

Abstract
In this review we discuss recent insights obtained from well-characterized model systems into the factors that determine the orientation and tilt angles of transmembrane peptides in lipid bilayers. We will compare tilt angles of synthetic peptides with those of natural peptides and proteins, and we will discuss how tilt can be modulated by hydrophobic mismatch between the thickness of the bilayer and the length of the membrane spanning part of the peptide or protein. In particular, we will focus on results obtained on tryptophan-flanked model peptides (WALP peptides) as a case study to illustrate possible consequences of hydrophobic mismatch in molecular detail and to highlight the importance of peptide dynamics for the experimental determination of tilt angles. We will conclude with discussing some future prospects and challenges concerning the use of simple peptide/lipid model systems as a tool to understand membrane structure and function.
Keywords: Transmembrane model peptides, Hydrophobic mismatch, Tryptophan anchoring, Tilt angle, Peptide orientation, Peptide motion
Introduction
Membrane proteins perform a wide range of vital functions in the cell, including signalling, transduction of energy, and transport of ions and solutes over the cell membrane. In spite of their obvious importance, knowledge on the structural properties of membrane proteins is still relatively sparse. Even less is known about the dynamical processes that are essential for functioning. By definition, membrane proteins need to undergo conformational changes to perform their function. These changes often include internal motions such as tilting, turning, or shifting of one or more α-helices or whole domains. Alternatively, secondary structure elements such as α-helices or β-sheets can (partially) fold or unfold, or adapt their structure. Finally, simple reorientation of one or more amino acid residues can occur. Since all these changes can be accompanied by a (partial) reorientation of the protein within the membrane, it can be expected that properties of the surrounding lipid environment may have a significant influence on the functioning of membrane proteins.
The functional activity of many membrane proteins indeed was shown to depend on membrane properties including membrane composition, fluidity, and thickness. Examples of such membrane proteins include the nicotinic acetylcholine receptor (AChR) (Barrantes 2004), the thermosensor protein DesK (Cybulski et al. 2002; Aguilar et al. 2001), cytochrome c oxidase (Montecucco et al. 1982), melibiose permease (Dumas et al. 2000), and different ATPases (Johannsson et al. 1981a, b; Caffrey and Feigenson 1981). In reconstitution studies on these latter membrane proteins, the activity was found to be highest when the proteins were reconstituted in bilayers with matching hydrophobic thickness. From these studies, it seems that many membrane proteins have an optimal bilayer thickness for functioning. Most probably, in bilayers with a nonoptimal thickness, the activity of these membrane proteins is altered by small changes in the structure or orientation of transmembrane segments of these proteins. Such changes will include variations in the helix tilt angle.
Functional importance of helix tilting in membrane proteins
An example of a membrane protein where changes in the tilt angle of transmembrane segments play an important role for the function is the mechanosensitive channel MscL. This well-studied channel protein prevents bacterial cells from bursting as a result of water transport into the cell in case of osmotic down shock (reviewed in Corry and Martinac 2008; Sukharev and Anishkin 2004). The regulation of MscL arises from changes in the lateral pressure of the membrane and is mediated by hydrophobic coupling between the lipids and the hydrophobic segments of membrane protein, resulting in the conformational changes needed to switch the channel to the open state. When switching to the open state, this channel undergoes a large concerted iris-like expansion combined with flattening of the whole channel, which involves large reorientations of the transmembrane α-helices (Betanzos et al. 2002).
In other proteins, small changes in the orientation of one or more α-helices are sufficient for proper functioning. For sensory rhodopsin II, a small outward tilting of TM6 (helix F) was observed (Wegener et al. 2000), which initiates a 20–30° turning of helix TM2 of the neighboring transducer complex (Wegener et al. 2001). For the related bacteriorhodopsin, an outward movement of TM6 (helix F) and a subsequent approach of TM7 (helix G) toward the proton channel were observed during the photo cycle (Radzwill et al. 2001). For melibiose permease, a symporter coupling uphill transport of melibiose and various other galactosides to the downhill transport of Na+, Li+ and H+ ions, the average helix tilt angle was observed to increase upon ion-binding from 26° to 36°, and to decrease again to 30° upon subsequent sugar binding (Dave et al. 2008). These examples demonstrate the importance of the orientation of transmembrane segments for the function of membrane proteins.
Much information on α-helix orientation in membrane proteins has been obtained from X-ray crystallography studies. A recent empirical study reported an average tilt angle of 24 ± 14° for the transmembrane α-helices of polytopic membrane proteins based on 46 qualified high-resolution structures available in 2006 (Ulmschneider et al. 2005), which agrees well with earlier reported values of 22 ± 12° based on 15 structures in 2001 (Ulmschneider and Sansom 2001) and 21 ± 10° based on only 2 structures, bacteriorhodopsin and cytochrome c oxidase, in 1997 (Bowie 1997). The low number of high-resolution structures available for membrane proteins is mainly due to the difficulties in growing crystals of these hydrophobic structures that are needed for structure elucidation using X-ray crystallography.
Although high-resolution structures are essential for understanding the structural changes required for functioning of membrane proteins, only limited information on the dynamics of these processes can be extracted from crystal structures. In addition, the use of X-ray crystallography does not allow to study the influence of the lipid environment on the orientation of the transmembrane segments. For this reason, alternative methods are being used to study structural and dynamic properties of proteins in lipid bilayers. These methods include solid-state NMR spectroscopy, EPR spectroscopy, CD spectroscopy, ATR-FTIR spectroscopy, and fluorescence spectroscopy. Using such alternative biophysical techniques, tilt angles of the transmembrane α-helices with respect to the membrane have been determined for a number of membrane proteins and peptides.
Orientation of transmembrane segments in membranes
For natural transmembrane peptides, tilt angles were found to vary significantly, as illustrated by the following examples performed in bilayers with acyl chain lengths ranging from 14 to 18 carbon atoms. From solid-state NMR measurements using the PISEMA method, a tilt angle of 38° was reported for the influenza A M2 channel (Wang et al. 2001), 26° for the membrane conformation of the major pVIII coat protein of fd filamentous bacteriophage (Marassi and Opella 2003), and 13° for wild-type phospholamban (Abu-Baker et al. 2007). For the channel-lining M2 segments from the d-subunit of the nicotinic acetylcholine receptor, a tilt angle of 12° was reported from PISEMA experiments (Opella et al. 1999) and a tilt angle of 14° from EPR studies (Inbaraj et al. 2007). For phospholemman, a tilt angle of 17° was found from ATR-FTIR spectroscopy experiments on the full length protein (Beevers and Kukol 2006), and a tilt angle of 7° was reported from NMR experiments on a phospholemman transmembrane region (Wong et al. 2008). Using fluorescence techniques, a tilt angle of 18° was determined for the major coat protein from bacteriophage M13 (Nazarov et al. 2007).
This variation in tilt angles raises questions about the determinants for the tilt of transmembrane helices. Is helix tilt mainly an intrinsic property of transmembrane helices, due for example to a specific distribution of amino acids around the helix axis or to a relatively large hydrophobic length of the helix? Or, in the case of oligomeric structures, is it determined by peptide–peptide interactions? Or is the tilt mainly imposed from the membrane, determined for example by its hydrophobic thickness? How does each of these factors contribute to determining the tilt angle? Addressing these questions requires highly systematic approaches. Years ago, this need initiated the design of simplified model systems of artificial transmembrane α-helical peptides, reconstituted into synthetic lipid bilayers. In such simple peptide/lipid model systems, both lipid and peptide composition can be easily and systematically varied, allowing elucidation of the basic principles of membrane protein organization and dynamics.
Use and design of transmembrane model peptides
Ideally, artificial transmembrane peptides should serve as mimics for transmembrane segments of membrane proteins. Therefore, the design of a proper transmembrane model peptide requires information on the general composition of membrane proteins. Empirical analysis of the amino acid structure in putative transmembrane segments of α-helical single-span proteins showed a nonrandom distribution of amino acids in transmembrane segments (Landolt-Marticorena et al. 1993; Arkin and Brunger 1998). The hydrophobic stretch is to a large percentage composed of hydrophobic residues such as isoleucine, leucine, valine, and alanine, with leucine being the most common residue. Putative helix initiating residues such as proline, asparagine, and serine were found to be enriched at the N-terminus of transmembrane segments. Aromatic residues such as tryptophan and tyrosine as well as charged residues such as lysine and arginine were found to have a positional preference for the interface region.
