

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited cause of kidney failure and accounts for ∼5% of end-stage renal disease (ESRD) population in the United States. The disorder is characterized by the focal and sporadic development of renal cysts, which increase in size and number with age. Mutations of PKD1 and PKD2 account for most of the cases. Although the clinical manifestations of both gene types overlap completely, PKD1 is associated with more severe disease than PKD2, with larger kidneys and earlier onset of ESRD. In general, renal ultrasonography is commonly used for the diagnosis of ADPKD and age-dependent criteria have been defined for subjects at-risk of PKD1. However, the utility of the PKD1 ultrasound criteria in the clinic setting is unclear since their performance characteristics have not been defined for the milder PKD2 and the gene type for most test subjects is unknown. Recently, highly predictive ultrasound diagnostic criteria have been derived for at-risk subjects of unknown gene type. Additionally, both DNA linkage or gene-based direct sequencing are now available for the diagnosis of ADPKD, especially in subjects with equivocal imaging results, a negative or indeterminate family history, or in younger at-risk individuals being evaluated as potential living-related kidney donors. Here, we review the clinical utilities and limitations of both imaging- and molecular-based diagnostic tests, and outline our approach for the evaluation of individuals suspected to have ADPKD.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD [MIM#173900]) is the most common form of monogenic, inherited kidney disease worldwide and affects 1 in 500 to 1000 births 1-3. The disorder is characterized by age-dependent development and progressive enlargement of renal cysts resulting in chronic renal failure typically in mid to late adulthood. Early in life individuals with ADPKD may not have detectable renal cysts by conventional imaging techniques. Later in life, however, the diagnosis is usually straightforward. In the setting of a positive family history, the hallmark radiographic findings of ADPKD are bilaterally enlarged kidneys with numerous cysts, frequently accompanied by liver cysts. There are, however, emerging indications for early identification of individuals affected by ADPKD. This review will explore strategies for pre-symptomatic diagnosis and their application in the care and management of individuals at risk for ADPKD.
Genetics of ADPKD: PKD1 versus PKD2
Genetic studies have indicated that most if not all cases of ADPKD are caused by mutations in two genes. It is estimated that about 85% of affected individuals from linkage-characterized European families have mutations in the PKD1 gene (on chromosome 16p13.3), while 15% have PKD2 gene mutations (on chromosome 4q21)4-6. Two recent studies from Toronto, Canada and Rochester, MN USA have documented a higher prevalence for PKD2 of 26% and 36%, respectively, suggesting that it may have been under-detected before the age of wide spread imaging7. A small fraction (∼1%) of all ADPKD families is not linked to either of the known gene loci, suggesting that there may be at least one more disease gene 8-10. However, confirmation of a disease locus in these families has been lacking, and the disease in one such family was found to result from two independently segregating PKD1 and PKD2 mutations 11. Thus, the existence of a third gene for ADPKD is questionable at the present time 12, 13.
As the name implies, ADPKD is inherited in an autosomal dominant fashion. Practically this means that each affected individual carries only one copy of a mutant PKD gene and the risk of passing that gene on to an offspring at birth is on average 50%. Although every cell in the body of an affected individual carries a PKD mutation, cyst formation is a focal process involving a small subset of cells. In a large proportion of cysts, there is evidence that somatic mutation results in loss of the wild type PKD gene 14, 15. This suggests that the decline of effective polycystin signaling below a critical threshold may lead to a series of cellular derangements that promote cyst formation.
As discussed in more detail elsewhere in this issue, the PKD1 protein product, polycystin-1 (PC1), is a large protein of unknown function that physically interacts with the PKD2 protein, polycystin-2 (PC2), which functions as a non-specific cation channel 16-19. Recent studies suggest that the polycystin complex resides in the primary cilia of renal tubular cells where it is thought to regulate intracellular calcium levels in response to external stimuli. Not surprisingly mutations of either PKD1 or PKD2 result in an identical clinical profile including extrarenal cysts (e.g. liver, pancreas spleen), hernias, cardiac valvular abnormalities and intracranial aneurysms 6.
The main distinction between individuals with PKD1 versus PKD2 mutations is that the latter is associated with less severe disease. Studies comparing the two groups of patients have demonstrated that as a group, individuals with PKD2 mutations have a milder phenotype by every criterion including later age at diagnosis, decreased prevalence of hypertension and delayed onset of ESRD (median age: ∼54 vs. ∼74) 20. This reflects a lower cyst burden in PKD2 as shown by the Consortium of Radiological Imaging Study of PKD (CRISP), which provided serial observations of total renal and cystic volumes by MRI in 156 PKD1 and 27 PKD2 patients over a three-year period 21, 22. At any given age, both the cyst number and total kidney volume were significantly lower in the PKD2 cohort when compared with their PKD1 counterparts.
These marked differences in disease severity between PKD1 and PKD2 have practical implications for diagnosis since imaging modalities may have a higher rate of false negative results in younger individuals from PKD2 families. Conversely, patients with PKD1 have a higher probability of being clinically diagnosed after 20 years of age. As a result, the differences in pre-test probability of disease between the two gene types needs to be adjusted accordingly to ensure proper evaluation of the performance characteristics of any imaging-based diagnostic test for ADPKD (Figure 1).
Figure 1.
