

Abstract
Membrane proteins exhibit different affinities for different lipid species, and protein–lipid selectivity regulates the membrane composition in close proximity to the protein, playing an important role in the formation of nanoscale membrane heterogeneities. The sensitivity of Förster resonance energy transfer (FRET) for distances of 10 Å up to 100 Å is particularly useful to retrieve information on the relative distribution of proteins and lipids in the range over which protein–lipid selectivity is expected to influence membrane composition. Several FRET-based methods applied to the quantification of protein–lipid selectivity are described herein, and different formalisms applied to the analysis of FRET data for particular geometries of donor–acceptor distribution are critically assessed.
Keywords: FRET, Protein–lipid interactions, Membrane proteins, Fluorescence
Introduction
Preferential interactions of proteins with selected lipids are able to drive enrichment of the bilayer around the protein in these lipids and impoverishment in others, creating local heterogeneities that can potentially extend to several lipid shells around the protein. Some superficial membrane proteins demonstrate specific binding to some lipid classes, a phenomenon that can control protein recruitment to the membrane and activate signaling cascades (Czech 2000). In addition, transmembrane proteins display differential interactions with lipids of different acyl-chain lengths due to packing constraints in the lipid–protein hydrophobic interface, which have a significant effect on the activity of several proteins. Membrane proteins also have been shown to present binding sites for lipids in hydrophobic pockets away from the protein–lipid interface, and binding of specific lipids to such sites is essential for activity in several cases (Lee 2003).
Interactions between membrane proteins and lipids have been generally addressed using electron spin resonance (ESR) (Marsh and Horváth 1998). This technique can discriminate between immobilized lipids (near the protein interface) and mobile lipids in the bulk, and has been the main approach in these type of studies. Fluorescence static or collisional quenching methods (London and Feigenson 1981; Everett et al. 1986; O’Keeffe et al. 2000; Williamson et al. 2002) can also provide a similar structural type of information. These techniques are able to probe the lipid environment in direct contact with the protein, but are insensitive to the presence of lipids displaced from the protein–lipid interface. On the other hand, Förster resonance energy transfer (FRET) is sensitive to distances up to 100 Å, and like all fluorescence methodologies offers the maximum sensitivity among spectroscopic techniques. In this way, and as will be shown in this review, it is an excellent tool for the study of protein–lipid interactions.
FRET between two molecules (donor and acceptor) exhibits a sixth-power dependence on the distance between them. The propensity for FRET to occur for a given donor–acceptor molecular pair is generally described by the Förster radius (R 0), the distance from the acceptor at which the probability of donor deactivation by energy transfer is 50%. Typical values for R 0 lie between 20 and 60 Å. Due to the high sensitivity of FRET to changes in distances around R 0, it is possible, through the use of chemical labeling or intrinsic fluorophores, to directly measure the binding of lipids to proteins, or protein-mediated enrichment of particular lipids in the membrane.
In this review we focus on the problem of quantifying the interaction of membrane proteins with biologically relevant lipid molecules making use of FRET methodologies. In the field of protein–lipid interactions, FRET has also been extensively used to characterize and quantify binding of lipids to soluble proteins (Petrescu et al. 2001; Gadella and Wirtz 1991; Dansen et al. 1999) and partition of membrane proteins to lipid membranes (Romoser et al. 1996; Corbin et al. 2004) and particular lipid phases (Wang et al. 1988), most notably to liquid-ordered (raft-like) phases (Zacharias et al. 2002). However, we will not discuss these particular FRET applications.
Apart from the use of FRET as a molecular ruler (Stryer and Haugland 1967), the information retrieved from its application in biological sciences is often qualitative, neglecting the full potential of FRET in the study of molecular interactions. This is particularly true for studies of interactions in biomembranes, due to the additional level of complexity introduced by the possibility of energy transfer to multiple acceptors, including nonbound molecules. In fact, as a consequence of partition of donors and acceptors to the lipid bilayer, the concentration of acceptors around each donor increases dramatically, resulting in a nonzero probability of energy transfer to many nonbound, nearby acceptors. However, the availability of a large amount of precise information on the properties of biomembranes and fluorescent lipid probes, as well as accurate numerical and analytical solutions to the problem of FRET in planar distributions of acceptors, surmounts these problems. We will critically present and discuss some of these formalisms, as well as the simplifications required for their application and the respective usefulness to the problem of quantifying protein–lipid selectivity.
FRET donors and acceptors
The aromatic amino acids tyrosine and tryptophan have been extensively employed as donors in FRET experiments (Pap et al. 1993; Antollini et al. 1996; Antollini and Barrantes 1998, 2002; Levi et al. 2000; Bonini et al. 2002; Levi et al. 2003; Fernandes et al. 2006a, 2007) (Table 1). Tryptophan fluorescence is more commonly used due to its superior extinction coefficient and quantum yield compared with tyrosine (Lakowicz 2006). Separation of tryptophan and tyrosine fluorescence is straightforward by selective tryptophan excitation at 295–310 nm (Lakowicz 2006). Although the use of intrinsic fluorophores can be advantageous, it often results in low sensitivity. In cases where higher sensitivity is desired, chemical derivatization of proteins with brighter donors or acceptors is an alternative. Some fluorophores commonly used in protein–lipid FRET studies are included in Table 1.