In particular these aromatic and charged residues at the interface can be expected to have a large influence on the orientation of transmembrane segments because their interactions with the membrane will be much more specific than those of the hydrophobic amino acids in the lipid acyl chain region. Indeed, a common structural motif observed in many membrane proteins is a belt of aromatic residues located at the membrane-water interface. These are present for example in the α-helical potassium channel KcsA (Doyle et al. 1998; Williamson et al. 2003) (see Fig. 1a) and the β-barrel maltoporin (Seshadri et al. 1998). Such interfacial tryptophans are believed to anchor membrane proteins in the membrane, where interaction with the surrounding lipids will help the proteins to adopt the proper orientation in the membrane.
Fig. 1.
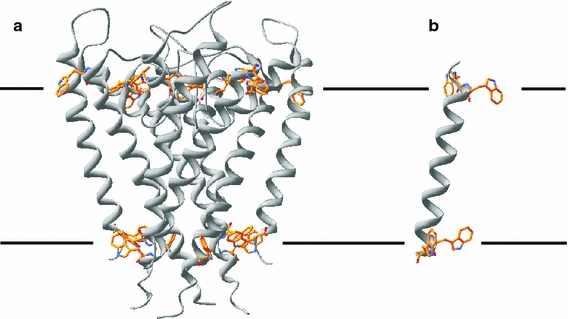
Structures of a the transmembrane parts of the potassium channel KscA (PDB entry 1J95) and b the transmembrane model peptide WALP23, which is used to mimic transmembrane segments of membrane proteins. Only tryptophans are shown with explicit side chains, illustrating the belt of aromatic residues present in the tetrameric KcsA
These structural elements characteristic for many membrane proteins were considered in the design of different model transmembrane peptides. An overview of transmembrane model peptides used in the literature is given in Table 1. Such designed model peptides offer several advantages over the use of natural transmembrane peptides. Most importantly, many specific properties, such as hydrophobicity, hydrophobic length, or nature of flanking residues, can be systematically varied. Moreover, suitable labels can easily be incorporated during peptide synthesis, allowing the use of a wide range of biophysical techniques. For these reasons, synthetic peptides are becoming increasingly useful in the development and improvement of biophysical techniques to characterize membrane proteins/peptides (Andronesi et al. 2004; Lemaitre et al. 2004) and as models to understand the basic principles of peptide/protein-lipid interactions (Killian and Nyholm 2006; Shahidullah and London 2008; Yano et al. 2002; Liu et al. 2002; Mall et al. 2000). In particular the WALP (depicted in Fig. 1b) and KALP peptides have been used for these purposes. These model peptides are composed of a hydrophobic stretch of alternating leucines and alanines flanked at both ends by a pair of tryptophans and lysines, respectively.
Table 1.
Amino acid sequences of tryptophan and/or lysine-flanked transmembrane model peptides used in literature
| Model peptide | Amino acid sequence | References |
|---|---|---|
| Peptides flanked by tryptophans | ||
| WALPx, x = 2m + n + 6 | Ac-GWW(LA)mLnWWA-amidea, m = {3, 5, 6, 7, 8, 10, 12}, n = {0, 1} | Killian et al. (1996), de Planque et al. (1998, 1999, 2001, 2003), Demmers et al. (2001), Rinia et al. (2002), Strandberg et al. (2002), Petrache et al. (2002), Morein et al. (2002), van der Wel et al. (2002, 2007); Kol et al. (2003), Weiss et al. (2003), Andronesi et al. (2004), Ganchev et al. (2004), Strandberg et al. (2004), van Duyl et al. (2005), Özdirekcan et al. (2005, 2007), Im and Brooks (2005), Sparr et al. (2005a, b), Siegel et al. (2006), Holt et al. (2008) |
| GWALP23 | Ac-GGALW(LA)6LWLAGA-ethanolamine | Vostrikov et al. (2008) |
| WLP23 | Ac-GWWL17WWA-amide | Özdirekcan et al. (2005), Esteban-Martin and Salgado (2007a) |
| WL22 | Ac-WWL18WW-amide | Esteban-Martin and Salgado (2007b) |
| WA22 | Ac-WWA18WW-amide | Esteban-Martin and Salgado (2007b) |
| Peptides flanked by tryptophans and lysines | ||
| LW | Ac-KKWWL8AL8WWKK-amide | Fastenberg et al. (2003) |
| W-L22-W | Ac-KKWL22WKK-amide | Liu et al. (2002) |
| KWALP23 | Ac-GKALW(LA)6LWLAKA-amide | Daily et al. (2008) |
| Peptides flanked by lysines | ||
| KALPx, x = 2m + 7 | Ac-GKK(LA)mLWWA-amide, m = {6, 8, 10, 12} | de Planque et al. (2001), Strandberg et al. (2002), Morein et al. (2002), Kol et al. (2003), van Duyl et al. (2005), Özdirekcan et al. (2005), Kandasamy and Larson (2006) |
| (LA)12 | Ac-KK(LA)12KK-amide | Zhang et al. (1995a, b, 2001) |
| KLP23 | Ac-GKKL17KKA-amide | Özdirekcan et al. (2005), Esteban-Martin and Salgado (2007a) |
| L24 | Ac-KKL24KK-amide | Axelsen et al. (1995), Subczynski et al. (1998), Liu et al. (2002) |
| P16/24 | Ac-KKGL16/24KKA-amide | Davis et al. (1983), Huschilt et al. (1985), Morrow et al. (1985), Huschilt et al. (1989), Zhang et al. (1992a, b, 1995c) |
| A24 | Ac-KKA24KK-amide | Lewis et al. (2001) |
| Peptides flanked by lysines that include a tryptophan in the hydrophobic stretch | ||
| p(LA)6/8 | Ac-KKG(LA)3/4W(LA)3/4KKA-amide | Vidal and McIntosh (2005), Krishnakumar and London (2007) |
| pLx, x = 2m | Ac-KKGLmWLmKKA-amide, m = {5, 6, 7, 8, 9, 10, 11} | Ren et al. (1999), Caputo and London (2003), Lew et al. (2003), Krishnakumar and London (2007), Shahidullah and London (2008) |
| pLmAm, m = {6, 7, 8} | Ac-KKGLmWAmKKA-amide | Krishnakumar and London (2007) |
| pA18/22 | Ac-KKA9/11WA9/11KK-amide | Shahidullah and London (2008) |
aIn some cases ethanolamine is used instead
As discussed above, an important parameter for the structural organization of membrane proteins is the orientation of their transmembrane segments within the lipid bilayer. One convenient way of addressing the question to what extent the orientation of transmembrane segments can be imposed from the membrane is by analysis of the tilt angle of transmembrane helices in model systems. In model systems, the hydrophobic length of the peptide and the bilayer thickness can be systematically varied, leading to a situation of hydrophobic mismatch.
Hydrophobic mismatch as driving force for helix reorientation
Hydrophobic mismatch occurs if the hydrophobic length of the membrane protein does not match the hydrophobic thickness of the membrane. To be more specific, positive hydrophobic mismatch describes the situations with too thin membranes, and negative hydrophobic mismatch the situations with too thick membranes. As already mentioned above, the activity of membrane proteins depends strongly on the hydrophobic thickness of the bilayer and is often decreased in nonmatching situations (for reviews see Andersen and Koeppe 2007; Jensen and Mouritsen 2004). In case of positive hydrophobic mismatch, a logical response to relieve the unfavorable exposure of hydrophobic amino acids to the aqueous phase seems to be increasing the tilt of the helix.
The response of the tilt angles of transmembrane segments to hydrophobic mismatch has been studied for a number of natural peptides and model peptides. So far, in all cases indeed an increase in tilt angle was observed upon decreasing bilayer thickness. However, in most cases this response was not sufficient to fully compensate for the mismatch. For example, 2H NMR experiments (discussed in more detail below) suggested that the tilt angle for WALP23 peptides increased from 5.2° in di-C14:0PC to 8.1° in di-C12:0PC, and for KALP23 peptides from 7.6° to 11.2° (Strandberg et al. 2004; Özdirekcan et al. 2005). Considering geometrics, this change in tilt angle would be too small to compensate for the changes in bilayer thickness.
Similar results were obtained for several natural peptides. For M13 coat protein, an increase from 18° in di-C20:1PC to 33° in di-C14:1PC was determined from fluorescence experiments using IAEDANS-labelled peptides (Koehorst et al. 2004). For alamethicin, a peptide antibiotic building pores in cell membranes, a decrease in tilt angle from 23° in di-C10:0PC to 13° in di-C18:0PC was found from EPR experiments employing TOAC, a spin-labelled amino acid analogue (Marsh et al. 2007). For both natural peptides, the changes in tilt are insufficient to compensate for changes in bilayer thickness.