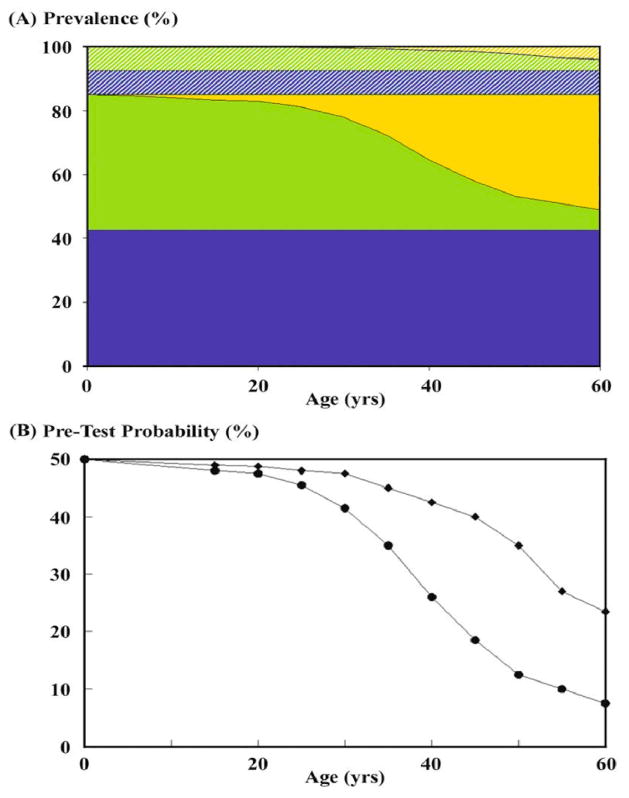
The pre-test probability of disease decreases with age in subjects born with 50% risk of ADPKD and differs between PKD1 and PKD2. (A) The prevalence of PKD1 (solid color) and PKD2 (strip color) are assumed to be 85% and 15%, respectively. At birth, the proportion of affected but clinically undiagnosed subjects (green color) is equal to that of at-risk but unaffected subjects (blue color). With increasing age the proportion of affected but clinically undiagnosed subjects (green color) diminishes as increasing proportion of the affected subjects are clinically diagnosed (yellow color). However, a higher proportion of the more severely affected PKD1 subjects will be clinically diagnosed (solid yellow color) compared to affected PKD2 subjects (strip yellow color). (B) The pre-test probability of disease among subjects born with 50% risk for PKD1 (circle) and PKD2 (diamond) diverges with increasing age (Data derived from Lancet 353: 103-7, 1999).
One question that is frequently asked is whether a family history of renal disease severity can be used to predict the ADPKD gene type (PKD1 vs. PKD2). Barua et al addressed this issue in a cohort of 481 affected individuals from 67 PKD1 and 23 PKD2 families 7. Despite significant variability in disease severity within individual families, the authors were able to devise predictive guidelines by focusing on cases where ESRD developed very late or very early. They found that the presence of “at least one family member who developed ESRD at or before 55 years of age” was highly predictive of PKD1 (positive predictive value 100%, sensitivity 72%). Similarly, having “at least one family member with ESRD at or after 70 years of age” was highly predictive of PKD2 (positive predictive value 100%, sensitivity 72%). Thus a detailed family history of renal disease severity in ADPKD may yield important prognostic information in over 70% of cases.
Distinguishing ADPKD from other Cystic Diseases
In a majority of cases, establishing a diagnosis of ADPKD is uncomplicated. The typical patient presents with enlarged cystic kidneys in the setting of a positive family history. Accompanying clinical manifestations may include extrarenal cysts in organs such as the liver, pancreas or spleen, inguinal hernias or intracranial aneurysms. In some cases, however, confounding aspects of a case may prompt the clinician to consider alternate diagnoses. In general renal cysts can be a manifestation of both hereditary and acquired disorders other than ADPKD (Table 1). A careful review of the history, clinical and radiologic investigations may reveal clinical features of these disorders that are atypical of ADPKD. We will discuss several of these scenarios below.
Table 1.
Differential Diagnosis of ADPKD
| Cystic Disorder | Prevalence | Inheritance | Distinguishing Renal Findings | Distinguishing Extrarenal Findings |
|---|---|---|---|---|
| Polycystic Liver Disease | Unknown | AD | Small number of renal cysts. | Predominantly liver cystic disease. |
| Autosomal Recessive Polycystic Kidney Disease | ∼1:20,000- | AR | Early in life kidneys cystic, enlarged and echogenic. With increasing age, kidneys are smaller with macroscopic cysts, nephrocalcinosis and/or small medullary calcifications common. | Oligohydramnios and pulmonary hypoplasia in utero, congenital hepatic fibrosis, Caroli's disease. |
| Tuberous Sclerosis | ∼1:10,000 | AD | Angiomyolipoma. Contiguous deletion of PKD1/TSC2 results in severe early onset PKD with ESRD typically occurring in the first 2 decades of life. | Skin lesions (facial angiofibromas, perungal fibroma, hypomelanotic macules, Shagreen patch); retinal hamartomas; seizures; mental retardation; cortical tuber; subependymal giant cell astrocytoma; cardiac rhabdomyoma; lymphangioleiomyomatosis. |
| Von Hippel-Lindau | ∼1:50,000 | AD | High risk of renal cell carcinomas. | Central nervous system (CNS) and retinal hemangioblastomas, pancreatic cysts, pancreatic endocrine tumors, pheochromocytoma. |
| Orofaciodigital Syndrome I | Very rare | X-linked, dominant | Embryonic male lethal, cleft palate, bifid tongue, hyperplastic frenula, hypertelorism, broadened nasal ridge, digital abnormalities including syndactyly, CNS malformations. | |
| Glomerulocystic disease | Rare | AD | Heterogeneous phenotypes: ultrasound may show hyperechogenic kidneys, renal hypoplasia/dysplasia, oligomeganephronia, multiple renal cysts, or glomerulocystic disease. | Mutations in the gene encoding HNF1-beta, maturity-onset diabetes of the young; genital tract abnormalities such as epididymal cysts, atresia of vas deferens and bicornuate uterus. |
| Medullary Cystic Kidney Disease | Unknown | AD | Interstitial fibrosis on renal biopsy. Rarely cysts in the corticomedullary junction; slowly progressive renal failure; small to normal size kidneys. | Hyperuricemia, gout. |
| Simple Renal Cysts | Common | -None-acquired | Number increases with age. Normal renal function; normal sized kidneys with smooth contour. | None. |
| Acquired Renal Cystic Disease | Common | -None-acquired | Associated with chronic renal insufficiency or ESRD. Multiple renal cysts associated with small to normal sized kidneys. | None |
AD=autosomal dominant; AR=autosomal recessive
Although ADPKD is primarily a disease of adulthood, there are rare instances of severe disease presenting in infancy or early childhood 23, 24. In some cases, this may be due to a contiguous gene syndrome involving a chromosomal deletion of both PKD1 and TSC2 (tuberous sclerosis) genes, which are located in close proximity on chromosome 16p13.3 25, 26. Patients with this syndrome may present during infancy with rapid progression to end stage renal disease. The absence of a family history is common in individuals with this type of mutation because their parents are somatic mosaics while other cases represent de novo mutations. Clinical features suggestive of tuberous sclerosis complex (TSC [MIM#19100]) such as angiomyolipomas or characteristic skin lesions may be absent in 30% of cases, however, leading to a misdiagnosis of this syndrome as ADPKD 26, 27. In other instances, especially when an early onset case occurs in the context of a family with typical ADPKD, adverse genetic and/or environmental modifiers are presumed to underpin the renal disease severity.