Table 1.
List of commonly used FRET pairs for study of protein–lipid interaction
| Donor fluorophore | Acceptor fluorophore | Förster radius (Å) |
|---|---|---|
| Trpa | Pyrene | 21–27 |
| Trpb | Laurdan | 29 |
| Trpc | cis/trans-Parinaric acid | 18l |
| Trpd | Cholestatrienol | 20m |
| Trpe | Dansyl | 24 |
| Trpf | Dehydroergosterol | 16 |
| Pyreneg | Rhodopsin retinal group | 34–35 |
| BODIPYh | Texas-Red | – |
| DCIAi | NBD | 39 |
| AEDANSj | NBD | 30–29n |
| GFPk | BODIPY | 50 (for EGFP)o |
BODIPY 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene, DCIA 7-diethylamino-3-((4′-(iodoacetyl)amino)phenyl)-4-methylcoumarin, NBD 7-nitro-2-1,3-benzoxadiazol-4-yl, AEDANS 5-((2-aminoethyl)amino)naphthalene-1-sulfonic acid, GFP green fluorescent protein, EGFP enhanced green fluorescent protein
aPap et al. (1993), Bonini et al. (2002), Levi et al. (2000)
bAntollini et al. (1996), Antollini and Barrantes (1998, 2002), Levi et al. (2003)
cNarayanaswami and McNamee (1993), Poveda et al. (2002)
dAlbert et al. (1996)
eWang et al. (1988)
fRaghuraman and Chattopadhyay (2004)
gPolozova and Litman (2000)
hGambhir et al. (2004), Nomikos et al. (2007)
iFernandes et al. (2004)
jFernandes et al. (2008)
kHughes et al. (2002)
lNemecz et al. (1991)
mHolt et al. (2008)
nFairclough and Cantor (1978)
oIlien et al. (2003)
Several fluorescently tagged lipids have been shown to mimic properties of their natural analogues and were extremely helpful in elucidating several problems in lipid trafficking and sorting (Maier et al. 2002). Nevertheless, for each different lipid probe, studies must be carried out to characterize its properties and confirm this mimicry. Problems observed with some cholesterol fluorescent derivatives are a good illustration of the need for careful selection of lipid probes (Loura et al. 2001). One important concern to bear in mind when studying protein–lipid selectivity is to avoid changing the particular lipid property we are interested in through the incorporation of the fluorescent tag, i.e., in case of studies on protein selectivity for particular acyl-chain characteristics (length or unsaturation), the fluorescent label should be inserted in the hydrophilic section of the molecule, and vice versa (Fernandes et al. 2004).
Measuring FRET efficiencies
FRET efficiencies are generally measured through quantification of the extent of donor fluorescence quenching introduced by the presence of the FRET acceptor. Therefore, two measurements are required (with and without acceptor). The acceptor species is not necessarily fluorescent. If it is fluorescent, FRET efficiencies can also be calculated from the sensitized fluorescence emission, through measurement of acceptor fluorescence after donor excitation. However, this approach is slightly more complex, as several corrections must be introduced into the analysis of the fluorescence signal, namely direct acceptor fluorescence upon donor excitation, and the differences in extinction coefficients between donors and acceptors must be accounted for. Generally, FRET efficiencies obtained through the monitoring of donor fluorescence quenching are more accurate (Lakowicz 2006).
Transient-state fluorescent data presents several advantages relative to steady-state FRET measurements. Apart from the additional kinetic information that can be related to particular donor–acceptor distributions (Fernandes et al. 2006b), time-dependent data is much less prone to artifacts such as light scattering, inner-filter effects, bleaching or contamination with autofluorescence, and errors are significantly minimized. Minimization of errors might be crucial when selective enrichment of lipids around the protein is not able to cause dramatic differences in FRET efficiencies due to the use of donor–acceptor pairs with high Förster radius (R 0) (Fernandes et al. 2004). Fluorescent decays can then be integrated and the data analyzed as intensities if necessary.
Energy migration (or homotransfer) between donors is also a matter of concern when attempting to measure FRET efficiencies in lipid bilayers. When donor energy migration takes place, the concentration of this species should be kept to a minimum in the experiment as this phenomenon induces an increase in heterotransfer efficiencies (Subramaniam et al. 2003), i.e., the excitation travels from an initially excited donor that is too far away from the acceptor, and when a shorter distance is reached heterotransfer takes place.