So far, to our knowledge, there is only one example of a natural peptide for which full compensation for hydrophobic mismatch has been observed. Using PISEMA experiments, for virus protein U (Vpu) from HIV-1 incorporated into bilayers of ether-linked di-C10:0PC/di-C10:0PG (9:1), a tilt angle of 51° was found, which systematically decreased upon increasing the bilayer thickness to 18° in ether-linked di-C18:1PC/di-C18:1PG (Park and Opella 2005), thereby fully compensating for the change in bilayer thickness. This finding is even more striking considering that relatively high peptide-to-lipid ratios were used and that the peptide most likely is present in an oligomeric form. Thus it seems that, at least in this specific case, surrounding lipids are much stronger determinants for peptide orientation than adjacent peptides. Another possibly relevant feature of the system is that the peptide misses interfacial anchoring residues at one end, which might influence its ability to adapt to mismatch.
From these experiments, it is clear that the membrane has an influence on the tilt of protein transmembrane segments but that peptide composition is important as well. However, the specific reasons for the differences in adaptation to hydrophobic mismatch are not clear yet. A complicating factor is the notion that a change in the tilt angle of transmembrane segments is not the only possible mechanism to compensate for hydrophobic mismatch. If, for a given protein, alternative mechanisms would be more favorable, there would be no reason to expect complete adaptation to a positive mismatch just by a change in tilt angle of the transmembrane segment. Below we will provide examples of such alternative mechanisms, which show that indeed many responses can occur.
Alternative/additional mechanisms to adapt to mismatch
The consequences of hydrophobic mismatch have been investigated extensively, in particular for WALP and KALP peptides. Figure 2 illustrates several possible adaptations for the case of a positive mismatch situation, which all have been observed experimentally. Besides helix tilt (a), another option is that the lipid acyl chains in direct vicinity to the peptide stretch to accommodate the peptide (b). This was in fact observed for WALP peptides by 2H NMR experiments on acyl chain deuterated lipids (de Planque et al. 1998), but not for KALP peptides (de Planque et al. 2003). Another possibility to alleviate the consequences of hydrophobic mismatch is peptide self-association (c). This behavior was observed for WALP peptides (Sparr et al. 2005a; de Planque et al. 2001; Killian et al. 1996) as well as for lysine-flanked peptides (Ren et al. 1999). Yet another possible adaptation is deformation of the backbone to decrease the effective length of the peptide (d). In principle this could be accomplished by formation of a π-helix, but this has not been observed experimentally. Alternatively, kinking or flexing of the transmembrane helix may occur as a response of the peptide backbone to positive mismatch (e). 2H NMR results on WALP peptides and analogues thereof indicated a kink in the helix as response to a too-thin bilayer (Strandberg et al. 2004; Daily et al. 2008). A similar reaction was found for the TM1 segment of lactose permease, which was observed to flex in addition to tilting to satisfy the hydrophobic mismatch (Yeagle et al. 2007). Finally, another consequence to extreme mismatch conditions could be that peptides switch to a non-transmembrane state, i.e., bound to the membrane interface (f), or are even excluded from the membrane. This has been documented extensively for lysine-flanked model peptides of different hydrophobic lengths and compositions (Krishnakumar and London 2007; Webb et al. 1998). Also for WALP peptides it was shown that when mismatches are too large, the peptides incorporate less well (de Planque et al. 2001; Killian et al. 1996).
Fig. 2.
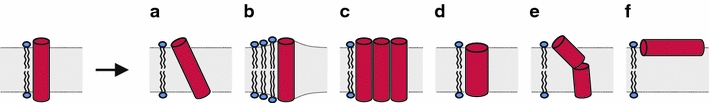
Schematic representation of possible adaptations in case of a too long transmembrane segment, i.e., positive hydrophobic mismatch conditions: a helix tilting, b acyl chain stretching, c oligomerization, d backbone deformation/distortion, e backbone kinking/flexing, f switching to a non-transmembrane state, i.e., binding to the membrane interface
Thus, many alternative responses of protein transmembrane segments to mismatch are possible besides tilting, and it is far from clear what determines the extent to which each of these adaptations may occur for a given protein. Considering the fact that in particular the WALP peptides adapt to hydrophobic mismatch in several different ways, one would expect that for these peptides tilting would be at most a partial response to mismatch and hence not sufficient to allow complete adaptation to bilayer thickness. Although WALP peptides can be considered fairly simple systems as compared to natural membrane proteins and peptides, characterization of the tilt of these model peptides has not been straightforward. We will now present a case study of the WALP peptides that illustrates the difficulties in determining tilt angles of peptides in membranes and the important role of peptide dynamics.
Implications of peptide dynamics on the determination of tilt angles
In general, solid-state NMR approaches are particularly suitable to obtain detailed information on the structural properties of transmembrane peptides in lipid bilayers. For example, helix tilt and rotation angles (van der Wel et al. 2002; Strandberg et al. 2004; Özdirekcan et al. 2005) as well as deviations from α-helical structure (Strandberg et al. 2004; Daily et al. 2008) can be investigated by analysis of a set of quadrupolar splittings obtained from peptides that include a deuterium-labelled alanine with the GALA method, or from PISEMA experiments on uniformly 15N-labelled peptides by analysis of the characteristic wheel-like patterns, which provides direct information on the orientation of the transmembrane helices (Traaseth et al. 2006; Park and Opella 2005; Marassi and Opella 2000).
However, with both these NMR methods, assumptions have to be made regarding the dynamics of the peptides. Importantly, a high mobility results in difficulties in interpretation of the results and can lead to an underestimation of the tilt angle (Özdirekcan et al. 2007; Esteban-Martin and Salgado 2007a). The importance of obtaining knowledge on motional properties of peptides is illustrated by comparison of results obtained by solid-state NMR and MD simulations on WALP peptides. 2H NMR experiments in combination with the GALA method indicated that WALP peptides have a relatively small tilt angle (<10°) in different types of bilayers (Özdirekcan et al. 2005). In contrast, MD studies on the same systems suggested much larger tilt angles, fluctuating from around 20–40° (Özdirekcan et al. 2007). As a potential explanation for this discrepancy, it was suggested that the WALP peptide can adopt different rotational angles but that there is an energy barrier between the preferred rotational states, such that ‘hopping’ motions occur. From the MD trajectories, the authors back-calculated the NMR quadrupolar splittings and they concluded that if such motions indeed would occur, this would lead to an underestimation of the tilt angles determined by 2H NMR experiments.
The extent to which transmembrane peptides undergo motional averaging will not only depend on peptide composition and properties of the lipid environment, but also on the extent to which the peptides self-associate. Most of the natural transmembrane peptides investigated with solid-state NMR methods are believed to exist as homo-oligomers in the functional unit; good examples are the influenza M2 channel (Wang et al. 2001; Cady et al. 2007), phospholemman (Wong et al. 2008), or phospholamban (Abu-Baker et al. 2007). Interactions among the subunits of these helical bundles can be expected to largely restrict the motions of the transmembrane segments. Especially oscillations around the helix axis will be small since such motions would alter the contact interfaces to neighboring helices. Thus, the orientation parameters of transmembrane segments participating in oligomers will be much less affected by motional averaging when investigated with solid-state NMR methods.
WALP peptides were shown to have no tendency to self-associate even at high peptide-to-lipid ratios, and only under large mismatch conditions a slightly higher tendency to self-associate was found (Sparr et al. 2005a). However, even for peptides that do not have a tendency to oligomerize, peptide-peptide interactions may occur if the samples contain a high peptide-to-lipid ratio. For example, in order to achieve sufficient sensitivity, PISEMA spectra of uniformly 15N-labelled peptides are recorded on oriented bilayers at high peptide-to-lipid ratios, e.g., of 1:16 (Wang et al. 2001) or 1:20 (Vostrikov et al. 2008). In contrast, 2H NMR experiments on peptides with single deuterium-labelled alanines by using the GALA method can be performed on unoriented bilayers at low peptide-to-lipid ratios of 1:100 (Strandberg et al. 2004), thereby avoiding side effects of peptide–peptide interactions on the orientation of the transmembrane peptides. Therefore, in these experiments, large peptide motions, i.e., oscillations around the helix axis, are much more probable, which would lead to averaging of the quadrupolar splittings.
It is clear that in order to unambiguously determine tilt angles of transmembrane peptides under different experimental conditions, information on their dynamics is required. WALP peptides in PC bilayers seem like an ideal test case for this because these systems have been characterized extensively already by many different techniques (Killian 2003; Killian and Nyholm 2006). Moreover, it is important to solve the discrepancy described above, and to reliably characterize these model peptides in order to allow using them as simple and well characterized model systems, for example for the validation of new techniques and calibration of existing methods. This holds not only for validation of solid-state NMR methods, but also for MD simulation techniques, which at present are limited by relatively short timescales and the requirement for many input parameters that need to be validated by experimental results. Indeed, to our knowledge MD simulations so far have not succeeded in reproducing the experimentally observed quadrupolar splittings.