Isolated autosomal dominant polycystic liver disease (PCLD [MIM174050]), which is discussed in another article in this issue, may also be mistaken for mild ADPKD 28, 29. PCLD is caused by mutations in a set of genes that are distinct from PKD1 and PKD2 30-32. In its classic form one can distinguish PLD by liver cysts in the absence of cystic kidney disease. However, on occasion, these patients may have a few renal cysts prompting the clinician to consider a diagnosis of ADPKD but as a general rule these individuals have predominantly polycystic liver disease and do not develop end stage renal disease.
Occasionally ADPKD can be confused with autosomal recessive polycystic kidney disease (ARPKD [MIM263200]), which is caused by mutations in the fibrocystin/polyductin gene on chromosome 6 33, 34. Individuals with ARPKD usually present in the neonatal period with enlarged, echogenic kidneys. With the advent of molecular diagnosis, however, adult forms of ARPKD have been described 33. These cases may be confused with ADPKD by virtue of their demographic characteristics ie, onset in adulthood. In addition to the absence of a family history, distinguishing features of older individuals with ARPKD include small kidneys with a few macrocysts and prominent hepatic disease characterized by congenital hepatic fibrosis and/or Caroli's disease rather than liver cystic disease 33, 35.
Autosomal dominant medullary cystic kidney disease (MCKD, [MIM603860]) is another disorder that may present in adulthood with renal dysfunction and occasionally renal cysts. MCKD is caused by mutations in the uromodulin gene (encoding Tamm-Horsfall protein) on chromosome 16 36. A second locus for MCKD on chromosome 1 has also been reported. In contrast to ADPKD, MCKD is characterized by tubular interstitial fibrosis with normal to small sized kidneys usually accompanied by early hyperuricemia and gout. Medullary cysts may be detected in some individuals late in the course of disease but are frequently absent 37.
There are several additional syndromic inherited disorders that may present with renal cystic disease including Von Hippel-Lindau [MIM19330] syndrome, orofaciodigital syndrome-I [MIM311200] and glomerulocystic disease [MIM137920] (Table 1). In each case, however, the spectrum of associated clinical findings is unique and distinguishes the disorder from ADPKD 38-40.
Although ADPKD is an inherited disease, it is not uncommon for individuals with the classic clinical findings of this disorder to report that they know of no other affected family members. Although one might consider alternative diagnoses, several series have suggested that a significant percentage of affected individuals in fact lack a family history 2, 41, 42. This may be due to the presence of a de novo mutation (estimated to occur in <10% of cases) or alternatively a failure to diagnose cystic kidney disease in mildly affected family members 41. This is probably more common in the case of PKD2 families that tend to exhibit a milder disease process. Review of parental imaging studies or autopsy reports, if available, may be helpful in establishing a family history.
Simple renal cysts and acquired renal cystic disease are non-inherited forms of cystic kidney disease that may also be seen in adults 43, 44. Simple renal cysts increase in frequency with age and are detected quite commonly with sensitive imaging methods such as CT or MRI 45, 46. For example, in one MRI based series, ∼11% of individuals 18-29 years of age, ∼51% subjects 30-44 years of age and ∼93% of subjects 45-59 years of age had at least one renal cyst 46. This supports the assumption that extrapolation of ultrasound based criteria (see below) to MRI is likely to result in false positive diagnoses. Acquired renal cystic disease is typically detected in individuals with longstanding kidney disease and can be found in up to 20% of patients with ESRD 44. Although patients with acquired cystic disease have multiple and bilateral renal cysts, kidney size is usually normal or small. This is in contrast to individuals with ESRD in the setting of ADPKD whose kidneys tend to be large.
Can ADPKD be detected using standard imaging Techniques?
Ultrasound is the most commonly used imaging modality for screening individuals at risk for ADPKD. This is based on its safety and modest cost in comparison with CT and MRI. In most moderate to advanced cases, ultrasound easily detects the classic finding of ADPKD including multiple, bilateral renal cysts and liver cysts as well. In younger patients with early stage PKD, however, the diagnosis may not be obvious since smaller cysts are more likely to escape sonographic detection, especially for patients with milder PKD2 (Figure 2).
Figure 2.
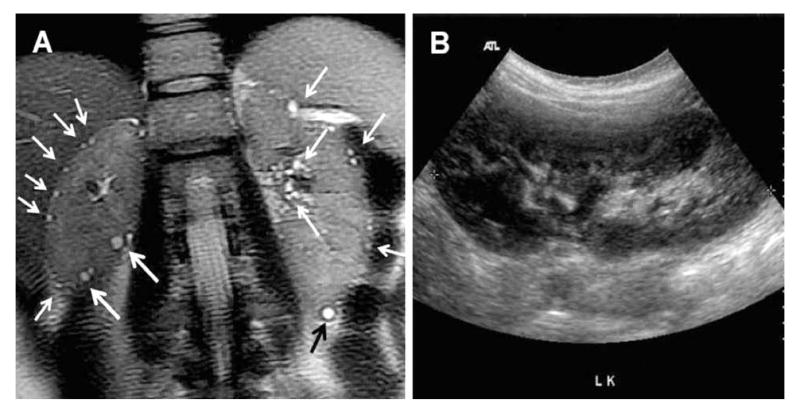
(A) T2 weighted breath-held MRI from a 28 year-old female at-risk of ADPKD performed without contrast injection in one breath-hold showing multiple tiny cysts in a cortical and perihilar distribution (white arrows) in both kidneys (≫10/kidney). (B) Corresponding ultrasound through both kidneys was normal. Only the left kidney ultrasound is shown. DNA testing subsequently detected a truncating PKD2 mutation in her and her other affected family members.