Another problem that must be avoided when choosing fluorescent probes for quantitative FRET measurements is diffusion of the donor molecule during its excited-state lifetime. Fluorophores presenting very long lifetimes will not be static after excitation, and this excited-state diffusion will lead to a bias towards higher FRET efficiencies as the probability of the donor residing at a closer distance from the acceptor during its lifetime is increased relative to the static situation (Thomas et al. 1978). Diffusion coefficients of lipids in lipid bilayers are generally on the order of 10–8 cm2/s, while protein diffusion coefficients can be even lower; in that case, for donor lifetimes higher than a few hundred nanoseconds, FRET can no longer be described by the static regime (Thomas et al. 1978). Most commonly used fluorophores present lifetimes lower than this limit and FRET data obtained from donors and acceptors in lipid bilayers are associated with a static system.
Qualitative FRET studies
Qualitative FRET studies for protein–lipid binding proved to be useful in the characterization and discrimination of lipid binding sites of the acetylcholine receptor (AChR). Antollini and Barrantes (1998) measured the effect of adding different lipids on the FRET efficiencies from the tryptophan residues of this protein to the fatty-acid derivative, 6-lauroyl-2-dimethylaminonaphthalene (laurdan). When the 18:1 fatty acid was added, a maximum decrease in FRET of 60% was observed. Addition of cholesterol led to a 35% decrease in FRET, while 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) was slightly less effective (25%). Adding cholesterol and DOPC together led to a decrease in FRET identical to that observed with the 18:1 fatty acid, suggesting the presence of two types of lipid binding sites in the protein, inaccessible to either cholesterol or phospholipids, while always accessible to fatty acids (Antollini and Barrantes 1998)
Also for AChR, Narayanaswami and McNamee (1993) showed through FRET measurements from the Trp residues to cis- and trans-parinaric acid that the receptor accommodated preferentially the cis-parinaric acid in its vicinity, suggesting preferential interaction of the protein with less rigid lipids. In another study using the fluorescent sterol cholestatrienol as the FRET acceptor from tryptophan residues in rhodopsin, it was shown that the protein exhibited greater affinity for cholesterol than for ergosterol, since the former was much more effective in decreasing the efficiency of FRET to cholestatrienol, likely through competition for a common binding site in the protein (Albert et al. 1996).
Hughes et al. (2002), using fluorescence lifetime imaging (FLIM), showed that an isoform of phospholipase D (PLD1b) tagged with GFP in HeLa cells, which is responsible for the conversion of phosphocholines (PC) to phosphatidic acid (PA), was susceptible to FRET from BODIPY-labeled PC molecules, but BODIPY-phosphoethanolamine (PE) was not able to induce a change in GFP lifetimes, suggesting preferential interaction of PLD1b with PC lipids. Catalytic mutants of PLD1b did not experience FRET from BODIPY-PC, reinforcing this conclusion.
Qualitative FRET studies can also be the only correct option when highly complex protein–lipid structures are formed such as in protein-mediated membrane fusion. In these cases, unless highly organized structures are formed, such as the lamellar structures induced by lysozyme (Coutinho et al. 2008), no analytical solution for FRET is adequate and recovery of very detailed information from FRET is impossible. That was the case for the interaction of the N-terminal amphipathic alpha-helix of a Bin–Amphiphysin–Rvs (BAR) domain with lipid bilayers (Fernandes et al. 2008).
Absence of noninteracting species in FRET analysis
Several strategies are available to avoid the problem of energy transfer to (or from) multiple and noninteracting lipids. Frequently, lipids bind to nonannular binding sites in the protein with very high affinity (Lee 2003; Hunte 2005). In these cases, it is desirable to limit the amount of donor/acceptor lipids in the medium to increase the sensitivity of the assay and, if donor/acceptor concentration is low enough, the probability of energy transfer to nonbound acceptors can be neglected (Fig. 1a). This dilution can be achieved by using an excess of nonlabeled lipid (Gambhir et al. 2004; Nomikos et al. 2007) or of detergent micelles (Pap et al. 1993; Levi et al. 2000, 2003). Nevertheless, the application of this strategy for FRET studies with protein and lipid species is restricted, as the difference between association constants obtained for different lipid species does not often surpass one order of magnitude, especially for annular binding sites (Lee 2003; Marsh 2008).
Fig. 1.