Recent developments in the “tilt story”
The importance of peptide dynamics for calculations of peptide rotation and tilt by solid-state NMR methods have led several groups to focus in more detail on motional properties of transmembrane peptides. Strandberg et al. reanalyzed experimental 2H NMR data published for WALP23 peptides (Strandberg et al. 2004) using different peptide models with increasing complexity of peptide dynamics. The authors compared the results obtained with the previous, quasi-static model that included peptide motion using a global order parameter S (Fig. 3a) with the results of more advanced dynamic models including explicit oscillations Δρ and/or wobbling-in-a-cone motions Δτ (Fig. 3b). The different motion models were found to match the data with similar deviations in terms of rmsd, but yielded very different tilt angles (Strandberg et al. 2009). In an accompanying publication, the same group of authors investigated the effect of peptide dynamics on PISEMA experiments (Esteban-Martin et al. 2009). For this purpose, PISEMA spectra were generated from trajectories of an earlier MD study on WLP23 (Esteban-Martin and Salgado 2007a). Analysis of these data with the different peptide models yielded similar tilt angles for both the quasi-static and the dynamic peptide models. However, the virtual PISEMA data could be fitted best with a peptide model including explicit oscillations and wobbling.
Fig. 3.
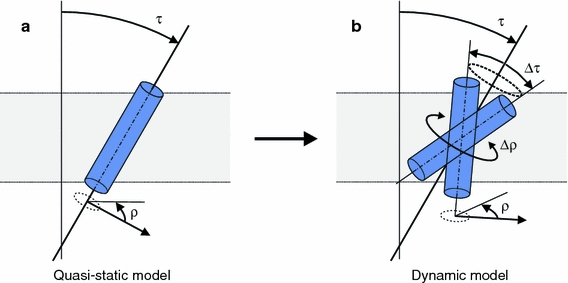
a The quasi-static model describes the orientation of the peptide by a tilt angle τ and a rotation angle ρ, with limited motional averaging described by a global order parameter S. b In the dynamic model, the previous model is extended by an oscillation Δρ around the helix axis and by a wobbling-in-a-cone motion Δτ
In an attempt to explore the differences between the established solid-state NMR methods, Vostrikov et al. compared the results of 2H NMR and PISEMA experiments obtained on a related model peptide, a GWALP peptide (Vostrikov et al. 2008). In this study, the experimental data sets were separately analyzed using the conventional, quasi-static peptide model. Both the 2H NMR and PISEMA data sets yielded very similar tilt angles of 12.6° and 10.8°, respectively. Closer inspection of the fits to the experimental data shows some deviations of the 15N data from the fitted PISEMA wheel, especially for larger dipolar couplings, which may suggest an underestimation of the tilt angle (see discussion in Esteban-Martin et al. 2009). Since the GWALP peptide is a close relative of the WALP peptides flanked at both ends by only a single tryptophan, it may exhibit significant motions around the helical axis. Together with the notion that analysis of tilt angles by using either 2H NMR or PISEMA data can be complicated due to motional averaging, it is very well possible that the use of the conventional, quasi-static peptide model may not be sufficient in this case.
Very recently, a new strategy based on solid-state NMR methods has been developed, called MACADAM (multiple anisotropic constraints and dynamic analysis of membrane peptides) (A. Holt, L. Rougier, V. Réat, F. Jolibois, O. Saurel, J. Czaplicki, J.A. Killian, A. Milon, unpublished data). This new approach combines nuclear interactions with different orientations with respect to the helix axis, which allows determination of both orientation and dynamics of transmembrane segments in membranes. The experimental data were analyzed using a dynamical model, which extended the previous “quasi-static” model by oscillations around the helix axis and wobbling-in-a-cone motions, similar to the dynamical models utilized in the recent work of Strandberg et al. (2009). A tilt angle of ~21° was obtained, which is much larger than the tilt angle of around 5° determined from 2H NMR experiments in combination with the GALA method. Furthermore, a rotation angle of ~150° was observed, which agrees well with the results reported previously from 2H NMR experiments.
In addition, an alternative, complementary approach based on steady-state fluorescence spectroscopy for the investigation of the orientation of transmembrane peptides in membranes was applied (Holt et al. 2009). For this study, WALP23 peptides including cysteine replacements in different positions along the helix were labelled with the environment-sensitive label BADAN. The fluorescence spectra of this label reflect the local polarity at the position of the label, which was used to determine the orientation of the peptide in the bilayer. For WALP23 inserted in di-C18:1PC, a tilt angle of ca. 24° and a rotation angle of 136° were found, which agree well with the results of the new MACADAM approach. Furthermore, as with the GALA method, only small changes in tilt angle were observed upon changing the hydrophobic thickness of the bilayer, i.e., 24.8° and 19.8° in bilayers of di-C16:1PC and di-C20:1PC, respectively. These findings suggest that WALP23 peptides do not fully compensate for hydrophobic mismatch by tilting, in agreement with the notion that other adaptations occur simultaneously (Killian and Nyholm 2006).
Functional importance of tryptophan anchoring
From the adaptations to hydrophobic mismatch, it is clear that WALP peptides interact strongly with the surrounding lipids and it is likely that the flanking tryptophan residues play a role in this interaction. Tryptophans in interfacial positions of membrane proteins are thought to couple transmembrane segments to the membrane in a very particular manner; on one hand tryptophans prefer a distinct location at the membrane-water interface (Yau et al. 1998; Persson et al. 1998; de Planque et al. 2003), but on the other hand tryptophans in interfacial positions were also shown to inhibit tilting of transmembrane segments (Chiang et al. 2005). This, at first sight contradictory, behavior of tryptophan-flanked transmembrane segments may have two reasons.
First, interfacial tryptophans were shown to order lipid acyl chains in the vicinity of transmembrane segments when experiencing positive hydrophobic mismatch (de Planque et al. 2002), which may also involve a more or less pronounced concerted response of lipids in more remote lipid layers around the transmembrane segments. Such a stretching of the lipids leads to a more ordered and condensed bilayer around the transmembrane peptide, which in turn may hinder tilting. Second, tilting may entail a different and more unfavorable rotamer conformation and/or localization of the tryptophan side chain in the membrane. It was found before that tryptophans prefer certain rotameric conformations and locations at the membrane interface (Yau et al. 1998; Persson et al. 1998; Chamberlain and Bowie 2004). The latter may be important for maintaining the correct localization and orientation of tryptophans at the membrane/water interface, assuring the proper orientation and insertion of membrane proteins.
Future prospects
The work on establishing properties of even the most simple model systems of a synthetic peptide (in this case WALP) in artificial lipid systems has proven to be quite challenging. We are now finally beginning to approach the situation that the properties of the systems are becoming fully understood and that the characterization is thorough and reliable under a wide range of experimental conditions. The availability of such a thoroughly characterized model system of a synthetic transmembrane peptide in lipid bilayers opens up many perspectives for future studies.
First, one can apply this knowledge to investigate fundamental questions regarding the complex interaction mechanisms of membrane proteins and lipids within the membrane by addressing the importance of specific peptide or lipid properties for the peptide/lipid interplay. As an example, studies on the behavior of model membranes containing either WALP or KALP peptides have yielded valuable insights on the possible roles of tryptophans and lysines as flanking residues. They showed that tryptophans strongly anchor transmembrane segments to the lipid/water interface and that they resist being pulled away from the interface either towards the hydrophobic core of the bilayer or towards the aqueous phase (de Planque et al. 2003). In contrast, lysines have a long flexible side chain, which does not resist being pulled into the aqueous phase, and which can snorkel to the membrane/water interface when the backbone is pulled into the hydrophobic part of the bilayer (Kandasamy and Larson 2006; Strandberg and Killian 2003). Hence, lysines as flanking residues may allow membrane proteins to readily adapt to varying membrane thickness, without significant distortion of either the surrounding lipids or the protein backbone. Options for future studies include for example finding out basic principles of how the overall hydrophobicity affects the tendency of a peptide to adjust its tilt angle to the bilayer thickness, or how the flanking residues and the amino acid distribution around the helix axis influence the preferred direction of tilt of a peptide.
Second, the availability of simple, well characterized and reliable model systems is important for validation and/or calibration of new techniques. WALP peptides have been used for improving MD simulations, and for testing various new methods including NMR approaches (Vogel et al. 2003; Lemaitre et al. 2004; Andronesi et al. 2004), EPR approaches (Nielsen et al. 2005), and computational techniques (Im and Brooks 2005; Bond et al. 2007; Ulmschneider et al. 2009).