Studies published in the 1990's by Ravine and colleagues established age graded ultrasound criteria for PKD1 47. The basic principle, given that sporadic cysts occur more frequently with age, was that more cysts were required to make a diagnosis of ADPKD in older individuals. Thus the presence of “two unilateral or bilateral cysts” was sufficient to confirm the diagnosis in at-risk subjects between 15-29 years, but at least “two cysts per kidney” and “four cysts per kidney” were required for diagnosis of at-risk subjects aged 30-59 years and 60 years of age or older, respectively. Although these criteria were designed to be used in individuals at risk for PKD1, in practice many clinicians have extrapolated them to all at-risk subjects, regardless of their underlying gene type. The concern here is that since PKD2 is typically less severe, these criteria might not perform as well. Thus there is a need for highly predictive criteria that can be applied to either ADPKD gene type.
To address this issue, Pei and associates have recently completed a multicenter study of 577 individuals from 58 PKD1 families and 371 individuals from 39 PKD2 families who underwent ultrasound screening 48. In addition, the authors used either DNA linkage or mutation analysis to determine the gene type and disease status of each study subject. The performance characteristics of various criteria were re-examined for individuals from both PKD1 and PKD2 families (Table 2A). As expected, the Ravine criteria resulted in a higher rate of false negative results when applied to individuals at risk for PKD2. Surprisingly, even though incidental simple renal cysts were thought to be vanishingly rare in young individuals, they found that 2.1% of the genetically unaffected population younger than 30 years of age had 1-2 renal cysts. These latter findings suggested that a more stringent criterion might be advantageous in this group of patients.
The authors also analyzed the performance characteristics of different ultrasound criteria in at-risk individuals of unknown gene type using a statistical re-sampling method termed “bootstrapping” to simulate the PKD1 and PKD2 case mix (ie, PKD1:PKD2 = 85:15). The recommended criteria are summarized in Table 2A. Specifically, for at-risk individuals 15 to 39 years of age, the presence of “three or more unilateral or bilateral renal cysts” was associated with a positive predictive value (PPV) of 100%, but a sensitivity (SEN) of 81.7% and 95.5% for the 15-29 and 30-39 age groups, respectively. For at-risk individuals aged 40 to 59 years, the presence of “at least two cysts in each kidney” was associated with a PPV of 100% and SEN of 90%. Finally, for at-risk individuals 60 years or older, the presence of “at least four cysts in each kidney” had a PPV and SEN of 100%.
Table 2.
Table 2(a). Ultrasound criteria for diagnosis of ADPKD*
| Age | PKD1 | PKD2 | Unknown ADPKD Gene Type |
|---|---|---|---|
| 15-30 | ≥3 cysts** | ||
| PPV=100% | PPV=100% | PPV=100% | |
| SEN=94.3% | SEN=69.5% | SEN=81.7% | |
| 30-39 | ≥3 cysts** | ||
| PPV=100% | PPV=100% | PPV=100% | |
| SEN=96.6% | SEN=94.9% | SEN=95.5% | |
| 40-59 | ≥2 cysts in each kidney | ||
| PPV=100% | PPV=100% | PPV=100% | |
| SEN=92.6% | SEN=88.8% | SEN=90% | |
| Table 2(b). Ultrasound criteria for exclusion of ADPKD* | |||
| Age | PKD1 | PKD2 | Unknown ADPKD Gene Type |
| 15-30 | ≥1 cyst | ||
| NPV=99.1% | NPV=83.5% | NPV=90.8% | |
| SPEC=97.6% | SPEC=96.6% | SPEC=97.1% | |
| 30-39 | ≥1 cyst | ||
| NPV=100% | NPV=96.8% | NPV=98.3% | |
| SPEC=96% | SPEC=93.8% | SPEC=94.8% | |
| 40-59 | ≥1 cyst | ||
| NPV=100% | NPV=100% | NPV=100% | |
| SPEC=93.9% | SPEC=93.7% | SPEC=93.9% | |
Derived from J Am Soc Nephrol 20: 205-212;
Unilateral or bilateral.
PPV (positive predictive value); SEN (sensitivity); NPV (negative predictive value); SPEC (specificity) – all values presented are mean estimates. For 95% confident intervals, please refer to the above paper.
Similarly, the investigators examined ultrasound diagnostic criteria for disease exclusion, an issue of importance for evaluating potential living-related kidney donors who are at-risk for ADPKD (Table 2b). Their study confirmed the limited ability of ultrasound to rule out disease in at-risk subjects less than 30 years of age with unknown gene type where the negative predictive value (NPV) associated with no renal cysts was 90.8% (in other words 10% of at-risk subjects with a negative ultrasound in this age group have ADPKD). The utility of ultrasound for disease exclusion improved with age such that the NPV associated with no renal cysts increased to 98.3% in at-risk subjects of unknown gene type aged 30 to 39 years. The NPVs varied between the two gene types, but were higher in subjects at-risk for PKD1 versus PKD2.
In those younger individuals where ultrasound might yield equivocal or indeterminate results, a negative CT or magnetic resonance imaging (MRI) may provide further assurance that they are unaffected but it must be noted that this approach has not been validated by any systematic study. It would be inappropriate to extrapolate ultrasound criteria to CT or MRI since more simple cysts may be detected by these modalities as a result of their enhanced detection sensitivity. Future studies are needed to validate the use of these more sensitive imaging modalities in the younger subjects at-risk of ADPKD.