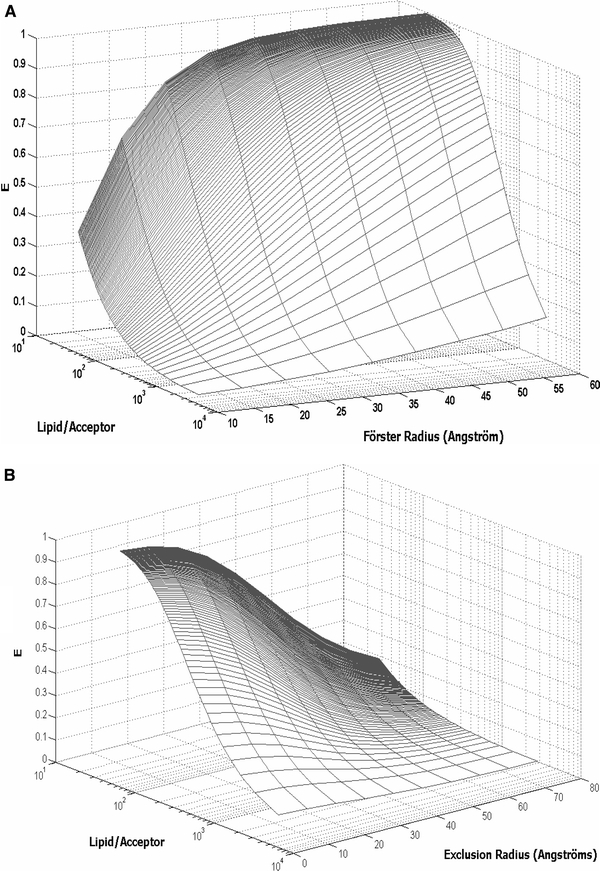
Simulations for energy transfer efficiencies between donors and acceptors in lipid bilayers (Eqn. 6). Donors and acceptors are assumed to be located at similar depths in the bilayers (l = 6 Å), and energy transfer is calculated for a single plane of acceptors (energy transfer to the second leaflet is not accounted for); 72 Å2 was taken as the area occupied by each lipid. a Dependence of energy transfer efficiencies on R 0 and acceptor concentration (no acceptor exclusion area is accounted for). b Dependence of energy transfer efficiencies on the radius of the circular area around the donor from which acceptors are excluded (a value of 40 Å was used for R 0)
This strategy was applied to the study of phosphatidyl-d-myo-inositol 4,5-bisphosphate (PIP2) sequestration by the basic effector domain of myristoylated alanine-rich C kinase substrate (MARCKS) (Gambhir et al. 2004) and by a basic peptide from phospholipase C-ξ (Nomikos et al. 2007). Controls were performed to ensure that the concentration of BODIPY-labeled PIP2 was low enough that the probability of occurrence of donor–acceptor pairs in the absence of specific interactions was negligible. Using 1% N-(6-tetramethylrhodaminethiocarbamoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (TRITC-PE) (from total lipid content), a lipid species that is not sequestrated by the protein (and therefore whose distribution around the protein is random), energy transfer to the Texas Red labeled MARCKS peptide was still substantial (Gambhir et al. 2004). The Förster radii (R 0) for FRET to Texas Red from rhodamine or BODIPY are very similar, and comparable energy transfer efficiencies are expected for random distribution of the two labeled lipids. A concentration of 0.1% was then chosen for the BODIPY-labeled lipid, as at this concentration no FRET was detected using the TRITC-PE probe. In these conditions, all FRET observed from BODIPY-PIP2 to the Texas Red labeled peptide could be assigned to sequestration of PIP2 lipids by the peptide. In order to calculate a dissociation constant for this interaction, the value for the energy transfer efficiency in the protein–lipid complex was assumed to be 100%, i.e., complete quenching of the donor-labeled lipid after interaction of the protein. This approximation will result in underestimation of binding, and the dissociation constant retrieved can be considered as an upper limit for its real value (Gambhir et al. 2004; Nomikos et al. 2007).
Protein dimensions are often comparable to or larger than typical R 0 values (20–60 Å). The presence of a transmembrane protein in the membrane leads to an exclusion of lipids from a significant fraction of the area around the protein axis. In that case, FRET efficiencies between protein and lipid species are smaller than would be expected from donors and acceptors with negligible size, especially for donor/acceptor pairs presenting low R 0 (Fig. 1b). If the protein is modeled as a cylinder, this lipid exclusion effect can be described by a protein exclusion radius (R e). In several protein–lipid selectivity studies it is assumed that, due to large R e values, FRET efficiencies to or from unbound lipid species can be neglected when R 0 < R e (Pap et al. 1993; Antollini et al. 1996; Levi et al. 2000, 2003; Bonini et al. 2002). Using this rationale, the properties of protein–lipid interaction can be extracted from energy transfer data even at high concentrations of the interacting species, allowing for quantification of binding affinities in the absence of strong and specific lipid binding sites, which is the case of lipid binding to annular sites in the protein. However, as is clearly visible from Fig. 1b, significant energy transfer will still occur to acceptors located past the first shell of lipids around the protein, unless R 0 ≪ R e and acceptor concentration is kept low. Moreover, if a fraction of the donor or acceptor groups in the protein are located in the periphery of the molecule, this lipid exclusion effect becomes less significant.