Third, one can use artificial model systems to investigate molecular details of the possible role of transmembrane protein segments in functional properties of membrane proteins. Examples are the influence of transmembrane segments on lipid transbilayer movement (Kol et al. 2001, 2003), the importance of the length of the transmembrane segment for partitioning into liquid-ordered domains (Ren et al. 1999; van Duyl et al. 2002; Vidal and McIntosh 2005), the involvement of protein transmembrane segments in signalling (Matthews et al. 2006), or the sensing of membrane fluidity by protein transmembrane segments (Mansilla and de Mendoza 2005).
As the investigations using designed model systems continue, and as more and more insight is obtained about the molecular nature of peptide/lipid interactions and the factors that can influence this, we will gradually improve our understanding of properties of membrane proteins and how they are influenced by the lipid environment. Because such model systems allow testing of an almost infinite number of parameters in lipid and peptide composition as well as environmental conditions, challenges will lie in selecting those parameters that allow direct testing of a working hypothesis or that will provide answers to general questions that are important for understanding protein/lipid interactions. The ultimate challenge will be to extend this research towards understanding and predicting properties and lipid interactions of multi-spanning proteins in membranes. An initial step towards this goal would require the use of more sophisticated model systems, for example with two or more covalently coupled transmembrane peptides whose properties can be systematically altered. In the end, the knowledge obtained on such systems may help to obtain a complete understanding of the principles that govern structure, function, and dynamic properties of this important but elusive class of proteins in lipid bilayers and how they can be modulated by the lipid environment.
Acknowledgments
This work was supported by a Marie Curie Early Stage Research Training Fellowship from the European Community’s Sixth Framework Program to A.H. (Biomem-MEST-CT 2004-007931).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- di-C10:0PC
1,2-Didecanoyl-sn-glycero-3-phosphocholine
- di-C12:0PC
1,2-Dilauroyl-sn-glycero-3-phosphocholine
- di-C14:0PC
1,2-Dimyristoyl-sn-glycero-3-phosphocholine
- di-C18:0PC
1,2-Distearoyl-sn-glycero-3-phosphocholine
- di-C14:1PC
1,2-Dimyristoleoyl-sn-glycero-3-phosphocholine
- di-C16:1PC
1,2-Dipalmitoleoyl-sn-glycero-3-phosphocholine
- di-C18:1PC
1,2-Dioleoyl-sn-glycero-3-phosphocholine
- di-C20:1PC
1,2-Dieicosenoyl-sn-glycero-3-phosphocholine
- di-C10:0PG
1,2-Didecanoyl-sn-glycero-3-phospho-(1’-sn-glycerol)
- di-C18:1PG
1,2-Dioleoyl-sn-glycero-3-phospho-(1’-sn-glycerol)
- EPR
Electron paramagnetic resonance
- CD
Circular dichroism
- NMR
Nuclear magnetic resonance
- ATR-FTIR
Attenuated total reflection-Fourier transform infrared
- GALA
Geometric analysis of labelled alanines
- PISEMA
Polarization inversion spin exchange at magic angle
- MD
Molecular dynamics
- BADAN
6-Bromoacetyl-2-dimethylaminonaphthalene
- IEADANS
5-((((2-Iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid
- TOAC
4-Amino-4-carboxy-2,2,6,6-tetramethyl-piperidino-1-oxyl
Footnotes
The more you see: spectroscopy in molecular biophysics.
Contributor Information
Andrea Holt, Email: a.holt@nki.nl.
J. Antoinette Killian, Phone: +31-30-2533342, FAX: +31-30-2533969, Email: j.a.killian@uu.nl.
References
- Abu-Baker S, Lu JX, Chu S, Shetty KK, Gor’kov PL, Lorigan GA. The structural topology of wild-type phospholamban in oriented lipid bilayers using 15N solid-state NMR spectroscopy. Protein Sci. 2007;16:2345–2349. doi: 10.1110/ps.072977707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar PS, Hernandez-Arriaga AM, Cybulski LE, Erazo AC, de Mendoza D. Molecular basis of thermosensing: a two-component signal transduction thermometer in Bacillus subtilis. EMBO J. 2001;20:1681–1691. doi: 10.1093/emboj/20.7.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OS, Koeppe RE., II Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- Andronesi OC, Pfeifer JR, Al-Momani L, Özdirekcan S, Rijkers DTS, Angerstein B, Luca S, Koert U, Killian JA, Baldus M. Probing membrane protein orientation and structure using fast magic-angle-spinning solid-state NMR. J Biomol NMR. 2004;30:253–265. doi: 10.1007/s10858-004-3452-3. [DOI] [PubMed] [Google Scholar]
- Arkin IT, Brunger AT. Statistical analysis of predicted transmembrane alpha-helices. Biochim Biophys Acta. 1998;1429:113–128. doi: 10.1016/s0167-4838(98)00225-8. [DOI] [PubMed] [Google Scholar]
- Axelsen PH, Kaufman BK, McElhaney RN, Lewis RNAH. The infrared dichroism of transmembrane helical polypeptides. Biophys J. 1995;69:2770–2781. doi: 10.1016/S0006-3495(95)80150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrantes F. Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res Rev. 2004;47:71–95. doi: 10.1016/j.brainresrev.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Beevers AJ, Kukol A. Secondary structure, orientation, and oligomerization of phospholemman, a cardiac transmembrane protein. Protein Sci. 2006;15:1127–1132. doi: 10.1110/ps.051899406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betanzos M, Chiang CS, Guy HR, Sukharev S. A large iris-like expansion of a mechanosensitive channel protein induced by membrane tension. Nat Struct Biol. 2002;9:704–710. doi: 10.1038/nsb828. [DOI] [PubMed] [Google Scholar]
- Bond PJ, Holyoake J, Ivetac A, Khalid S, Sansom MS. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J Struct Biol. 2007;157:593–605. doi: 10.1016/j.jsb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Bowie JU. Helix packing in membrane proteins. J Mol Biol. 1997;272:780–789. doi: 10.1006/jmbi.1997.1279. [DOI] [PubMed] [Google Scholar]
- Cady SD, Goodman C, Tatko CD, DeGrado WF, Hong M. Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: a 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle. J Am Chem Soc. 2007;129:5719–5729. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- Caffrey M, Feigenson GW. Fluorescence quenching in model membranes. 3. Relationship between calcium adenosinetriphosphatase enzyme activity and the affinity of the protein for phosphatidylcholines with different acyl chain characteristics. Biochemistry. 1981;20:1949–1961. doi: 10.1021/bi00510a034. [DOI] [PubMed] [Google Scholar]
- Caputo GA, London E. Cumulative effects of amino acid substitutions and hydrophobic mismatch upon the transmembrane stability and conformation of hydrophobic alpha-helices. Biochemistry. 2003;42:3275–3285. doi: 10.1021/bi026697d. [DOI] [PubMed] [Google Scholar]
- Chamberlain AK, Bowie JU. Analysis of side-chain rotamers in transmembrane proteins. Biophys J. 2004;87:3460–3469. doi: 10.1529/biophysj.104.044024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CS, Shirinian L, Sukharev S. Capping transmembrane helices of MscL with aromatic residues changes channel response to membrane stretch. Biochemistry. 2005;44:12589–12597. doi: 10.1021/bi050750r. [DOI] [PubMed] [Google Scholar]
- Corry B, Martinac B. Bacterial mechanosensitive channels: experiment and theory. Biochim Biophys Acta. 2008;1778:1859–1870. doi: 10.1016/j.bbamem.2007.06.022. [DOI] [PubMed] [Google Scholar]
- Cybulski LE, Albanesi D, Mansilla MC, Altabe S, Aguilar PS, de Mendoza D. Mechanism of membrane fluidity optimization: isothermal control of the Bacillus subtilis acyl-lipid desaturase. Mol Microbiol. 2002;45:1379–1388. doi: 10.1046/j.1365-2958.2002.03103.x. [DOI] [PubMed] [Google Scholar]
