

Abstract
During recent decades, bacteriophages have been at the cutting edge of new developments in molecular biology, biophysics, and, more recently, bionanotechnology. In particular filamentous viruses, for example bacteriophage M13, have a virion architecture that enables precision building of ordered and defect-free two and three-dimensional structures on a nanometre scale. This could not have been possible without detailed knowledge of coat protein structure and dynamics during the virus reproduction cycle. The results of the spectroscopic studies conducted in our group compellingly demonstrate a critical role of membrane embedment of the protein both during infectious entry of the virus into the host cell and during assembly of the new virion in the host membrane. The protein is effectively embedded in the membrane by a strong C-terminal interfacial anchor, which together with a simple tilt mechanism and a subtle structural adjustment of the extreme end of its N terminus provides favourable thermodynamical association of the protein in the lipid bilayer. This basic physicochemical rule cannot be violated and any new bionanotechnology that will emerge from bacteriophage M13 should take this into account.
Keywords: M13 bacteriophage, Major coat protein, Site-directed labelling, Membrane protein anchoring, Bionanotechnology
Introduction
Viruses, in particular bacterial viruses or bacteriophages, are the most abundant biological entities in the environment—estimates range form 1030 to 1032 in total—and play a key role in controlling biological systems (Kutter and Sulakvelidze 2005). The advance in molecular biology over the past 50 years has been built in a large part on the study of bacteriophages. Restriction endonucleases, which form the basis of molecular cloning, were developed following studies of phage infection. Many phage enzymes provide tools for the molecular biologist studying cell replication, transcription, translation, and transport pathways. The technique of phage display has provided powerful methodology for identification and optimisation of ligands for antibodies and other biomolecules. In environmental studies bacteriophages have been widely applied as tracers and indicators of pollution, and for monitoring and validating biological filters (Trevor and Richard 2000). They are also a continuing challenge to fermentation and, in particular, the dairy industry, where phage infection can lead to commercial disasters. The most recent application of bacteriophages in novel engineered materials puts them at the forefront of new nanotechnology devices.
From the perspective of nanotechnology, viruses can be regarded as efficient nanomachines, producing numerous copies of themselves. In fact, viral reproduction leads to a wealth of wonderful nanostructures. Examples of icosahedral (spherical) viruses are shown in Fig. 1. In looking at these nanoarchitectures, one could ask the question: what are the molecular forces and processes that account for the assembly of such structures? On the other hand, with the increasing expansion of the field of nanotechnology, one could ask: what can we learn from virus assembly in biology for creating new technologies on a nanolevel? Of course, both questions go hand-in-hand, because new advances in bionanotechnology always will require a deep understanding of individual molecular building blocks and their structures, assembly properties, and dynamic behaviour (Zhang 2003).
Fig. 1.
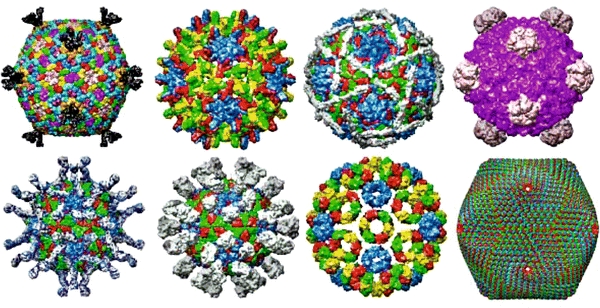
Various virus structures—a wealth of nanoshapes and nanoarchitectures for icosahedral (spherical) viruses. Courtesy: http://www.viperdb.scripps.edu/ (Carrillo-Tripp et al. 2009)
During recent decades, bacteriophages have also been at the cutting edge of new developments in biophysics. For example, filamentous bacteriophages served as model systems for the development and implementation of spectroscopic methods suitable for biological supramolecular assemblies. Coat proteins from filamentous phages are small (about 50 amino acid residues) and can be readily prepared in the laboratory. Furthermore, the coat proteins have two primary roles during the replication cycle—as membrane proteins and as principal structural elements of the phage particles. How can a small coat protein accomplish such widely different roles in the bacteriophage replication cycle? Which essential features of the protein enable it to do so? These questions will be the main focus of this review. By learning about protein structures in different environments, we will also be in a position to fully understand their most recent nanotechnological applications, which have revolutionized the science of nanobiology and bionanotechnology.
Filamentous bacteriophages
Filamentous phages comprise a family of viruses that have about ten genes. The relative simplicity of these viruses and the ease with which they can be genetically manipulated have made them extremely fruitful models to study macromolecular structure and interactions. All filamentous bacteriophage strains have a similar virion structure and life cycle. The filamentous phages best studied biochemically, genetically, and biophysically are M13 bacteriophage and its closest relative fd bacteriophage; both are F-specific filamentous phages that infect Escherichia coli bacteria (Lopez and Webster 1983, 1985; Makowski 1992, 1994; Model and Russel 1988; Russel 1991).
M13 bacteriophage causes chronic infections, and infected cells continue to grow and divide, although at a lower rate than normal. The phage is a long thread-shaped particle and has a diameter of 6.5 nm and is 900 nm long. The flexible filament contains a circular, single-stranded viral DNA genome, having 6,407 nucleotides, that is protected by a long cylindrical protein coat. This coat is made up of approximately 2,700 copies of the major coat protein (gp8; Fig. 2) capped by minor coat proteins (3–5 copies each) gp3 and gp6, or gp7 and gp9 at both ends (Webster and Lopez 1985). The major coat proteins form a tube around the viral DNA, in an overlapping helical array (Fig. 3a). They are oriented such that the N terminus is located at the outside of the coat and the C terminus interacts with the DNA at the inside of the coat. The hydrophobic domain of the major coat protein is located in the central part of the protein, and it interlocks the coat protein with its neighbouring coat proteins in the viral particle.
Fig. 2.
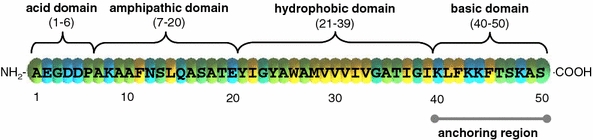
Primary structure of M13 major coat protein with classification of the important domains (Table 1). The colour coding is based on amino acid residue hydrophobicity scales (White and Wimley 1999) with the yellow colour corresponding to hydrophobic residues, green to neutral, and blue to charged residues. Anchoring of the protein at the membrane–water interface is provided by the C-terminal lysine residues and phenylalanines
Fig. 3.