Pap et al. (1993) presented a formalism to study lipid binding to protein kinase C when R 0 was small enough that only donor–acceptor bound species were expected to contribute to FRET. The authors measured Trp fluorescence quenching due to FRET to different pyrene-labeled phospholipids in mixed micelles and determined binding constants by assuming that binding of a pyrene lipid to a site in the protein contributed additively to the observed donor quenching. Levi et al. (2000, 2003) employed a similar reasoning to quantify selectivity constants for the lipid-plasma membrane calcium pump interaction. According to this model, donor quenching due to FRET can be described by the following relationship:
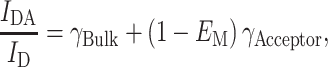 |
1 |
where I DA and I D are fluorescence intensities measured in the presence and absence of acceptors (respectively), γ Bulk is the fraction of binding sites occupied by bulk (nonfluorescent) lipid, and γ Acceptor is the fraction of binding sites occupied by acceptors. Application of this model allows for the direct recovery of the fraction of binding sites occupied by acceptors, and of relative binding constants if acceptor and bulk lipid concentrations are known. Despite the attractive simplicity and elegance of this approach, the application of the formalism described above in this form is inaccurate as FRET efficiencies are not additive (i.e., E values for a donor with two identical acceptors is not twice the value of E when only one acceptor is present) and Eq. 1 is only approximately valid when very small FRET efficiencies (<10%) are obtained when all available sites in the protein surface are occupied by acceptors, which is highly unlikely even for R 0 ≪ R e.
Gutiérrez-Merino derived a more general FRET procedure based on analytical expressions for the average rate of energy transfer, <k T>, presented in two papers (Gutiérrez-Merino 1981a, 1981b). The first one establishes the general approach and its application to phase separation in binary phospholipid mixtures (Gutiérrez-Merino 1981a); it is specifically addressed in the article in this issue about FRET study of membrane lateral heterogeneity (Fernandes et al. 2009, companion paper). The concept was then applied to the problem of random/nonrandom distribution and aggregation state of membrane proteins [assuming FRET from a donor in the protein to phospholipids labeled with acceptor (Gutiérrez-Merino 1981b)]. The model describes the relationship between <k T> and the geometrical and thermodynamic parameters describing the aggregation of proteins. As described in the companion paper (Fernandes et al. 2009, companion paper), again this simple and elegant treatment is limited by the major assumption of considering FRET only to neighboring acceptor molecules, and also by the indirect relationship between the calculated <k T> and the experimental FRET efficiency. Notwithstanding, this analytical approach was later extended to calculate the dependence of <k T> and E on the position of the donor in the membrane protein with respect to the plane of acceptors (Gutierrez-Merino et al. 1987) and later still applied to the study of protein–lipid selectivity (Antollini et al. 1996). In the latter study, the lipid annulus around the oligomeric transmembrane AChR, in liposomes prepared from the endogenous lipids present in the AChR-rich membrane (PC being the predominant phospholipid), was studied using FRET from the protein Trp residues to laurdan. The donor Trp residues were modeled as lying in a ring inside the perimeter of the transmembrane portion of AChR, and two topological parameters, H (transverse distance between the donor and acceptor chromophores) and r (minimum donor–acceptor distance) are considered. Figure 2 illustrates the topology considered for this system.
Fig. 2.
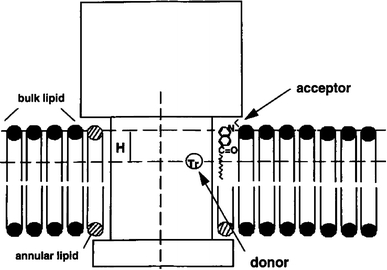
Topographical relationship between the membrane-bound AChR, surrounding lipid molecules, and laurdan in cross-sectional representation. H is the distance between the plane of the donor and that of the acceptor. Reprinted with permission from Antollini et al. (1996); copyright 1996, Biophysical Society
In addition to the geometrical parameters, one crucial introduction to the model was that of an interaction parameter K r, which represents the apparent dissociation constant of laurdan for the lipid belt region, that is, the ratio of the dissociation constant of laurdan over that of the unlabeled lipid. K r < 1 implies preferential location of laurdan in the lipid belt region, whereas K r > 1 denotes laurdan’s exclusion from this region. From spectral data, the Förster radius for the tryptophan–laurdan pair was found to be R 0 = 2.9 nm. Fixing this value, H was allowed to vary between 0 and 1 nm based on previous results, and it was found that within this range of H values an exclusion distance of r = 1.4 ± 0.1 nm provided an adequate fit to the data, together with K r ≅ 1. Because the data are not analyzed globally (i.e., the parameters are not optimized simultaneously), it could of course be argued whether a different set of (H, r, K r) values might fit the data equally well. Naturally, as with all formalisms for lipid selectivity, this method works best if good estimates are known for H and r, and the sole optimizing parameter is K r. The same methodology was applied to a fluorescent derivative of sphingomyelin (N-[10-(1-pyrenyl)decanoyl]sphingomyelin), which was found to exhibit moderate selectivity for the annular region, with K r ≅ 0.55 (Bonini et al. 2002). Upon sphingomyelinase digestion of the membrane, FRET efficiency increased by about 50%, indicating that the resulting pyrenyl-ceramide species has higher affinity for the