DNA Testing in ADPKD
One take home message is that there will always be equivocal cases where a clinical diagnosis of ADPKD cannot be established with certainty on the basis of imaging. In these cases, DNA based diagnostic strategies can be considered depending on the clinical situation 49-51.
There are two methods for ADPKD DNA testing, which are both available on a fee-for-service basis: linkage analysis and direct mutation screening. In both cases blood is collected and sent to a clinical laboratory where DNA is extracted from peripheral white blood cells for analysis. The most up to date information about which laboratories are currently providing DNA diagnosis for ADPKD can be obtained from GeneTests (www.genetests.org), a valuable web-based resource that is funded by the National Institutes of Health. The web site provides a comprehensive list of academic and commercial facilities, in the United States and abroad, that offer testing on either a clinical or research basis. The two methods of DNA analysis will be reviewed and the limitations discussed.
In linkage analysis, the segregation of chromosomal markers flanking the disease gene is examined and compared within a family where the clinical status (ie, affected or unaffected) of each individual is known 52. By examining several markers, a haplotype (or a pattern of marker genotypes on the same chromosome) that segregates with the disease can be determined. This haplotype is then compared to that of the individual seeking testing and the probability of having the disease is assessed. The calculated genetic risk is weighted by the prior probability that his/her disease is linked to the locus being examined, for example 85% for PKD1 or 15% for PKD2. Linkage to both PKD1 and PKD2 must be assayed to fully understand the test results. The major advantage of this approach is that when a large family with multiple affected members (preferably 4 or more) is available, it is almost always possible to determine if the at-risk subject is an obligate carrier without the need to identify the pathogenic mutation (Figure 3).
Figure 3.
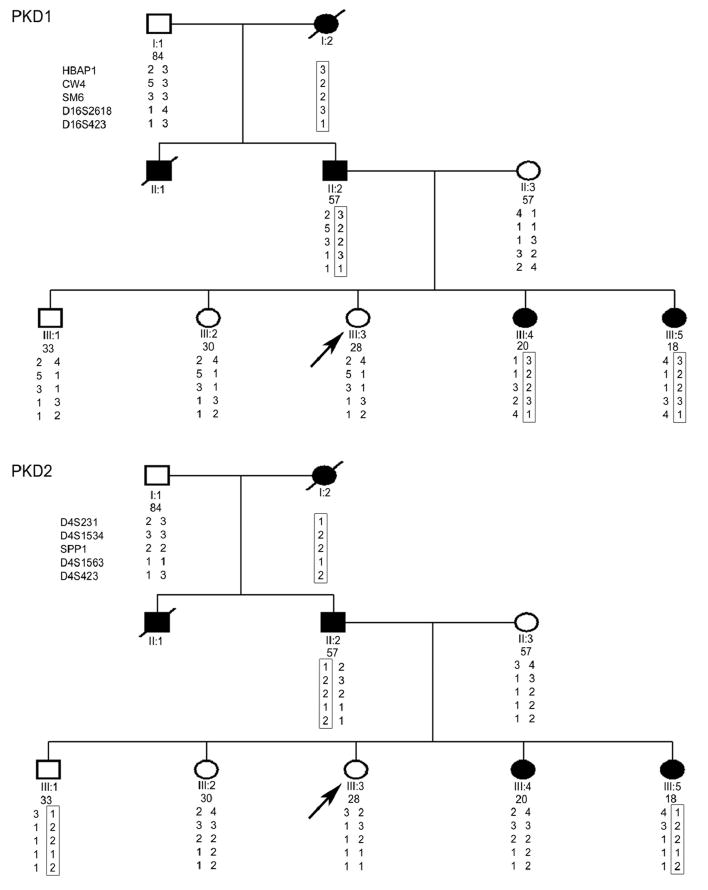
Molecular genetic evaluation of III:3, who had a negative renal ultrasound, as a potential living-related kidney donor. Direct sequencing of PKD1 and PKD2 in her affected father (II:2) identified two unclassifiable PKD1 variants (Q739R and del2763M) of uncertain clinical significance. Although III:3 did not carry these variants, the mutation analysis was considered as indeterminate. Haplotype analysis, on the other hand, showed that only the PKD1 haplotype 3-2-2-3-1 (upper panel), but no single PKD2 haplotype (lower panel), co-segregated with the affected family members indicating that this family was PKD1-linked. Since III:3 did not carry this putative PKD1 disease haplotype, she was considered as unaffected (adapted from C J Am Soc Nephrol 3: 146-52, 2008).
There are several limitations to linkage testing; the most important being that no information can be obtained from testing the proband alone. A rough rule of thumb is that a minimum of 3 affected family members in two generations are required, but additional affected and unaffected family members can strengthen the accuracy with which the disease related haplotype is ascertained. Linkage testing cannot be used if a family is small, if family members refuse to participate, or if the proband is suspected to have a de novo mutation. In addition a definitive clinical diagnosis must be established in those individuals who agree to participate. Practically, the clinician should review the imaging studies for each family member participating in the analysis to make sure that the clinical diagnosis is correct. As of this writing there is only one laboratory in the United States offering linkage testing for ADPKD. The cost (regardless of the number of samples sent) is ∼$2500 with results returning in about 3 weeks.