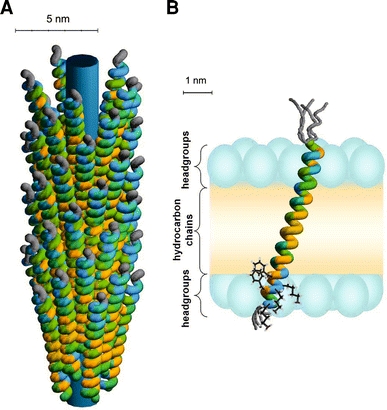
a Schematic illustration of the phage-bound model for the major coat protein of bacteriophage M13. The colour coding of the amino acid residues is based on a hydrophobicity scale (Fig. 2). Unstructured protein regions are indicated in grey. The inner cylinder indicates the viral DNA. b Structure and membrane embedding of M13 coat protein in fully hydrated vesicles of 18:1PC (and mixed phospholipid systems with C18 acyl chains), based upon recent site-directed labelling spectroscopy (Koehorst et al. 2004; Nazarov et al. 2007; Stopar et al. 2006b; Vos et al. 2005, 2007). The protein is effectively anchored with the C-terminal domain at the membrane–water interface by three “snorkelling” lysines (Lys40, Lys43, and Lys44) and two “anti-snorkelling” phenylalanines (Phe42 and Phe45). The size of the membrane regions is obtained from the literature, with the positions of the carbonyls serving as borders for the headgroup region (Ridder et al. 2002; White and Wimley 1999). The phospholipid headgroups are indicated with blue ellipsoids and the hydrocarbon chain region is coloured in yellow. The protein forms a mainly α-helical conformation tilted at 18° to the membrane normal. The first nine amino acid residues, which encompass the hydrophilic anchor (Table 1), are unstructured (as indicated by different possible gray structures). The membrane-bound protein structure does not differ much from the native α-helical structure of the protein in bacteriophage M13 in a. The protein survives the membrane-bound state by a simple tilt mechanism based on anchoring of its C-terminal domain at the membrane–water interface and a subtle structural adjustment at the extreme end of the N-terminal domain
Throughout the reproductive life cycle, the major coat protein is involved in various molecular processes that take place in different environments of the cell. During the infectious entry, the major coat protein is stripped from the phage particle and is deposited in the inner host membrane. Viral DNA enters the cell and is converted into a double-stranded replicative form by host enzymes. Progeny DNA is replicated by a rolling circle mechanism, and is assembled with the viral replication–assembly protein gp5 into an elongated intracellular nucleoprotein complex. The replication-assembly protein (covering and protecting the viral DNA inside the cell) is then replaced by coat proteins (covering and protecting the viral DNA outside the cell) in the cell membrane, where the virion is extruded through the membrane with the help of viral and host proteins. The new coat protein is synthesized as a procoat protein, a precursor of the major coat protein containing an additional amino acid leader sequence necessary for insertion into the cytoplasmic membrane. The procoat molecule is inserted into the membrane and subsequently the extra leader sequence is clipped off by a host cell leader peptidase. The resulting mature transmembrane coat protein is stored in the inner membrane prior to its use in the phage assembly process. After termination of phage assembly, the filamentous phage is released into the medium, and is ready to attack a new host cell. It is most remarkable that macromolecular assembly and disassembly do not cause the host membrane irreparable damage. The pivotal entity in this process is the major coat protein, which is a small 50-amino-acid-residue protein (Fig. 2) responsible for protein–protein, protein–lipid, and protein–DNA interactions during macromolecular assembly and disassembly (Russel and Model 2006; Spruijt et al. 1999).
Protein structure in filamentous phage and in a membrane-bound environment
The structure of the phage-bound coat protein has been known for many years, on the basis of results from X-ray fibre diffraction experiments (Fig. 3a; Glucksman et al. 1992; Marvin et al. 1994). This structure is an almost perfect α-helix, leaving 4–5 flexible unstructured amino acid residues in the N terminus that protrude from the phage coat into the aqueous phase. Clearly, the function of this negatively charged N-terminal protein domain is to keep the phage particle in solution. Any insert in the N-terminus in a genetically engineered phage should retain this function. The lysine-rich C terminus interacts non-specifically with the viral DNA phosphate groups by charge neutralization (Table 1; Marvin et al. 1994). This enables the phage particle to be elongated simply by inserting more DNA in the viral genome. Each coat protein subunit can be closely approximated by a single gently curved α-helix measuring about 1 by 7 nm. The diffraction data show that the α-helix axis is at a small angle to the virion axis. In a detailed molecular model it can be seen that this angle is very precisely constrained by the neighbouring subunits, because they form an overlapping interdigitated helical array with extensively interlocked side chains. The coat protein subunits have a continuous 19-residue apolar domain in the interior of the sequence (Table 1), and are held together in the virion by hydrophobic interactions between these domains. Mutagenesis studies suggest that small apolar residues are highly conserved in the apolar domain. Recently the phage-bound structure of the coat protein was further refined by solid-state NMR (Zeri et al. 2003) and cryo-electron microscopy (Wang et al. 2006) studies. Results from solid-state NMR confirmed earlier models from X-ray fibre diffraction studies, whereas the cryo-electron microscopy studies suggested heterogeneity of the structure when the coat protein is part of the phage. However, this conclusion is at odds with early X-ray work, and re-interpretation of the cryo-electron microscopy data led Straus et al. to conclude that this perceived heterogeneity is, in fact, a result of errors in the construction of the model (Straus et al. 2008).
Table 1.