protein than the parental sphingomyelin derivative. On the other hand, a FRET analysis using the described methodology was instrumental in a multitechnique (infrared and fluorescence spectroscopies, differential scanning calorimetry) study which showed that AChR causes formation of specific phosphatidic-acid-rich lipid domains, which include the protein (Poveda et al. 2002). Egg phosphatidylcholine (egg-PC)/1,2-dimirystoyl-sn-glycero-phosphatidic acid (DMPA)/cholesterol (2:1:1) vesicles were prepared in absence of protein, and no evidence of domain formation was verified in these conditions. However, reconstitution of AChR in this lipid mixture leads to enhanced selectivity of the protein for gel-phase probe trans-parinaric acid (K r = 0.6 ± 0.1), denoting enrichment of gel-phase lipid DMPA in the vicinity of AChR. This is a specific effect for phosphatidic acid, which is not observed for other phospholipid classes (phosphatidylcholine, phosphatidylserine, and phosphatidylglycerol). In particular, when the FRET experiment was carried out replacing DMPA with 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), uniform acceptor distribution was inferred (K r = 1.0 ± 0.1, negative control). On the other hand, in the absence of protein, all the phospholipid classes, including phosphatidic acid, exhibit ideal mixing behavior. Because PA and cholesterol have been implicated in functional modulation of the reconstituted AChR, the authors raised the hypothesis that such a specific modulatory role could be mediated by domain segregation of these lipid classes.
FRET analysis including contributions from bulk acceptors
The model introduced by Fernandes et al. (2004) is not subject to the limitations mentioned for the formalisms described above. Through the use of detailed information concerning the position of donor and acceptor labels, the authors showed that it was possible to recover accurate selectivity constants for lipid binding in the annular shell of lipids around the M13 major coat protein (MCP). The problem of lipid selectivity by the M13 MCP has been extensively addressed through electron spin resonance (ESR) techniques (Wolfs et al. 1989; Peelen et al. 1992), and selectivity constants were recovered for the interaction of the protein with different phospholipids. M13 MCP is composed of a single transmembrane segment and as such is expected to immobilize around 10–12 phospholipids in its immediate vicinity or annular binding sites (Marsh and Horváth 1998; Cornea et al. 1997). However, due to MCP aggregation, only five annular binding sites were identified by protein molecule when using ESR (Wolfs et al. 1989). Optimization of the protein purification procedure allowed monomeric MCP to be obtained after membrane incorporation (Spruijt et al. 1989) but failed to produce long-lived immobilization of the annular shell of lipids (Sanders et al. 1992). This type of immobilization is essential in order to distinguish the ESR spectra of immobilized spin-labeled lipids from the labeled lipids probing the bulk lipid (unaffected by the protein).
The FRET model described by Fernandes et al. (2004) assumed that two types of acceptor were present, one located in the annular shell around the protein (annular binding sites) and the other outside of this area. The distribution of the latter population was random and unaffected by the presence of the protein. The donor fluorescence decay curve has FRET contributions from both populations:
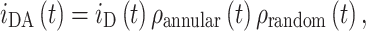 |
2 |
where i DA(t) and i D(t) are the donor decays in the presence and absence of acceptors, and ρ annular(t) and ρ random(t) are the FRET contributions from annular lipids and lipids outside the annular shell. A hexagonal geometry was assumed for the protein–lipid packing, and 12 identical annular binding sites were introduced into the model (six per each lipid monolayer). The probability of each of these sites to be occupied by an acceptor (μ) depends on the acceptor molar fraction and on a relative selectivity constant (K S) which reports the relative affinity of the labeled and unlabeled phospholipid.
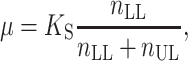 |
3 |
where n LL is the concentration of labeled lipid, and n UL is the concentration of unlabeled lipid. M13 MCP was labeled with a donor fluorophore (coumarin) in an amino-acid residue located in the center of the transmembrane region of the protein (Spruijt et al. 1996). As such, the plane of acceptors from each lipid leaflet is expected to be at the same distance from the donor, eliminating the requirement for distinction of each leaflet in the FRET formalisms. Using a binomial distribution the probability of each occupation number (0–12 sites occupied simultaneously by labeled lipid) is calculated, and the FRET contribution arising from energy transfer to annular lipids is given by
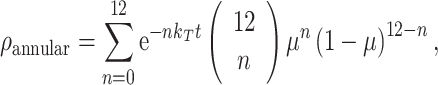 |
4 |
where k T is the energy transfer rate for an acceptor located in an annular site:
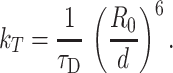 |
5 |
τD is the donor lifetime in the absence of acceptor, and d is the distance from the donor (in the protein) to the acceptor inside the annular lipid shell. The value of d was calculated from published data on the position of the acceptor fluorophores (NBD) in the labeled phospholipids incorporated in lipid bilayers (Abrams and London 1993; Màzeres et al. 1996). This detailed evaluation of the contribution of the different number of acceptors bound to each protein is of great importance, as it allows extension of the applicability of the FRET modeling to significant values of E, unlike FRET models previously applied to the problem of protein–lipid selectivity, as commented above.