Direct mutation analysis is another option for molecular diagnosis in ADPKD. The commercially available test (in the US offered by Athena Diagnostics) involves direct DNA sequencing of the entire coding regions of both PKD1 and PKD2, including intron/exon boundaries 53, 54. The cost is dependent on the entity being billed and varies between $3650-$4880 with a minimum turn around time of about 6-8 weeks. Theoretically the advantage of direct mutation analysis is that one only needs a blood sample from a single patient. This is indeed the case if one identifies a mutation that is predicted to disrupt either PC-1 or PC-2. This class of mutation consists of those alterations such as: 1) deletions or insertions that shift the reading frame of the message 2) nucleotide substitutions that result in stop codons and therefore premature truncation of the protein or 3) nucleotide substitutions that directly alter a consensus splice site that would likely result in the creation of a new message. The test result form typically refers to these mutations as “previously reported disease-associated mutations” or “predicted disease-associated mutations”. The major limitation of direct DNA testing, however, is that definitive disease-associated mutations are found in only about 40-60% of cases. Likely disease-associated mutations may be detected in an additional 26-37% of patients 55, 56. This category is comprised largely of non-synonymous missense variants that typically result in a single amino acid alteration (referred to as unknown amino acid change in the results report form). Although the pathogenicity of some missense mutations has been established (ADPKD mutation database: http://pkdb.mayo.edu/), the disease-causing potential of other variants is uncertain. A variety of algorithms (such as Polyphen; http://genetics.bwh.harvard.edu/pph/) can be used to assess the likelihood that an amino acid substitution is deleterious to the protein function but this may not provide sufficient certainty for clinical decision making. Therefore, in the absence of a valid functional assay, the clinical utility of this category of mutations remains uncertain.
Finally up to 4% of the patients with ADPKD may have gross gene rearrangements that are missed by direct sequencing strategies 57. High throughput screening methods for this type of mutation using a multiplex ligation-dependent probe amplification (MLPA) assay have been developed but are not yet commercially available. In general, for gene-based mutation screening we recommend testing a definitively affected family member first to identify any potentially disease-causing variants to be tested in the at-risk subject of interest.
Why Establish a Diagnosis of ADPKD?
One might reasonably ask “Why establish a pre-symptomatic diagnosis of ADPKD?” At the present time there are no therapies proven to delay cyst progression and complications such as hypertension are treated as they arise irrespective of the underlying cause. Moreover, establishing a diagnosis may have negative implications such as restricting access to insurance. However, we believe that there are emerging indications for presymptomatic diagnosis of ADPKD in at risk individuals.
Over the past several years, much progress has been made in understanding the signaling pathways that are altered when polycystin signaling falls below a critical threshold. Moreover, the results from several laboratories studying conditional murine models of Pkd1 inactivation support the notion that there may be a significant lag time between the disruption of these pathways and frank cyst formation 58, 59. This early phase, prior to fatal cystic distortion of renal architecture, may provide an optimal time for intervention and would conceivably require accurate pre-symptomatic diagnosis. There are several trials currently underway that are examining the efficacy of therapies such as tolvaptan, octreotide and rapamycin in early to middle stages of disease and so preventive therapies for ADPKD may not be too far off 18.
Another relevant indication for early identification of ADPKD is clearing prospective kidney donors from affected families 49, 51. Many patients with ADPKD reach ESRD in mid-adulthood with few co-morbidities and therefore are good candidates for renal transplantation. Since deceased donor organs are in short supply, living related donation should be considered if disease could be excluded in the prospective donor. The agenda in this scenario is a dual one: to exclude disease in younger individuals without cysts but also to ensure that older at risk individuals are not disqualified from donating because of sporadic, age related renal cysts. Algorithms for screening donors from ADPKD families have been proposed. Since most transplant centers define donor anatomy via CT or MRI, this is often the first step. DNA testing is usually pursued in those with equivocal imaging studies such as those prospective donors with a few renal cysts below age-specific diagnostic cut-offs.
There are other plausible reasons why individuals from ADPKD families might want to establish their disease status early on. Extra renal complications such as intracranial aneurysms cluster in certain ADPKD families and the current recommendations are to screen individuals from these families via MR angiography 54, 60. In these families, it may be desirable to establish a diagnosis earlier rather than later in order to decide who might benefit from screening. Finally the knowledge of disease status might impact family planning decisions for some individuals.
A Diagnostic Algorithm for ADPKD
Figure 4 illustrates our approach in the evaluation of a patient suspected to have ADPKD. As indicated in this algorithm, obtaining a detailed family history is a key step in the diagnosis not only to confirm the autosomal dominant pattern of inheritance but also to aid in determining underlying gene type. The knowledge of gene type helps in the selection of appropriate ultrasound criteria especially for the purpose of disease exclusion. In general, the presence of “three unilateral or bilateral renal cysts” in at-risk subjects aged 15-39 years or “two cysts in each kidney” in at-risk subjects aged 40-59 years can be considered sufficient for the diagnosis of ADPKD regardless of their underlying gene type (PPV=100%) (Table 2a). For older at-risk subjects aged 60 years or older, the presence of “at least four cysts in each kidney” is sufficient for diagnosis of ADPKD regardless of the gene type. The sensitivity (ie, the proportion of the affected subjects with the positive test result) of these criteria, however, will differ between the different gene types. On the other hand, individuals with an affected relative who developed ESRD at 55 years or younger are at risk of PKD1 and therefore, PKD1-specific ultrasound criteria should be applied for earlier disease exclusion. In this case, the absence of any renal cyst by 30 years of age provides almost complete certainty for disease exclusion. By contrast, in the absence of such a family history indicative of PKD1, the ultrasound criteria for unknown genotype can be used and ADPKD cannot be excluded until 40 years of age.
Figure 4.
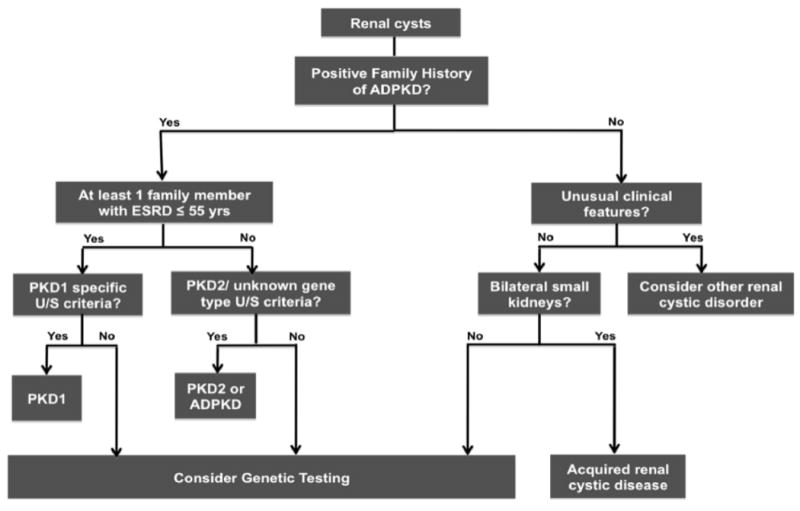
Diagnostic algorithm for the evaluation of an individual suspected to have ADPKD. See text for details.