Classification of important domains in M13 major coat protein
| Amino acid residue | Classification | Phage-bound state | Membrane-bound state |
|---|---|---|---|
| 1–6 | Acid | The negatively charged residues dictate bacteriophage solubility in aqueous solutiona | Hydrophilic anchor that contains negatively charged amino acid residues (Glu2, Asp4, and Asp5) that will extend into the aqueous phaseb |
| 7–20 | Amphipathic | Covers and shields the hydrophobic surface of underlying protein units (the coat proteins pack like roof tiles)a | Membrane–water interface domainb |
| 21–39 | Hydrophobic | These residues promote tight protein–protein interactions and thus bacteriophage stabilitya | Transmembrane domainb |
| 40–50 | Basic | The lysine residues are involved in DNA-binding by charge neutralization; the phenylalanines participate in intercalating protein–protein interactionsa | Membrane–water interface anchor by snorkellingc of the lysine residues to the phospholipid headgroup region together with anti-snorkelling of the phenylalanines with the hydrocarbon chain region of the membraneb |
The highly organized crystal-like environment of the major coat protein in the phage particle enables detailed structural and topological information to be obtained. On the other hand, determination of the membrane-bound protein structure turned out to be more challenging. This is mainly because of technical problems of conventional high-resolution structural techniques, for example high-resolution solution NMR and X-ray crystallography, which cannot handle the large size of a proteolipid complex and the lack of high-quality crystals of the protein in lipid bilayers, respectively. To overcome these problems, solid-state NMR spectroscopy was applied on several occasions. The major conclusion from solid-state NMR was a proposal for a two-helix protein structure in the lipid bilayer, with a transmembrane and an amphipathic helix perpendicular to each other (L-shape protein) (McDonnell et al. 1993; Opella et al. 2008; Shon et al. 1991). Although convincing, the model suffered from the fact that the coat protein needed to be reconstituted in oriented lipid bilayers that distort, primarily, the topology of the N terminus, forcing it to bend on the lipid bilayer. These protein–lipid systems do not leave enough space between the bilayers, which perturbs the topology of the N-terminal protein domain (Vos et al. 2005, 2009). Solution NMR spectroscopy for the protein reconstituted in lipid bilayers is not possible. There have, however, been a few attempts to study the coat protein solubilized in micellar detergent systems. This resulted in a model in which the transmembrane helix of the protein is flanked by an N-terminus revolving around a hinge region provided by amino acid residue 20 (Papavoine et al. 1998). Some of the possible structures allowed by modelling of the nuclear Overhauser enhancement (NOE) constraints conform to an L-shaped protein. There are, however, other more extended structures that deviate from such a structure. This model suggests a much more flexible protein structure, reflecting a more dynamical detergent system (Vos et al. 2005, 2009).
The major coat protein is a monotopic single-helix membrane-spanning protein that during the phage life cycle should be inserted into the membrane, have a stable thermodynamic association with the membrane, and then leave the membrane during the assembly process. There is probably no single secondary structure of the major coat protein that would be optimally suited to all the processes during the replication cycle. If one assumes that the transmembrane helix is aligned with the lipid bilayer normal, then an L-shaped protein conformation is probably the best topology for a membrane inserted protein, because of the optimal hydrophobic interactions. There are, however, strong experimental arguments against orientation of the transmembrane helix parallel to the lipid bilayer normal (Koehorst et al. 2004). Experimental data from both NMR (Glaubitz et al. 2000; Marassi and Opella 2003; Opella et al. 2008) and fluorescence spectroscopy (Koehorst et al. 2004; Nazarov et al. 2006; Vos et al. 2005, 2007) suggest a tilted orientation of the protein. Taking tilt into consideration, one should probably abandon the notion of a perfect L-shaped protein in a lipid bilayer. Because determination of protein topology in lipid bilayers is especially important for virus assembly and for the capacity of the protein to serve as a vehicle for peptide display, we have developed new biophysical tools to study the protein structure, topology, and dynamics in lipid bilayers.
Site-directed labelling of M13 coat protein
Among the spectroscopic methods, site-directed techniques have great potential as alternative tools for determination of the structure of membrane proteins. The site-directed labelling (SDL) techniques are based on site-directed mutagenesis, which include the substitution of an amino acid residue by a cysteine, in combination with site-specific labelling. The structure of the membrane-bound protein can be studied by using site-directed spin labelling (SDSL) in conjunction with ESR spectroscopy or site-directed fluorescence labelling (SDFL), together with fluorescent spectroscopy and Förster resonance energy transfer (FRET). The major coat protein of bacteriophage M13 is ideally suited for SDL technology because it has no cysteine residue in its primary sequence. The usefulness of the SDL technology was tested by producing several bacteriophage particles containing single-cysteine coat protein mutants (Spruijt et al. 1996). This approach was limited, however, because of the viability requisite of the phage mutants, and only a few amino acid positions could be replaced by a cysteine and subsequently labelled with a spin label or fluorescent probe. Nevertheless, these studies provided insight in the location and embedment of the hydrophobic domain of the coat protein when inserted in phospholipid bilayers and detergent systems (Bashtovyy et al. 2001; Stopar et al. 1996, 1997a, b, 1998, 2002, 2003).
To enhance the flexibility of the cysteine mutagenesis the gene of the M13 major coat protein was cloned into an expression vector enabling enhanced expression of the coat protein without any limitations because of phage viability. Using the QuikChange site-directed mutagenesis procedure (Stratagene, La Jolla, CA, USA) unique cysteine residues at different positions along the primary sequence of the major coat protein were introduced (Spruijt et al. 2004). Such a mutagenesis approach is basically unlimited and at the moment 40 out of the 50 amino acid residues of the major coat protein have been substituted by cysteines (Koehorst et al. 2004; Nazarov et al. 2007; Spruijt et al. 2004; Stopar et al. 2006b; Vos et al. 2005, 2007). Replacement of essential amino acid residues, principally aromatic and charged amino acid residues, was avoided, as this could give rise to anomalous results with regard to the behaviour of the protein in membranes. In general, replacement of Ala, Gly, Ser, Met, and Thr for Cys were non-problematic. In fact, the best strategy for cysteine mutation might be a straightforward high-throughput approach in which, in the course of the protein mutant purifications and spectroscopic experiments, and subsequent data analysis and molecular modelling, samples that cannot be handled well or show peculiar effects can be removed from the data series. In addition, several double mutants were prepared, and elongations of the transmembrane protein domain (Meijer et al. 2001; Spruijt et al. 2000), as reviewed by Stopar et al. (2006a) and Vos et al. (2009).
