, Cai W, Koelsch KH, Holley DC, Rose AS, Olang F, Lineswala JP, Holloway WG, Gerdes JM, Behforouz M, Beall HD. J. Med. Chem. 2008;51:3104. doi: 10.1021/jm701066a. [DOI] [PubMed] [Google Scholar]
- 17.Behforouz M, Cai W, Mohammadi F, Stocksdale MG, Gu Z, Ahmadian M, Baty DE, Etling MR, Al-Anzi CH, Swiftney TM, Tanzer LR, Merriman RL, Behforouz NC. Bioorg. Med. Chem. 2007;15:495. doi: 10.1016/j.bmc.2006.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ernster L. Meth. Enzymol. 1967;10:309. [Google Scholar]
- 19.Winski SL, Koutalos Y, Bentley DL, Ross D. Cancer Res. 2002;62:1420. [PubMed] [Google Scholar]
- 20.Talalay P, Dinkova-Kostova AT. Meth. Enzymol. 2004;382:355. doi: 10.1016/S0076-6879(04)82019-6. [DOI] [PubMed] [Google Scholar]
- 21.Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. Chem.-Biol. Interact. 2000;129:77. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 22.Danson S, Ward TH, Butler J, Ranson M. Cancer. Treat. Rev. 2004;30:437. doi: 10.1016/j.ctrv.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Schlager JJ, Powis G. Int. J. Cancer. 1990;45:403. doi: 10.1002/ijc.2910450304. [DOI] [PubMed] [Google Scholar]
- 24.Mikami K, Naito M, Ishiguro T, Yano H, Tomida A, Yamada T, Tanaka N, Shirakusa T, Tsuruo T. Jpn. J. Cancer Res. 1998;89:910. doi: 10.1111/j.1349-7006.1998.tb00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beall HD, Murphy AM, Siegel D, Hargreaves RHJ, Butler J, Ross D. Mol. Pharmacol. 1995;48:499. [PubMed] [Google Scholar]
- 26.Plumb JA, Gerritsen M, Workman P. Br. J. Cancer. 1994;70:1136. doi: 10.1038/bjc.1994.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robertson N, Haigh A, Adams GE, Stratford IJ. Eur. J. Cancer. 1994;30A:1013. doi: 10.1016/0959-8049(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 28.Beall HD, Winski S, Swann E, Hudnott AR, Cotterill AS, O’Sullivan N, Green SJ, Bien R, Siegel D, Ross D, Moody CJ. J. Med. Chem. 1998;41:4755. doi: 10.1021/jm980328r. [DOI] [PubMed] [Google Scholar]
- 29.Swann E, Barraja P, Oberlander AM, Gardipee WT, Hudnott AR, Beall HD, Moody CJ. J. Med. Chem. 2001;44:3311. doi: 10.1021/jm010884c. [DOI] [PubMed] [Google Scholar]
- 30.Fryatt T, Pettersson HI, Gardipee WT, Bray KC, Green SJ, Slawin AMZ, Beall HD, Moody CJ. Bioorg. Med. Chem. 2004;12:1667. doi: 10.1016/j.bmc.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 31.Behforouz M, Zarrinmayeh H, Ogle ME, Riehle TJ, Bell FW. J. Heterocycl. Chem. 1988;25:1627. [Google Scholar]
- 32.Winski SL, Hargreaves RH, Butler J, Ross D. Clin. Cancer Res. 1998;4:3083. [PubMed] [Google Scholar]
- 33.Behforouz M, Haddad J, Cai W, Gu Z. J. Org. Chem. 1998;63:343. [Google Scholar]
- 34.Snyder H, Matteson D. J. Am. Chem. Soc. 1957;79:2217. [Google Scholar]