When no family history of ADPKD is apparent, screening parents or review of their autopsy results if available may uncover occult disease, especially in small families with milder PKD2. Molecular genetic testing, however, is indicated in the evaluation of at-risk individuals with equivocal imaging results, younger at-risk individuals being evaluated for living kidney donation, and individuals with atypical or de novo renal cystic disease. In the not too distant future, the role of molecular genetic testing may expand if the ADPKD gene type influences choice of effective disease-modifying treatments that are currently being tested.
There are a number of other disorders discussed above that may mimic ADPKD (Table 1). In most cases, however, additional renal (eg, angiomyolipomas, renal cell carcinomas) and/or extra-renal (eg, facial and periungal fibromas, pancreatic cysts) findings may provide clues in identifying these syndromic disorders. Individuals with findings atypical for ADPKD should undergo further evaluation, including additional imaging, referral for medical genetics consultation and molecular testing. On the other hand, the finding of small kidneys bilaterally should alert to the possibility of acquired cystic disease.
Acknowledgments
This work was supported by R01DK70617 and R01GM073704 to T.W. and by CIHR MOP grant 77806 to Y.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- 2.Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. Am J Kidney Dis. 1983;2:630–639. doi: 10.1016/s0272-6386(83)80044-4. [DOI] [PubMed] [Google Scholar]
- 3.Davies F, Coles GA, Harper PS, Williams AJ, Evans C, Cochlin D. Polycystic kidney disease re-evaluated: a population-based study. Q J Med. 1991;79:477–485. [PubMed] [Google Scholar]
- 4.Peters DJ, Sandkuijl LA. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol. 1992;97:128–139. doi: 10.1159/000421651. [DOI] [PubMed] [Google Scholar]
- 5.Dobin A, Kimberling WJ, Pettinger W, Bailey-Wilson JE, Shugart YY, Gabow P. Segregation analysis of autosomal dominant polycystic kidney disease. Genet Epidemiol. 1993;10:189–200. doi: 10.1002/gepi.1370100305. [DOI] [PubMed] [Google Scholar]
- 6.Parfrey PS, Bear JC, Morgan J, et al. The diagnosis and prognosis of autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323:1085–1090. doi: 10.1056/NEJM199010183231601. [DOI] [PubMed] [Google Scholar]
- 7.Barua M, Cil O, Paterson AD, et al. Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol. 2009;20:1833–1838. doi: 10.1681/ASN.2009020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daoust MC, Reynolds DM, Bichet DG, Somlo S. Evidence for a third genetic locus for autosomal dominant polycystic kidney disease. Genomics. 1995;25:733–736. doi: 10.1016/0888-7543(95)80020-m. [DOI] [PubMed] [Google Scholar]
- 9.de Almeida S, de Almeida E, Peters D, et al. Autosomal dominant polycystic kidney disease: evidence for the existence of a third locus in a Portuguese family. Hum Genet. 1995;96:83–88. doi: 10.1007/BF00214191. [DOI] [PubMed] [Google Scholar]
- 10.Turco AE, Clementi M, Rossetti S, Tenconi R, Pignatti PF. An Italian family with autosomal dominant polycystic kidney disease unlinked to either the PKD1 or PKD2 gene. Am J Kidney Dis. 1996;28:759–761. doi: 10.1016/s0272-6386(96)90261-9. [DOI] [PubMed] [Google Scholar]
- 11.Pei Y, Paterson AD, Wang KR, et al. Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am J Hum Genet. 2001;68:355–363. doi: 10.1086/318188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paterson AD, Pei Y. PKD3-to be or not to be? Nephrol Dial Transplant. 1999;14:2965–2966. doi: 10.1093/ndt/14.12.2965. [DOI] [PubMed] [Google Scholar]
- 13.Paterson AD, Pei Y. Is there a third gene for autosomal dominant polycystic kidney disease? Kidney Int. 1998;54:1759–1761. doi: 10.1046/j.1523-1755.1998.00166.x. [DOI] [PubMed] [Google Scholar]
- 14.Watnick T, He N, Wang K, et al. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet. 2000;25:143–144. doi: 10.1038/75981. [DOI] [PubMed] [Google Scholar]
- 15.Pei Y, Watnick T, He N, et al. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1999;10:1524–1529. doi: 10.1681/ASN.V1071524. [DOI] [PubMed] [Google Scholar]
- 16.Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 17.Hughes J, Ward CJ, Peral B, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 18.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 20.Hateboer N, v Dijk MA, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353:103–107. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 21.Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17:3013–3019. doi: 10.1681/ASN.2006080835. [DOI] [PubMed] [Google Scholar]
- 22.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 23.Kaariainen H. Polycystic kidney disease in children: a genetic and epidemiological study of 82 Finnish patients. J Med Genet. 1987;24:474–481. [PMC free article] [PubMed] [Google Scholar]
- 24.Zerres K, Rudnik-Schoneborn S, Deget F. Childhood onset autosomal dominant polycystic kidney disease in sibs: clinical picture and recurrence risk. German Working Group on Paediatric Nephrology (Arbeitsgemeinschaft fur Padiatrische Nephrologie. J Med Genet. 1993;30:583–588. doi: 10.1136/jmg.30.7.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brook-Carter PT, Peral B, Ward CJ, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome. Nat Genet. 1994;8:328–332. doi: 10.1038/ng1294-328. [DOI] [PubMed] [Google Scholar]