Using SDFL studies employing FRET between fluorescence-labelled cysteines strategically placed along a protein primary sequence and tryptophan it was possible to make a molecular ruler for measuring distances between the amphipathic and transmembrane helix. Combining FRET fluorescence data with molecular modelling clearly demonstrated an extended or L-shaped structure of the protein when embedded in the lipid bilayer. The amino acid residues 10–46 form a continuous α-helix in 18:1PC lipid bilayers leaving a few unstructured residues in the N and C-terminal domain (Fig. 3b) (Koehorst et al. 2004; Nazarov et al. 2007; Stopar et al. 2006b; Vos et al. 2005, 2007). Interestingly, the membrane-bound protein structure as obtained from SDL is not dramatically different from the phage-bound structure. Although, on average, the membrane-bound coat protein is well described by a continuous α-helix, a small kink around residue 20 was identified in the helical protein model (Nazarov et al. 2007). This finding denotes a smooth protein bend. In addition, small deformations of the α-helical structure from residues 38–50 were found (Vos et al. 2007). These helical deformations could enable efficient incorporation into the phage particle on detaching these residues from the C-terminal membrane–water interface (Vos et al. 2007).
The L-shaped structure obtained from SDFL deviates from the L-shape obtained from solid-state NMR structural analysis of lipid bilayers (Marassi and Opella 2003; Opella et al. 2008) and from solution NMR structural analysis (Almeida and Opella 1997; Papavoine et al. 1998). The differences are most probably related to different sample preparation. For the purpose of the solid-state NMR experiments lipid vesicles were oriented and, in addition, a much lower L/P ratio (i.e., higher protein concentration) was used. To utilize solution NMR, the protein must be incorporated into detergent micelles that do not provide a characteristic bilayer environment. Micelles have a curved surface, forcing the N-terminal domain of the protein to bend back on this surface, thus providing a variety of protein shapes, including L and U-shapes and extended structures (Vos et al. 2005, 2009). Obviously the structure of the protein is strongly affected by the environment surrounding it, e.g., micelles, vesicles, or oriented membranes (Vos et al. 2005). As a result, extreme caution is necessary because external molecular forces on the protein could disturb its structure (Vos et al. 2009). This consideration is most important for the N-terminal domain of the protein that protrudes from the membrane (Stopar et al. 2006a). The major coat protein seems to be internally flexible and responsive to environmental constraints, a property that explains the different structural models that emerged from solid-state and solution NMR spectroscopy, and a “stress-free lipid bilayer” environment as is used in SDFL. In conclusion, the newly developed technique of SDL enabled the study of the structure of M13 coat protein in a “stress free” membrane environment and the structure obtained by FRET does not resemble the solid-state and solution NMR structures, but is more in line with the X-ray model of the phage-bound protein (Vos et al. 2009).
Membrane embedding by anchoring and tilt
Although a tilt of membrane domains is allowed in membrane proteins and peptides (Lomize et al. 2006; Ulmschneider et al. 2006; Van der Wel et al. 2002), it is much more common in polytopic membrane proteins, where a single helix is stabilised by tertiary contact with neighbouring protein segments. In monotopic membrane proteins or peptides with a pronounced hydrophobic transmembrane helix, for example M13 major coat protein, a tilt is possible only if strong additional anchoring elements are provided at the N and C-terminal ends of the helix. Strong anchoring of the C-terminal domain of the major coat protein at the membrane–water interface is provided by snorkelling of the lysine residues in the phospholipid headgroup region (Fig. 3b). Together with anti-snorkelling of the phenylalanines in the hydrocarbon chain region of the membrane (Fig. 3b), lysine residues provide a strong anchoring system that allows only a small vertical motion of the protein in the lipid bilayer (Table 1). Another important factor in membrane embedment and protein tilt is the length of the hydrophobic helical segment of the protein relative to the hydrophobic thickness of the membrane. Both of these properties have been varied experimentally in studies of the M13 coat protein (Koehorst et al. 2004; Meijer et al. 2001; Spruijt et al. 2000, 2004; Stopar et al. 2006b; Vos et al. 2007). By changing the membrane thickness the length of the helical transmembrane domain is altered, as a variable number of amino acid residues of the N-terminal domain can take part into the transmembrane helix. For instance, in thick phospholipid bilayers (22:1PC) the unstructured N-terminal domain is seven amino acid residues long whereas in thin membranes (14:1PC) the length of the unstructured N-terminus increases to 14 residues (Stopar et al. 2006b; Vos et al. 2007).
A crucial element enabling the protein to stably associate with a membrane is its ability to tilt in the lipid bilayer by concerted action of all the anchoring elements. The tilt of the protein relative to the membrane normal depends on the thickness of the membrane, because the hydrophobic domain of the protein tends to be in optimal contact with the hydrophobic core of the membrane. For example in thick phospholipid bilayers (20:1PC) the tilt angle is 19° and in thin bilayers (14:1PC) it increases to 33° (Koehorst et al. 2004). Thus, a tilt of the protein provides an additional mechanism to regulate its membrane embedment and to adjust to hydrophobic mismatch situations (Killian 1998). The protein does not undergo major structural rearrangements in response to hydrophobic mismatch. Aggregation at high protein concentrations (Fernandes et al. 2003), minor conformational adaptations of the N-terminus, and adjustment of the tilt angle are the main responses of the coat protein to conditions of hydrophobic mismatch (Vos et al. 2007).
Implications for bionanotechnology
Because of the wealth of information known about M13 coat protein and the coat proteins of related phages, the membrane-bound state of the protein has served as a model–reference system in many spectroscopic studies. For example, new solid-state NMR schemes, for example two-dimensional PISEMA (polarization inversion spin exchange at the magic angle) (Marassi and Opella 2003; Opella et al. 2008), were developed with the help of the major coat protein. Also new approaches in the analysis of SDSL data, as initiated by Štrancar et al. (Kavalenka et al. 2005; Stopar et al. 2005, 2006b; Štrancar et al. 2009), were based on data obtained from membrane-embedded M13 coat protein. The same applies to use of the BADAN (6-bromoacetyl-2-dimethylaminonaphthalene) fluorescent label in steady-state (Koehorst et al. 2008) and time-resolved fluorescence spectroscopy (Koehorst et al. 2009). This illustrates that not only the structure of the membrane-embedded M13 coat protein has been resolved, but it has also given a strong impetus for developing new tools in biophysics and spectroscopy.