- Daily AE, Greathouse DV, van der Wel PCA, Koeppe RE., II Helical distortion in tryptophan- and lysine-anchored membrane-spanning a-helices as a function of hydrophobic mismatch: a solid-state deuterium NMR investigation using the geometric analysis of labeled alanines method. Biophys J. 2008;94:480–491. doi: 10.1529/biophysj.106.097543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave N, Lórenz-Fonfría VA, Leblanc G, Padrós E. FTIR spectroscopy of secondary-structure reorientation of melibiose permease modulated by substrate binding. Biophys J. 2008;94:3659–3670. doi: 10.1529/biophysj.107.115550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JH, Clare DM, Hodges RS, Bloom M. Interaction of a synthetic amphiphilic polypeptide and lipids in a bilayer structure. Biochemistry. 1983;22:5298–5305. [Google Scholar]
- de Planque MRR, Greathouse DV, Koeppe RE, II, Schäfer H, Marsh D, Killian JA. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane alpha-helical peptides and gramicidin A. Biochemistry. 1998;37:9333–9345. doi: 10.1021/bi980233r. [DOI] [PubMed] [Google Scholar]
- de Planque MRR, Kruijtzer JAW, Liskamp RMJ, Marsh D, Greathouse DV, Koeppe RE, II, de Kruijff B, Killian JA. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane alpha-helical peptides. J Biol Chem. 1999;274:20839–20846. doi: 10.1074/jbc.274.30.20839. [DOI] [PubMed] [Google Scholar]
- de Planque MRR, Goormaghtigh E, Greathouse DV, Koeppe RE, II, Kruijtzer JAW, Liskamp RMJ, de Kruijff B, Killian JA. Sensitivity of single membrane-spanning alpha-helical peptides to hydrophobic mismatch with a lipid bilayer: effects on backbone structure, orientation, and extent of membrane incorporation. Biochemistry. 2001;40:5000–5010. doi: 10.1021/bi000804r. [DOI] [PubMed] [Google Scholar]
- de Planque MRR, Boots JWP, Rijkers DTS, Liskamp RMJ, Greathouse DV, Killian JA. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane alpha-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 2002;41:8396–8404. doi: 10.1021/bi0257686. [DOI] [PubMed] [Google Scholar]
- de Planque MRR, Bonev BB, Demmers JAA, Greathouse DV, Koeppe RE, II, Separovic F, Watts A, Killian JA. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 2003;42:5341–5348. doi: 10.1021/bi027000r. [DOI] [PubMed] [Google Scholar]
- Demmers JAA, van Duijn E, Haverkamp J, Greathouse DV, Koeppe RE, II, Heck AJR, Killian JA. Interfacial positioning and stability of transmembrane peptides in lipid bilayers studied by combining hydrogen/deuterium exchange and mass spectrometry. J Biol Chem. 2001;276:34501–34508. doi: 10.1074/jbc.M101401200. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Dumas F, Tocanne JF, Leblanc G, Lebrun MC. Consequences of hydrophobic mismatch between lipids and melibiose permease on melibiose transport. Biochemistry. 2000;39:4846–4854. doi: 10.1021/bi992634s. [DOI] [PubMed] [Google Scholar]
- Esteban-Martin S, Salgado J. The dynamic orientation of membrane-bound peptides: bridging simulations and experiments. Biophys J. 2007;93:4278–4288. doi: 10.1529/biophysj.107.113043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban-Martin S, Salgado J. Self-assembling of peptide/membrane complexes by atomistic molecular dynamics simulations. Biophys J. 2007;92:903–912. doi: 10.1529/biophysj.106.093013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban-Martin S, Strandberg E, Fuertes G, Ulrich AS, Salgado J. Influence of whole-body dynamics on 15N PISEMA NMR spectra of membrane proteins: a theoretical analysis. Biophys J. 2009;96:3233–3241. doi: 10.1016/j.bpj.2008.12.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fastenberg ME, Shogomori H, Xu X, Brown DA, London E. Exclusion of a transmembrane-type peptide from ordered-lipid domains (rafts) detected by fluorescence quenching: extension of quenching analysis to account for the effects of domain size and domain boundaries. Biochemistry. 2003;42:12376–12390. doi: 10.1021/bi034718d. [DOI] [PubMed] [Google Scholar]
- Ganchev DN, Rijkers DTS, Snel MME, Killian JA, de Kruijff B. Strength of integration of transmembrane alpha-helical peptides in lipid bilayers as determined by atomic force spectroscopy. Biochemistry. 2004;43:14987–14993. doi: 10.1021/bi048372y. [DOI] [PubMed] [Google Scholar]
- Holt A, de Almeida RFM, Nyholm TKM, Loura LMS, Daily AE, Staffhorst RWHM, Rijkers DTS, Koeppe RE, II, Prieto M, Killian JA. Is there a preferential interaction between cholesterol and tryptophan residues in membrane proteins? Biochemistry. 2008;47:2638–2649. doi: 10.1021/bi702235k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt A, Koehorst RBM, Rutters-Meijneke T, Gelb MH, Rijkers DT, Hemminga MA, Killian JA. Tilt and rotation angles of a transmembrane model peptide as studied by fluorescence spectroscopy. Biophys J. 2009;97:2258–2266. doi: 10.1016/j.bpj.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huschilt JC, Hodges RS, Davis JH. Phase equilibria in an amphiphilic peptide-phospholipid model membrane by deuterium nuclear magnetic resonance difference spectroscopy. Biochemistry. 1985;24:1377–1386. [Google Scholar]
- Huschilt JC, Millman BM, Davis JH. Orientation of alpha-helical peptides in a lipid bilayer. Biochim Biophys Acta. 1989;979:139–141. doi: 10.1016/0005-2736(89)90534-8. [DOI] [PubMed] [Google Scholar]
- Im W, Brooks CL., III Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc Natl Acad Sci USA. 2005;102:6771–6776. doi: 10.1073/pnas.0408135102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbaraj JJ, Laryukhin M, Lorigan GA. Determining the helical tilt angle of a transmembrane helix in mechanically aligned lipid bilayers using EPR spectroscopy. J Am Chem Soc. 2007;129:7710–7711. doi: 10.1021/ja071587l. [DOI] [PubMed] [Google Scholar]
- Jensen M, Mouritsen OG. Lipids do influence protein function—the hydrophobic matching hypothesis revisited. Biochim Biophys Acta. 2004;1666:205–226. doi: 10.1016/j.bbamem.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Johannsson A, Keightley CA, Smith GA, Richards CD, Hesketh TR, Metcalfe JC. The effect of bilayer thickness and n-alkanes on the activity of the (Ca2+ + Mg2+)-dependent ATPase of sarcoplasmic reticulum. J Biol Chem. 1981;256:1643–1650. [PubMed] [Google Scholar]
- Johannsson A, Smith G, Metcalfe J. The effect of bilayer thickness on the activity of (Na+ + K+)-ATPase. Biochim Biophys Acta. 1981;641:416–421. doi: 10.1016/0005-2736(81)90498-3. [DOI] [PubMed] [Google Scholar]
- Kandasamy SK, Larson RG. Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys J. 2006;90:2326–2343. doi: 10.1529/biophysj.105.073395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian JA. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 2003;555:134–138. doi: 10.1016/s0014-5793(03)01154-2. [DOI] [PubMed] [Google Scholar]
- Killian JA, Nyholm TKM. Peptides in lipid bilayers: the power of simple models. Curr Opin Struct Biol. 2006;16:473–479. doi: 10.1016/j.sbi.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Killian JA, Salemink I, de Planque MRR, Lindblom G, Koeppe RE, II, Greathouse DV. Induction of nonbilayer structures in diacylphosphatidylcholine model membranes by transmembrane alpha-helical peptides: importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry. 1996;35:1037–1045. doi: 10.1021/bi9519258. [DOI] [PubMed] [Google Scholar]
- Koehorst RBM, Spruijt RB, Vergeldt FJ, Hemminga MA. Lipid bilayer topology of the transmembrane a-helix of M13 major coat protein and bilayer polarity profile by site-directed fluorescence spectroscopy. Biophys J. 2004;87:1445–1455. doi: 10.1529/biophysj.104.043208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol MA, de Kroon AIPM, Rijkers DTS, Killian JA, de Kruijff B. Membrane-spanning peptides induce phospholipid flop: a model for phospholipid translocation across the inner membrane of E. coli. Biochemistry. 2001;40:10500–10506. doi: 10.1021/bi010627+. [DOI] [PubMed] [Google Scholar]
- Kol MA, van Laak ANC, Rijkers DTS, Killian JA, de Kroon AIPM, de Kruijff B. Phospholipid flop induced by transmembrane peptides in model membranes is modulated by lipid composition. Biochemistry. 2003;42:231–237. doi: 10.1021/bi0268403. [DOI] [PubMed] [Google Scholar]