The FRET contribution from energy transfer to acceptors randomly distributed outside the annular region in two different planes at the same distance (l) to the donor plane (from the center of the bilayer to both leaflets) is given by Davenport et al. (1985) as
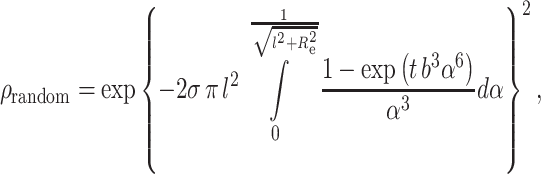 |
6 |
where b = (R 0/l)2τ−1/3D, σ is the acceptor density in each leaflet, and R e is now the distance between the protein axis and the second lipid shell (exclusion distance for bulk acceptors) (Fig. 3). l is the unlabeled lipid bilayer thickness, and the value assigned for R e was 16 Å, assuming radii of 5 and 4.5 Å for the protein and phospholipid, respectively. The value σ must be corrected for the presence of labeled lipid in the annular region, which therefore is not part of the randomly distributed acceptors pool. After i DA is calculated from Eqs. 2–6, E is readily obtained through numerical integration of the simulated decay and, during fitting of this model to the experimental data, the only unknown value is K S.
Fig. 3.
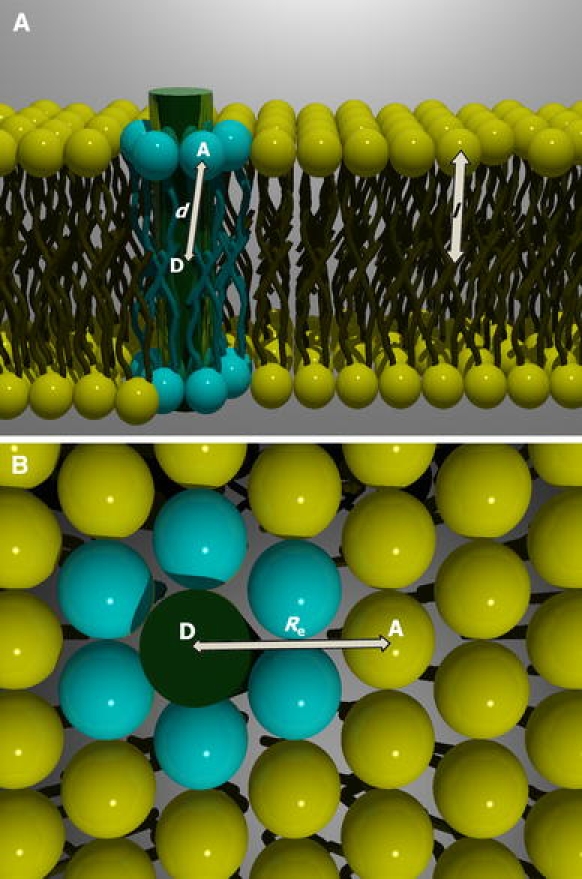
Molecular model for the FRET analysis according to the model of Fernandes et al. (2004): (a) side view and (b) top view. Protein–lipid organization presents a hexagonal geometry. Donor fluorophore from the mutant protein is located in the center of the bilayer, whereas the acceptors are distributed in the bilayer surface. Two different environments are available for the labeled lipids (acceptors): the annular shell surrounding the protein and the bulk lipid. Energy transfer to acceptors in direct contact with the protein has a rate coefficient dependent on the distance between donor and annular acceptor (Eq. 5). Energy transfer toward acceptors in the bulk lipid is given by Eq. 6
The model was applied to the analysis of two sets of experiments. In the first set, the same headgroup-labeled lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)-NBD, displaying perfect hydrophobic matching to the M13 MCP, was added to proteoliposomes presenting different lipid composition. The donor DCIA-labeled M13 MCP was shown to exhibit higher selectivity for the acceptor (NBD)-labeled lipid when it was incorporated in lipids presenting either positive or negative hydrophobic mismatch toward the protein, reflecting enrichment of hydrophobically matching lipid in the annular shell around the protein due to the energetic cost of hydrophobic mismatch stress (results are shown in Table 2). In a second set of experiments, the selectivity of the protein for different phospholipid headgroups was assessed using different phospholipid classes (PE, PC, phosphoglycerol- (PG), phosphoserine- (PS), and PA) labeled with NBD at one of the acyl chains (1-oleoyl-2-[12-[(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]dodecanoyl] (18:1-(12:0-NBD)- PE, PC, PG, PS, and PA)). These experiments were performed under conditions of hydrophobic matching in order to avoid influence of hydrophobic mismatch stress on the NBD–lipid distribution around the protein. The relative association constants [K S/K S(PC labeled lipid)] obtained from fitting of this model to the experimental data (Fig. 4; Table 2) were almost identical to the values obtained by Peelen et al. (1992) with ESR and the aggregated form of the protein.