More importantly still, the spectroscopic information can be put to good use in peptide display technology serving numerous applications (Kehoe and Kay 2005; Russel and Model 2006; Sidhu 2001; Smith and Petrenko 1997). Phage display is achieved by inserting a DNA sequence into a viral gene encoding a phage coat protein. The resulting fusion protein will display the protein product of the inserted DNA sequence on the surface of the phage particle, which also contains the encoding DNA. Phage displayed library members with particular functions can be isolated with simple selections and polypeptide sequences can be decoded from the encapsulated DNA. Depending on the application, fusion proteins of the major coat protein and the phage minor coat proteins can be made (Kehoe and Kay 2005; Russel and Model 2006). The success of the technology depends on the display efficiency of polypeptides on the phage surface.
Combinatorial libraries of polypeptides that can be fused to M13 coat proteins provide a valuable means of screening for binding candidates against different targets (Kehoe and Kay 2005). Phages are prospective probes in a new generation of sensors for food-safety control and environmental real-time monitoring. As elements of detectors for field use, they are superior to polyclonal and monoclonal antibodies, because they are inexpensive, highly specific, selective and strong binders, and resistant to unfavourable environmental conditions (Petrenko 2008). Recently, targets have been extended beyond biological molecules to a wide variety of inorganic molecules, in efforts to discover new biological systems capable of organizing and growing new materials. In addition to serving as the vehicle for displaying these ligands, the unique structure of M13 bacteriophage itself has been exploited as a biological template for nanotechnology, such as in the directed synthesis of semiconducting/magnetic nanowires and lithium ion battery electrodes (Nam et al. 2004, 2006). For instance, filamentous viruses enable the organization of various nanomaterials into periodically ordered hierarchical structures, such as viral rings and wires, that have electronic, optical and biotechnological applications (Merzlyak and Lee 2006). In materials science, this approach is used to create novel peptides with the capacity to bind to selected technical materials (Fischlechner and Donath 2007). In particular, M13 bacteriophage with coat proteins selected to bind to semiconducting and magnetic materials have been used as templates to grow and organize nanowires (Mao et al. 2004). In this respect the filamentous M13 bacteriophage is used as a scaffold for the synthesis of single-crystal ZnS and CdS, and chemically ordered CoPt and FePt nanowires. Nucleating peptides are incorporated into the M13 coat protein and thereby provide a template for the directed preparation of semiconducting and magnetic materials. Another potential application of a genetically engineered phage might be as a template material for tissue engineering and regenerative medicine. Such templates imitate the native extracellular matrix, which is composed of a fibrous protein mesh and provides cells with a physical support (Merzlyak and Lee 2006). The applications of bacteriophage M13 in biomolecular engineering are summarized in Table 2.
Table 2.
Biomolecular engineering and nanoapplications using M13 bacteriophage
| Topic | Description |
|---|---|
| Biorecognition | Phage-derived materials as biorecognition interface and for production of bioselective nanomateriala |
| Memory devices | Charge trapping in nanoparticles for data storageb |
| Nanoelectronics | Genetic engineering of self-assembling bacteriophages so they can be used to produce optical, conducting, semi-conducting, and magnetic materialsc, d, e, f, g |
| Phage display | Landscape phage and phage display technologyh, i, j |
| Self-assembly | Utilizing molecular self-assembly as an approach for fabricating supramolecular architectures (nanostructures)c, k, l |
| Tissue engineering | Building blocks in tissue engineering scaffoldsk, m |
aMerzlyak and Lee ( 2006); bTseng et al. (2006); cZhao and Zhang (2004); dSanford and Kumar (2005); eNam et al. (2006); fNam et al. (2004); gMao et al. (2004); hFischlechner and Donath (2007); iPetrenko (2008); jMerzlyak and Lee (2006); kZhang (2003); lWhitesides and Grzybowski (2002); mSanford and Kumar (2005)
All this and many more applications and patents that will be issued in the future are a direct consequence of the M13 virion architecture that enables precision building of ordered and defect-free two and three-dimensional structures on the nanometre scale. This is essential for the construction of next-generation optical, electronic, and magnetic materials and devices. These studies demonstrate that the major protein coat of M13 bacteriophage is extremely malleable, and can be used to engineer phage particles of desired functionality, assuming no fundamental biophysical principle is violated, in particular the ability of the protein to form a stable symmetrical coat and the ability of the protein to be inserted, anchored at a correct tilt angle, and assembled in the new virus particle. This technology is self-evolving in the sense that phage display can produce new proteins that in turn might improve the technology itself. With these new tools, imagination seems to be the only limit in design of new nanoarchitecture gadgets.
Conclusions
M13 major coat protein (and the major coat proteins from related bacteriophages) are effectively embedded in the membrane by a strong C-terminal interfacial anchor which together with a simple tilt mechanism and a subtle structural adjustment at the extreme end of its N terminus provides favourable thermodynamic association of the protein in the lipid bilayer.
Phage engineering and phage display products offer novel opportunities in various disciplines of bionanotechnology, for example molecular medicine, material science, and electric engineering. The directed evolution selection process enables an unprecedented level of novel protein material to be used as building blocks for nanoarchitecture in hybrid biophysical and chemical applications.
Outlook
Membrane-bound phage assembly is a complex process and currently not all molecular details are well resolved. Phage extrusion is based on an ATP-driven motor that adds coat proteins to the viral DNA in a steady and continuous manner to generate viral particles. One part of this system that has been elucidated in recent years is related to the role of the major coat protein in this process. Our recent findings indicate that the major coat proteins, being the main protein building blocks for the phage, do not significantly change shape during the membrane-bound assembly, thereby providing an efficient mechanism with low-energy consumption. Clearly, any nanotechnology that will emerge from bacteriophage M13 should take into account these basic physicochemical rules (Table 1).
At this moment, many aspects of the biogeneration of nanostructures are still unresolved, but it can be expected that in the coming decades new advances in biophysical techniques will increase our molecular insights. If we realize that we just are at the beginning of the new era of bionanotechnology, the future is bright for virus-based nanomachines.