- 26.Sampson JR, Maheshwar MM, Aspinwall R, et al. Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet. 1997;61:843–851. doi: 10.1086/514888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 28.Qian Q, Li A, King BF, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–171. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 29.Hoevenaren IA, Wester R, Schrier RW, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 2008;28:264–270. doi: 10.1111/j.1478-3231.2007.01595.x. [DOI] [PubMed] [Google Scholar]
- 30.Li A, Davila S, Furu L, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davila S, Furu L, Gharavi AG, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 32.Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–347. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- 33.Adeva M, El-Youssef M, Rossetti S, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD) Medicine (Baltimore) 2006;85:1–21. doi: 10.1097/01.md.0000200165.90373.9a. [DOI] [PubMed] [Google Scholar]
- 34.Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. quiz 55. [DOI] [PubMed] [Google Scholar]
- 35.Nicolau C, Torra R, Badenas C, et al. Sonographic pattern of recessive polycystic kidney disease in young adults. Differences from the dominant form. Nephrol Dial Transplant. 2000;15:1373–1378. doi: 10.1093/ndt/15.9.1373. [DOI] [PubMed] [Google Scholar]
- 36.Hart TC, Gorry MC, Hart PS, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39:882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bleyer AJ. Improving the recognition of hereditary interstitial kidney disease. J Am Soc Nephrol. 2009;20:11–13. doi: 10.1681/ASN.2007121330. [DOI] [PubMed] [Google Scholar]
- 38.Siroky BJ, Czyzyk-Krzeska MF, Bissler JJ. Renal involvement in tuberous sclerosis complex and von Hippel-Lindau disease: shared disease mechanisms? Nat Clin Pract Nephrol. 2009;5:143–156. doi: 10.1038/ncpneph1032. [DOI] [PubMed] [Google Scholar]
- 39.Gurrieri F, Franco B, Toriello H, Neri G. Oral-facial-digital syndromes: review and diagnostic guidelines. Am J Med Genet A. 2007;143A:3314–3323. doi: 10.1002/ajmg.a.32032. [DOI] [PubMed] [Google Scholar]
- 40.Woolf AS, Feather SA, Bingham C. Recent insights into kidney diseases associated with glomerular cysts. Pediatr Nephrol. 2002;17:229–235. doi: 10.1007/s00467-001-0819-5. [DOI] [PubMed] [Google Scholar]
- 41.Reed B, McFann K, Kimberling WJ, et al. Presence of de novo mutations in autosomal dominant polycystic kidney disease patients without family history. Am J Kidney Dis. 2008;52:1042–1050. doi: 10.1053/j.ajkd.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalgaard OZ. Bilateral polycystic disease of the kidneys; a follow-up of 284 patients and their families. Dan Med Bull. 1957;4:128–133. [PubMed] [Google Scholar]
- 43.Ravine D, Gibson RN, Donlan J, Sheffield LJ. An ultrasound renal cyst prevalence survey: specificity data for inherited renal cystic diseases. Am J Kidney Dis. 1993;22:803–807. doi: 10.1016/s0272-6386(12)70338-4. [DOI] [PubMed] [Google Scholar]
- 44.Tantravahi J, Steinman TI. Acquired cystic kidney disease. Semin Dial. 2000;13:330–334. doi: 10.1046/j.1525-139x.2000.00092.x. [DOI] [PubMed] [Google Scholar]
- 45.Carrim ZI, Murchison JT. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58:626–629. doi: 10.1016/s0009-9260(03)00165-x. [DOI] [PubMed] [Google Scholar]
- 46.Nascimento AB, Mitchell DG, Zhang XM, Kamishima T, Parker L, Holland GA. Rapid MR imaging detection of renal cysts: age-based standards. Radiology. 2001;221:628–632. doi: 10.1148/radiol.2213010178. [DOI] [PubMed] [Google Scholar]
- 47.Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824–827. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- 48.Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205–212. doi: 10.1681/ASN.2008050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang E, Samaniego-Picota M, McCune T, et al. DNA testing for live kidney donors at risk for autosomal dominant polycystic kidney disease. Transplantation. 2009;87:133–137. doi: 10.1097/TP.0b013e318191e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pei Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2006;1:1108–1114. doi: 10.2215/CJN.02190606. [DOI] [PubMed] [Google Scholar]
- 51.Zhao X, Paterson AD, Zahirieh A, He N, Wang K, Pei Y. Molecular diagnostics in autosomal dominant polycystic kidney disease: utility and limitations. Clin J Am Soc Nephrol. 2008;3:146–152. doi: 10.2215/CJN.03430807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burke W. Genetic testing. N Engl J Med. 2002;347:1867–1875. doi: 10.1056/NEJMoa012113. [DOI] [PubMed] [Google Scholar]
- 53.Phakdeekitcharoen B, Watnick TJ, Germino GG. Mutation analysis of the entire replicated portion of PKD1 using genomic DNA samples. J Am Soc Nephrol. 2001;12:955–963. doi: 10.1681/ASN.V125955. [DOI] [PubMed] [Google Scholar]
- 54.Watnick TJ, Piontek KB, Cordal TM, et al. An unusual pattern of mutation in the duplicated portion of PKD1 is revealed by use of a novel strategy for mutation detection. Hum Mol Genet. 1997;6:1473–1481. doi: 10.1093/hmg/6.9.1473. [DOI] [PubMed] [Google Scholar]
- 55.Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 56.Garcia-Gonzalez MA, Jones JG, Allen SK, et al. Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Mol Genet Metab. 2007;92:160–167. doi: 10.1016/j.ymgme.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Consugar MB, Wong WC, Lundquist PA, et al. Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int. 2008;74:1468–1479. doi: 10.1038/ki.2008.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13:1490–1495. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19:2351–2363. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pirson Y, Chauveau D, Torres V. Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2002;13:269–276. doi: 10.1681/ASN.V131269. [DOI] [PubMed] [Google Scholar]