- Krishnakumar SS, London E. Effect of sequence hydrophobicity and bilayer width upon the minimum length required for the formation of transmembrane helices in membranes. J Mol Biol. 2007;374:671–687. doi: 10.1016/j.jmb.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolt-Marticorena C, Williams KA, Deber CM, Reithmeier RAF. Nonrandom distribution of amino-acids in the transmembrane segments of human type I single span membrane proteins. J Mol Biol. 1993;229:602–608. doi: 10.1006/jmbi.1993.1066. [DOI] [PubMed] [Google Scholar]
- Lemaitre V, de Planque MRR, Howes AP, Smith ME, Dupree R, Watts A. Solid-state 17O NMR as a probe for structural studies of proteins in biomembranes. J Am Chem Soc. 2004;126:15320–15321. doi: 10.1021/ja0473283. [DOI] [PubMed] [Google Scholar]
- Lew S, Caputo GA, London E. The effect of interactions involving ionizable residues flanking membrane-inserted hydrophobic helices upon helix-helix interaction. Biochemistry. 2003;42:10833–10842. doi: 10.1021/bi034929i. [DOI] [PubMed] [Google Scholar]
- Lewis RNAH, Zhang YP, Hodges RS, Subczynski WK, Kusumi A, Flach CR, Mendelsohn R, McElhaney RN. A polyalanine-based peptide cannot form a stable transmembrane alpha-helix in fully hydrated phospholipid bilayers. Biochemistry. 2001;40:12103–12111. doi: 10.1021/bi010555m. [DOI] [PubMed] [Google Scholar]
- Liu F, Lewis RNAH, Hodges RS, McElhaney RN. Effect of variations in the structure of a polyleucine-based alpha-helical transmembrane peptide on its interaction with phosphatidylcholine bilayers. Biochemistry. 2002;41:9197–9207. doi: 10.1021/bi025661i. [DOI] [PubMed] [Google Scholar]
- Mall S, Broadbridge R, Sharma RP, Lee AG, East JM. Effects of aromatic residues at the ends of transmembrane alpha-helices on helix interactions with lipid bilayers. Biochemistry. 2000;39:2071–2078. doi: 10.1021/bi992205u. [DOI] [PubMed] [Google Scholar]
- Mansilla MC, de Mendoza D. The Bacillus subtilis desaturase: a model to understand phospholipid modification and temperature sensing. Arch Microbiol. 2005;183:229–235. doi: 10.1007/s00203-005-0759-8. [DOI] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003;12:403–411. doi: 10.1110/ps.0211503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D, Jost M, Peggion C, Toniolo C. Lipid chain-length dependence for incorporation of alamethicin in membranes: electron paramagnetic resonance studies on TOAC-spin labeled analogs. Biophys J. 2007;92:4002–4011. doi: 10.1529/biophysj.107.104026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EE, Zoonens M, Engelman DM. Dynamic helix interactions in transmembrane signaling. Cell. 2006;127:447–450. doi: 10.1016/j.cell.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Smith GA, Dabbeni-sala F, Johannsson A, Galante YM, Bisson R. Bilayer thickness and enzymatic activity in the mitochondrial cytochrome c oxidase and ATPase complex. FEBS Lett. 1982;144:145–148. doi: 10.1016/0014-5793(82)80588-7. [DOI] [PubMed] [Google Scholar]
- Morein S, Killian JA, Sperotto MM. Characterization of the thermotropic behavior and lateral organization of lipid-peptide mixtures by a combined experimental and theoretical approach: effects of hydrophobic mismatch and role of flanking residues. Biophys J. 2002;82:1405–1417. doi: 10.1016/S0006-3495(02)75495-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow MR, Huschilt JC, Davis JH. Simultaneous modeling of phase and calorimetric behavior in an amphiphilic peptide phospholipid model membrane. Biochemistry. 1985;24:5396–5406. doi: 10.1021/bi00341a018. [DOI] [PubMed] [Google Scholar]
- Nazarov PV, Koehorst RBM, Vos WL, Apanasovich VV, Hemminga MA. FRET study of membrane proteins: determination of the tilt and orientation of the N-terminal domain of M13 major coat protein. Biophys J. 2007;92:1296–1305. doi: 10.1529/biophysj.106.095026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen RD, Che K, Gelb MH, Robinson BH. A ruler for determining the position of proteins in membranes. J Am Chem Soc. 2005;127:6430–6442. doi: 10.1021/ja042782s. [DOI] [PubMed] [Google Scholar]
- Opella SJ, Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, Montal M. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat Struct Biol. 1999;6:374–379. doi: 10.1038/7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdirekcan S, Rijkers DTS, Liskamp RMJ, Killian JA. Influence of flanking residues on tilt and rotation angles of transmembrane peptides in lipid bilayers. A solid-state 2H NMR study. Biochemistry. 2005;44:1004–1012. doi: 10.1021/bi0481242. [DOI] [PubMed] [Google Scholar]
- Özdirekcan S, Etchebest C, Killian JA, Fuchs PFJ. On the orientation of a designed transmembrane peptide: toward the right tilt angle? J Am Chem Soc. 2007;129:15174–15181. doi: 10.1021/ja073784q. [DOI] [PubMed] [Google Scholar]
- Park SH, Opella SJ. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J Mol Biol. 2005;350:310–318. doi: 10.1016/j.jmb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Persson S, Killian JA, Lindblom G. Molecular ordering of interfacially localized tryptophan analogs in ester- and ether-lipid bilayers studied by 2H-NMR. Biophys J. 1998;75:1365–1371. doi: 10.1016/s0006-3495(98)74054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache HI, Zuckerman DM, Sachs JN, Killian JA, Koeppe RE, II, Woolf TB. Hydrophobic matching mechanism investigated by molecular dynamics simulations. Langmuir. 2002;18:1340–1351. [Google Scholar]
- Radzwill N, Gerwert K, Steinhoff HJ. Time-resolved detection of transient movement of helices F and G in doubly spin-labeled bacteriorhodopsin. Biophys J. 2001;80:2856–2866. doi: 10.1016/S0006-3495(01)76252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Lew S, Wang J, London E. Control of the transmembrane orientation and interhelical interactions within membranes by hydrophobic helix length. Biochemistry. 1999;38:5905–5912. doi: 10.1021/bi982942a. [DOI] [PubMed] [Google Scholar]
- Rinia HA, Boots JWP, Rijkers DTS, Kik RA, Snel MME, Demel RA, Killian JA, van der Eerden JPJM, de Kruijff B. Domain formation in phosphatidylcholine bilayers containing transmembrane peptides: specific effects of flanking residues. Biochemistry. 2002;41:2814–2824. doi: 10.1021/bi011796x. [DOI] [PubMed] [Google Scholar]
- Seshadri K, Garemyr R, Wallin E, von Heijne G, Elofsson A. Architecture of beta-barrel membrane proteins: analysis of trimeric porins. Protein Sci. 1998;7:2026–2032. doi: 10.1002/pro.5560070919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah K, London E. Effect of lipid composition on the topography of membrane-associated hydrophobic helices: stabilization of transmembrane topography by anionic lipids. J Mol Biol. 2008;379:704–718. doi: 10.1016/j.jmb.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel DP, Cherezov V, Greathouse DV, Koeppe RE, II, Killian JA, Caffrey M. Transmembrane peptides stabilize inverted cubic phases in a biphasic length-dependent manner: implications for protein-induced membrane fusion. Biophys J. 2006;90:200–211. doi: 10.1529/biophysj.105.070466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparr E, Ash WL, Nazarov PV, Rijkers DTS, Hemminga MA, Tieleman DP, Killian JA. Self-association of transmembrane alpha-helices in model membranes—importance of helix orientation and role of hydrophobic mismatch. J Biol Chem. 2005;280:39324–39331. doi: 10.1074/jbc.M502810200. [DOI] [PubMed] [Google Scholar]
- Sparr E, Ganchev DN, Snel MME, Ridder ANJA, Kroon-Batenburg LMJ, Chupin V, Rijkers DTS, Killian JA, de Kruijff B. Molecular organization in striated domains induced by transmembrane alpha-helical peptides in dipalmitoyl phosphatidylcholine bilayers. Biochemistry. 2005;44:2–10. doi: 10.1021/bi048047a. [DOI] [PubMed] [Google Scholar]
- Strandberg E, Killian JA. Snorkeling of lysine side chains in transmembrane helices: how easy can it get? FEBS Lett. 2003;544:69–73. doi: 10.1016/s0014-5793(03)00475-7. [DOI] [PubMed] [Google Scholar]
- Strandberg E, Morein S, Rijkers DTS, Liskamp RMJ, van der Wel PCA, Killian JA. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry. 2002;41:7190–7198. doi: 10.1021/bi012047i. [DOI] [PubMed] [Google Scholar]