Acknowledgments
This work was supported by contract no. QLG-CT-2000-01801 of the European Commission (MIVase—New Therapeutic Approaches to Osteoporosis: targeting the osteoclast V-ATPase).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- 14:1PC
1,2-Dimyristoleoyl-sn-glycero-3-phosphocholine
- 18:1PC
1,2-Dioleoyl-sn-glycero-3-phosphocholine
- 20:1PC
1,2-Dieicosenoyl-sn-glycero-3-phosphocholine
- 22:1PC
1,2-Dieurocoyl-sn-glycero-3-phosphocholine
- ESR
Electron spin resonance (or electron paramagnetic resonance)
- FRET
Förster (or fluorescence) resonance energy transfer
- gp8
Gene 8 product; the protein coded by viral gene number 8 (gp3, gp5, gp6, gp7, gp9 are defined in the same way)
- L/P
Lipid to protein molar ratio
- NMR
Nuclear magnetic resonance
- SDFL
Site-directed fluorescence labelling
- SDL
Site-directed labelling
- SDSL
Site-directed spin labelling
Footnotes
The more you see: spectroscopy in molecular biophysics.
References
- Almeida FCL, Opella SJ. Fd coat protein structure in membrane environments: structural dynamics of the loop between the hydrophobic trans-membrane helix and the amphipathic in-plane helix. J Mol Biol. 1997;270:481–495. doi: 10.1006/jmbi.1997.1114. [DOI] [PubMed] [Google Scholar]
- Bashtovyy D, Marsh D, Hemminga MA, Páli T. Constrained modeling of spin-labeled major coat protein mutants from M13 bacteriophage in a phospholipid bilayer. Protein Sci. 2001;5:979–987. doi: 10.1110/ps.43801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Tripp M, Shepherd CM, Borelli IA, Venkataraman S, Lander G, Natarajan P, Johnson JE, Brooks CL, III, Reddy VS. VIPERdb2: an enhanced and web API enabled relational database for structural virology. Nucl Acids Res. 2009;37:D436–D442. doi: 10.1093/nar/gkn840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes F, Loura LMS, Prieto M, Koehorst RBM, Spruijt RB, Hemminga MA. Dependence of M13 major coat protein oligomerization and lateral segregation on bilayer composition. Biophys J. 2003;85:2430–2441. doi: 10.1016/S0006-3495(03)74666-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischlechner M, Donath E. Viruses as building blocks for materials and devices. Angew Chem Int Edit. 2007;46:3184–3193. doi: 10.1002/anie.200603445. [DOI] [PubMed] [Google Scholar]
- Glaubitz C, Grobner G, Watts A. Structural and orientational information of the membrane embedded M13 coat protein by 13C-MAS NMR spectroscopy. Biochim Biophys Acta. 2000;1463:151–161. doi: 10.1016/S0005-2736(99)00195-9. [DOI] [PubMed] [Google Scholar]
- Glucksman MJ, Bhattacharjee S, Makowski L. Three-dimensional structure of a cloning vector: X-ray diffraction studies of filamentous bacteriophage M13 at 7 Å resolution. J Mol Biol. 1992;226:455–470. doi: 10.1016/0022-2836(92)90960-R. [DOI] [PubMed] [Google Scholar]
- Kavalenka AA, Filipič B, Hemminga MA, Štrancar J. Speeding up a genetic algorithm for EPR-based spin label characterization of biosystem complexity. J Chem Inf Model. 2005;45:1628–1635. doi: 10.1021/ci0501589. [DOI] [PubMed] [Google Scholar]
- Kehoe JW, Kay BK. Filamentous phage display in the new millennium. Chem Rev. 2005;105:4056–4072. doi: 10.1021/cr000261r. [DOI] [PubMed] [Google Scholar]
- Killian JA. Hydrophobic mismatch between proteins and lipids in membranes. Biochim Biophys Acta. 1998;1376:401–415. doi: 10.1016/s0304-4157(98)00017-3. [DOI] [PubMed] [Google Scholar]
- Koehorst RBM, Spruijt RB, Vergeldt FJ, Hemminga MA. Lipid bilayer topology of the transmembrane α-helix of M13 major coat protein and bilayer polarity profile by site-directed fluorescence spectroscopy. Biophys J. 2004;87:1445–1455. doi: 10.1529/biophysj.104.043208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehorst RBM, Spruijt RB, Hemminga MA. Site-directed fluorescence labeling of a membrane protein with BADAN: probing protein topology and local environment. Biophys J. 2008;94:3945–3955. doi: 10.1529/biophysj.107.125807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehorst RBM, Laptenok S, van Oort B, van Hoek A, Spruijt RB, van Stokkum IHM, van Amerongen H, Hemminga MA (2009) Profiling of dynamics in protein-lipid-water systems: A time-resolved fluorescence study of a model membrane protein, site-directed labelled with BADAN. Eur Biophys J (submitted) [DOI] [PMC free article] [PubMed]
- Kutter E, Sulakvelidze A. Bacteriophages: biology and applications. Boca Raton: CRC Press; 2005. [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Lopez J, Webster RE. Morphogenesis of filamentous bacteriophage f1: orientation of extrusion and production of polyphage. Virology. 1983;127:177–193. doi: 10.1016/0042-6822(83)90382-3. [DOI] [PubMed] [Google Scholar]
- Lopez J, Webster RE. Assembly site of bacteriophage f1 corresponds to adhesion zones between the inner and outer membranes of the host cell. J Bacteriol. 1985;163:1270–1274. doi: 10.1128/jb.163.3.1270-1274.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowski L. Terminating a macromolecular helix: structural model for the minor proteins of bacteriophage M13. J Mol Biol. 1992;228:885–892. doi: 10.1016/0022-2836(92)90872-H. [DOI] [PubMed] [Google Scholar]
- Makowski L. Phage display: structure, assembly and engineering of filamentous bacteriophage M13. Curr Opin Struct Biol. 1994;4:225–230. doi: 10.1016/S0959-440X(94)90312-3. [DOI] [Google Scholar]
- Mao C, Solis DJ, Reiss BD, Kottmann ST, Sweeney RY, Hayhurst A, Georgiou G, Iverson B, Belcher AM. Virus-based toolkit for the directed synthesis of magnetic and semiconducting nanowires. Science. 2004;303:213–217. doi: 10.1126/science.1092740. [DOI] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003;12:403–411. doi: 10.1110/ps.0211503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin DA. Filamentous phage structure, infection and assembly. Cur Opin Str Biol. 1998;8:150–158. doi: 10.1016/S0959-440X(98)80032-8. [DOI] [PubMed] [Google Scholar]
- Marvin DA, Hale RD, Nave C, Citterich MH. Molecular models and structural comparisons of native and mutant class-I filamentous bacteriophages Ff (fd, f1, M13), If1 and IKe. J Mol Biol. 1994;235:260–286. doi: 10.1016/S0022-2836(05)80032-4. [DOI] [PubMed] [Google Scholar]
- McDonnell PA, Shon K, Kim Y, Opella SJ. fd Coat protein structure in membrane environments. J Mol Biol. 1993;233:447–463. doi: 10.1006/jmbi.1993.1523. [DOI] [PubMed] [Google Scholar]
- Meijer AB, Spruijt RB, Wolfs CJAM, Hemminga MA. Membrane–anchoring interactions of M13 major coat protein. Biochemistry. 2001;40:8815–8820. doi: 10.1021/bi002956s. [DOI] [PubMed] [Google Scholar]
- Merzlyak A, Lee S-W. Phage as templates for hybrid materials and mediators for nanomaterial synthesis. Curr Opin Chem Biol. 2006;10:246–252. doi: 10.1016/j.cbpa.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Model P, Russel M. Filamentous bacteriophage. In: Calendar R, editor. The viruses: the bacteriophages. New York: Plenum Press; 1988. pp. 375–456. [Google Scholar]
- Nam KT, Peelle BR, Lee SW, Belcher AM. Genetically driven assembly of nanorings based on the M13 virus. Nano Lett. 2004;4:23–27. doi: 10.1021/nl0347536. [DOI] [Google Scholar]
- Nam KT, Kim D-W, Yoo PJ, Chiang C-Y, Meethong N, Hammond PT, Chiang Y-M, Belcher AM. Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes. Science. 2006;312:885–888. doi: 10.1126/science.1122716. [DOI] [PubMed] [Google Scholar]
- Nazarov PV, Koehorst RBM, Vos WL, Apanasovich VV, Hemminga MA. FRET study of membrane proteins: simulation-based fitting for analysis of membrane protein embedment and association. Biophys J. 2006;91:454–466. doi: 10.1529/biophysj.106.082867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarov PV, Koehorst RBM, Vos WL, Apanasovich VV, Hemminga MA. FRET study of membrane proteins: determination of the tilt and orientation of the N-terminal domain of M13 major coat protein. Biophys J. 2007;92:1296–1305. doi: 10.1529/biophysj.106.095026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella SJ, Zeri AC, Park SH. Structure, dynamics, and assembly of filamentous bacteriophages by nuclear magnetic resonance spectroscopy. Annu Rev Phys Chem. 2008;59:635–657. doi: 10.1146/annurev.physchem.58.032806.104640. [DOI] [PubMed] [Google Scholar]
- Papavoine CHM, Christiaans BEC, Folmer RHA, Konings RNH, Hilbers CW. Solution structure of the M13 major coat protein in detergent micelles: a basis for a model of phage assembly involving specific residues. J Mol Biol. 1998;282:401–419. doi: 10.1006/jmbi.1998.1860. [DOI] [PubMed] [Google Scholar]
- Petrenko VA. Landscape phage as a molecular recognition interface for detection devices. Microelectron J. 2008;39:202–207. doi: 10.1016/j.mejo.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder ANJA, van de Hoef W, Stam J, Kuhn A, de Kruijff B, Killian JA. Importance of hydrophobic matching for spontaneous insertion of a single-spanning membrane protein. Biochemistry. 2002;41:4946–4952. doi: 10.1021/bi0158674. [DOI] [PubMed] [Google Scholar]
- Russel M. Filamentous phage assembly. Mol Microbiol. 1991;5:1607–1613. doi: 10.1111/j.1365-2958.1991.tb01907.x. [DOI] [PubMed] [Google Scholar]
- Russel M, Model P. Filamentous phage. In: Calendar R, editor. The bacteriophages. 2. New York: Oxford University Press; 2006. pp. 146–160. [Google Scholar]
- Sanford K, Kumar M. New proteins in a materials world. Curr Opin Biotechnol. 2005;16:416–421. doi: 10.1016/j.copbio.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Shon K-J, Kim Y, Colnago LA, Opella SJ. NMR studies of the structure and dynamics of membrane-bound bacteriophage Pf1 coat protein. Science. 1991;252:1303–1305. doi: 10.1126/science.1925542. [DOI] [PubMed] [Google Scholar]
- Sidhu SS. Engineering M13 for phage display. Biomol Eng. 2001;18:57–63. doi: 10.1016/S1389-0344(01)00087-9. [DOI] [PubMed] [Google Scholar]
- Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- Spruijt RB, Wolfs CJAM, Verver JWG, Hemminga MA. Accessibility and environment probing using cysteine residues introduced along the putative transmembrane domain of the major coat protein of bacteriophage M13. Biochemistry. 1996;35:10383–10391. doi: 10.1021/bi960410t. [DOI] [PubMed] [Google Scholar]
- Spruijt RB, Wolfs CJAM, Hemminga MA. M13 phage. In: Creighton T, editor. The encyclopedia of molecular biology. New York: Wiley; 1999. pp. 1425–1429. [Google Scholar]
- Spruijt RB, Meijer AB, Wolfs CJAM, Hemminga MA. Localization and rearrangement modulation of the N-terminal arm of the membrane-bound major coat protein of bacteriophage M13. Biochim Biophys Acta. 2000;1509:311–323. doi: 10.1016/S0005-2736(00)00314-X. [DOI] [PubMed] [Google Scholar]
- Spruijt RB, Wolfs CJAM, Hemminga MA. Membrane assembly of M13 major coat protein: evidence for structural adaptation in the hinge region and a tilted transmembrane domain. Biochemistry. 2004;43:13972–13980. doi: 10.1021/bi048437x. [DOI] [PubMed] [Google Scholar]