- Strandberg E, Özdirekcan S, Rijkers DTS, van der Wel PCA, Koeppe RE, II, Liskamp RMJ, Killian JA. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H solid-state NMR. Biophys J. 2004;86:3709–3721. doi: 10.1529/biophysj.103.035402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandberg E, Esteban-Martin S, Salgado J, Ulrich AS. Orientation and dynamics of peptides in membranes calculated from 2H-NMR data. Biophys J. 2009;96:3223–3232. doi: 10.1016/j.bpj.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subczynski WK, Lewis RNAH, McElhaney RN, Hodges RS, Hyde JS, Kusumi A. Molecular organization and dynamics of 1-palmitoyl-2-oleoylphosphatidylcholine bilayers containing a transmembrane alpha-helical peptide. Biochemistry. 1998;37:3156–3164. doi: 10.1021/bi972148+. [DOI] [PubMed] [Google Scholar]
- Sukharev S, Anishkin A. Mechanosensitive channels: what can we learn from ‘simple’ model systems? Trends Neurosci. 2004;27:345–351. doi: 10.1016/j.tins.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Traaseth NJ, Buffy JJ, Zamoon J, Veglia G. Structural dynamics and topology of phospholamban in oriented lipid bilayers using multidimensional solid-state NMR. Biochemistry. 2006;45:13827–13834. doi: 10.1021/bi0607610. [DOI] [PubMed] [Google Scholar]
- Ulmschneider MB, Sansom MSP. Amino acid distributions in integral membrane protein structures. Biochim Biophys Acta. 2001;1512:1–14. doi: 10.1016/s0005-2736(01)00299-1. [DOI] [PubMed] [Google Scholar]
- Ulmschneider MB, Sansom MS, Di Nola A. Properties of integral membrane protein structures: derivation of an implicit membrane potential. Proteins Struct Funct Bioinf. 2005;59:252–265. doi: 10.1002/prot.20334. [DOI] [PubMed] [Google Scholar]
- Ulmschneider JP, Smith JC, Doux JPF, Killian JA, Ulmschneider MB. Peptide partitioning and folding into lipid bilayers. J Chem Theor Comp. 2009;5:2202–2205. doi: 10.1021/ct900256k. [DOI] [PubMed] [Google Scholar]
- van der Wel PCA, Strandberg E, Killian JA, Koeppe RE., II Geometry and intrinsic tilt of a tryptophan-anchored transmembrane alpha-helix determined by 2H NMR. Biophys J. 2002;83:1479–1488. doi: 10.1016/S0006-3495(02)73918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel PCA, Reed ND, Greathouse DV, Koeppe RE., II Orientation and motion of tryptophan interfacial anchors in membrane-spanning peptides. Biochemistry. 2007;46:7514–7524. doi: 10.1021/bi700082v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duyl BY, Rijkers DTS, de Kruijff B, Killian JA. Influence of hydrophobic mismatch and palmitoylation on the association of transmembrane alpha-helical peptides with detergent-resistant membranes. FEBS Lett. 2002;523:79–84. doi: 10.1016/s0014-5793(02)02939-3. [DOI] [PubMed] [Google Scholar]
- van Duyl BY, Meeldijk H, Verkleij AJ, Rijkers DTS, Chupin V, de Kruijff B, Killian JA. A synergistic effect between cholesterol and tryptophan-flanked transmembrane helices modulates membrane curvature. Biochemistry. 2005;44:4526–4532. doi: 10.1021/bi047937n. [DOI] [PubMed] [Google Scholar]
- Vidal A, McIntosh TJ. Transbilayer peptide sorting between raft and nonraft bilayers: comparisons of detergent extraction and confocal microscopy. Biophys J. 2005;89:1102–1108. doi: 10.1529/biophysj.105.062380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel A, Scheidt HA, Huster D. The distribution of lipid attached spin probes in bilayers: application to membrane protein topology. Biophys J. 2003;85:1691–1701. doi: 10.1016/S0006-3495(03)74599-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vostrikov VV, Grant CV, Daily AE, Opella SJ, Koeppe RE., II Comparison of “polarization inversion with spin exchange at magic angle” and “geometric analysis of labeled alanines” methods for transmembrane helix alignment. J Am Chem Soc. 2008;130:12584–12585. doi: 10.1021/ja803734k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb RJ, East JM, Sharma RP, Lee AG. Hydrophobic mismatch and the incorporation of peptides into lipid bilayers: a possible mechanism for retention in the Golgi. Biochemistry. 1998;37:673–679. doi: 10.1021/bi972441+. [DOI] [PubMed] [Google Scholar]
- Wegener AA, Chizhov I, Engelhard M, Steinhoff HJ. Time-resolved detection of transient movement of helix F in spin-labelled pharaonis sensory rhodopsin II. J Mol Biol. 2000;301:881–891. doi: 10.1006/jmbi.2000.4008. [DOI] [PubMed] [Google Scholar]
- Wegener AA, Klare JP, Engelhard M, Steinhoff HJ. Structural insights into the early steps of receptor-transducer signal transfer in archaeal phototaxis. EMBO J. 2001;20:5312–5319. doi: 10.1093/emboj/20.19.5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss TM, van der Wel PCA, Killian JA, Koeppe RE, II, Huang HW. Hydrophobic mismatch between helices and lipid bilayers. Biophys J. 2003;84:379–385. doi: 10.1016/S0006-3495(03)74858-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson IM, Alvis SJ, East JM, Lee AG. The potassium channel KcsA and its interaction with the lipid bilayer. Cell Mol Life Sci. 2003;60:1581–1590. doi: 10.1007/s00018-003-3172-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Beevers AJ, Kukol A, Dupree R, Smith ME. Solid-state 17O NMR spectroscopy of a phospholemman transmembrane domain protein: implications for the limits of detecting dilute 17O sites in biomaterials. Solid State Nucl Magn Reson. 2008;33:72–75. doi: 10.1016/j.ssnmr.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Yano Y, Takemoto T, Kobayashi S, Yasui H, Sakurai H, Ohashi W, Niwa M, Futaki S, Sugiura Y, Matsuzaki K. Topological stability and self-association of a completely hydrophobic model transmembrane helix in lipid bilayers. Biochemistry. 2002;41:3073–3080. doi: 10.1021/bi011161y. [DOI] [PubMed] [Google Scholar]
- Yau WM, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- Yeagle PL, Bennett M, Lemaître V, Watts A. Transmembrane helices of membrane proteins may flex to satisfy hydrophobic mismatch. Biochim Biophys Acta. 2007;1768:530–537. doi: 10.1016/j.bbamem.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Lewis RNAH, Hodges RS, McElhaney RN. FTIR spectroscopic studies of the conformation and amide hydrogen exchange of a peptide model of the hydrophobic transmembrane alpha-helixes of membrane proteins. Biochemistry. 1992;1992:11572–11578. doi: 10.1021/bi00161a041. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Lewis RNAH, Hodges RS, McElhaney RN. Interaction of a peptide model of a hydrophobic transmembrane alpha-helical segment of a membrane protein with phosphatidylcholine bilayers: differential scanning calorimetric and FTIR spectroscopic studies. Biochemistry. 1992;31:11579–11588. doi: 10.1021/bi00161a042. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Lewis RNAH, Henry GD, Sykes BD, Hodges RS, McElhaney RN. Peptide models of helical hydrophobic transmembrane segments of membrane proteins. 1. Studies of the conformation, intrabilayer orientation, and amide hydrogen exchangeability of ac-K2-(LA)12–K2-amide. Biochemistry. 1995;34:2348–2361. doi: 10.1021/bi00007a031. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Lewis RNAH, Hodges RS, McElhaney RN. Peptide models of helical hydrophobic transmembrane segments of membrane proteins. 2. Differential scanning calorimetric and FTIR spectroscopic studies of the interaction of ac-K2-(LA)12–K2-amide with phosphatidylcholine bilayers. Biochemistry. 1995;34:2362–2371. doi: 10.1021/bi00007a032. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Lewis RNAH, Hodges RS, McElhaney RN. Interaction of a peptide model of a hydrophobic transmembrane alpha-helical segment of a membrane protein with phosphatidylethanolamine bilayers: differential scanning calorimetric and fourier transform infrared spectroscopic studies. Biophys J. 1995;68:847–857. doi: 10.1016/S0006-3495(95)80261-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Lewis RNAH, Hodges RS, McElhaney RN. Peptide models of the helical hydrophobic transmembrane segments of membrane proteins: interactions of acetyl-K2-(LA)12–K2-amide with phosphatidylethanolamine bilayer membranes. Biochemistry. 2001;40:474–482. doi: 10.1021/bi002170u. [DOI] [PubMed] [Google Scholar]