- Stopar D, Spruijt RB, Wolfs CJAM, Hemminga MA. Local dynamics of the M13 major coat protein in different membrane-mimicking systems. Biochemistry. 1996;35:15467–15473. doi: 10.1021/bi961770j. [DOI] [PubMed] [Google Scholar]
- Stopar D, Jansen KAJ, Páli T, Marsh D, Hemminga MA. Membrane location of spin-labeled M13 major coat protein mutants determined by paramagnetic relaxation agents. Biochemistry. 1997;36:8261–8268. doi: 10.1021/bi970139v. [DOI] [PubMed] [Google Scholar]
- Stopar D, Spruijt RB, Wolfs CJAM, Hemminga MA. In situ aggregational state of M13 bacteriophage major coat protein in sodium cholate and lipid bilayers. Biochemistry. 1997;36:12268–12275. doi: 10.1021/bi970747a. [DOI] [PubMed] [Google Scholar]
- Stopar D, Spruijt RB, Wolfs CJAM, Hemminga MA. Mimicking initial interactions of bacteriophage M13 coat protein disassembly in model membrane systems. Biochemistry. 1998;37:10181–10187. doi: 10.1021/bi9718144. [DOI] [PubMed] [Google Scholar]
- Stopar D, Spruijt RB, Wolfs CJAM, Hemminga MA. Structural characterization of bacteriophage M13 solubilization by amphiphiles. Biochim Biophys Acta. 2002;1594:54–63. doi: 10.1016/s0167-4838(01)00281-3. [DOI] [PubMed] [Google Scholar]
- Stopar D, Spruijt RB, Wolfs CJAM, Hemminga MA. Protein–lipid interactions of bacteriophage M13 major coat protein. Biochim Biophys Acta. 2003;1611:5–15. doi: 10.1016/S0005-2736(03)00047-6. [DOI] [PubMed] [Google Scholar]
- Stopar D, Štrancar J, Spruijt RB, Hemminga MA. Exploring the local conformational space of a membrane protein by site-directed spin labeling. J Chem Inf Model. 2005;45:1621–1627. doi: 10.1021/ci0501490. [DOI] [PubMed] [Google Scholar]
- Stopar D, Spruijt RB, Hemminga MA. Anchoring mechanisms of membrane-associated M13 major coat protein. Chem Phys Lipids. 2006;141:83–93. doi: 10.1016/j.chemphyslip.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Stopar D, Štrancar J, Spruijt RB, Hemminga MA. Motional restrictions of membrane proteins: a site-directed spin labeling study. Biophys J. 2006;91:3341–3348. doi: 10.1529/biophysj.106.090308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Štrancar J, Kavalenka A, Ziherl P, Stopar D, Hemminga MA. Analysis of side chain rotational restrictions of membrane-embedded proteins by spin-label ESR spectroscopy. J Magn Reson. 2009;197:245–248. doi: 10.1016/j.jmr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- Strandberg E, Killian JA. Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett. 2003;544:69–73. doi: 10.1016/S0014-5793(03)00475-7. [DOI] [PubMed] [Google Scholar]
- Straus S, Scott W, Symmons M, Marvin D. On the structures of filamentous bacteriophage Ff (fd, f1, M13) Eur Biophys J. 2008;37:521–527. doi: 10.1007/s00249-007-0222-7. [DOI] [PubMed] [Google Scholar]
- Trevor M, Richard S. Bacteriophages and biotechnology: a review. J Chem Technol Biotechnol. 2000;75:6–17. doi: 10.1002/(SICI)1097-4660(200001)75:1<6::AID-JCTB157>3.0.CO;2-A. [DOI] [Google Scholar]
- Tseng RJ, Tsai C, Ma L, Ouyang J, Ozkan CS, Yang Y. Digital memory device based on tobacco mosaic virus conjugated with nanoparticles. Nat Nanotechnol. 2006;1:72–77. doi: 10.1038/nnano.2006.55. [DOI] [PubMed] [Google Scholar]
- Ulmschneider MB, Sansom MSP, Di Nola A. Evaluating tilt angles of membrane-associated helices: comparison of computational and NMR techniques. Biophys J. 2006;90:1650–1660. doi: 10.1529/biophysj.105.065367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Wel PCA, Strandberg E, Killian JA, Koeppe RE., II Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys J. 2002;83:1479–1488. doi: 10.1016/S0006-3495(02)73918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos WL, Koehorst RBM, Spruijt RB, Hemminga MA. Membrane-bound conformation of M13 major coat protein: a structure validations through FRET-derived constrains. J Biol Chem. 2005;280:38522–38527. doi: 10.1074/jbc.M505875200. [DOI] [PubMed] [Google Scholar]
- Vos WL, Schor M, Nazarov PV, Koehorst RBM, Spruijt RB, Hemminga MA. Structure of membrane-embedded M13 major coat protein is insensitive to hydrophobic stress. Biophys J. 2007;93:3541–3547. doi: 10.1529/biophysj.107.112698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos WL, Nazarov PV, Koehorst RBM, Spruijt RB, Hemminga MA. From ‘I’ to ‘L’ and back again: the odyssey of membrane-bound M13 protein. Trends Biochem Sci. 2009;34:249–255. doi: 10.1016/j.tibs.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Wang YA, Yu X, Overman S, Tsuboi M, Thomas JGJ, Egelman EH. The structure of a filamentous bacteriophage. J Mol Biol. 2006;361:209–215. doi: 10.1016/j.jmb.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Webster RE, Lopez J. Structure and assembly of the class I filamentous bacteriophage. In: Casjens S, editor. Virus structure and assembly. Boston: Jones and Bartelett; 1985. pp. 235–267. [Google Scholar]
- White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- Whitesides GM, Grzybowski B. Self-assembly at all scales. Science. 2002;295:2418–2421. doi: 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]
- Zeri AC, Mesleh MF, Nevzorov AA, Opella SJ. Structure of the coat protein in fd filamentous bacteriophage particles determined by solid-state NMR spectroscopy. Proc Natl Acad Sci USA. 2003;100:6458–6463. doi: 10.1073/pnas.1132059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zhang S. Fabrication of molecular materials using peptide construction motifs. Trends Biotechnol. 2004;22:470–476. doi: 10.1016/j.tibtech.2004.07.011. [DOI] [PubMed] [Google Scholar]