Table 2.
Labeled phospholipids relative association constants toward M13 major coat protein (adapted from Fernandes et al. 2004)
| Labeled phospholipid | Bilayer composition | K S | K S/K S(PC)a |
|---|---|---|---|
| DOPE-NBD | di(18:1)PC | 1.4 | – |
| DOPE-NBD | di(22:1)PC | 2.1 | – |
| DOPE-NBD | di(14:1)PC | 2.9 | – |
| (18:1-(12:0-NBD))-PE | di(18:1)PC | 2.0 | 1.0 |
| (18:1-(12:0-NBD))-PC | di(18:1)PC | 2.0 | 1.0 |
| (18:1-(12:0-NBD))-PG | di(18:1)PC | 2.3 | 1.1 |
| (18:1-(12:0-NBD))-PS | di(18:1)PC | 2.7 | 1.3 |
| (18:1-(12:0-NBD))-PA | di(18:1)PC | 3.0 | 1.5 |
a K S(PC) is the relative association constant of (18:1-(12:0-NBD))-PC
Fig. 4.
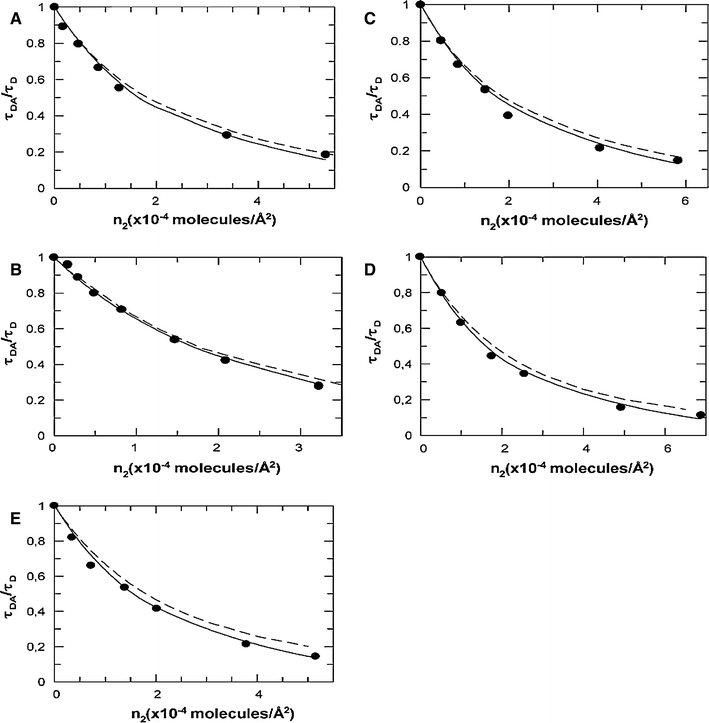
DCIA-labeled M13 MCP fluorescence quenching by energy transfer acceptor (18:1-(12:0-NBD)-PX), where “X” stands for the different headgroup structures, in pure bilayers of di18:1-PC. n 2 is the acceptor density for the whole bilayer. Continuous line shows theoretical simulations obtained from the annular model for protein–lipid interaction using the fitted K S; Dashed line shows simulations for random distribution of acceptors (K S = 1.0). (a) PC-labeled phospholipid (fitted K S = 2.0); (b) PE-labeled phospholipid (fitted K S = 2.0); (c) PG-labeled phospholipid (fitted K S = 2.3); (d) PS-labeled phospholipid (fitted K S = 2.7); and (e) PA-labeled phospholipid (fitted K S = 3). Reprinted with permission from Fernandes et al. (2004); copyright 2004, Biophysical Society
One important difference between the ESR and FRET techniques is that the latter is not dependent on lipid immobilization and therefore is not restricted to lipids adjacent to a given protein molecule. Not only labeled lipids in the first shell of lipids will be potential acceptors to a donor-labeled integral protein; acceptors in the other lipid shells surrounding the protein will also contribute to the final result. For that reason, this study apparently confirms the hypothesis that single transmembrane domains are only able to influence lipid composition in the first shell of lipids around it. It is likely, however, that larger proteins are able to induce the formation of lipid enrichment at larger distances from the protein.
Due to the low selectivity character of the protein–annular lipid interaction, and the large R 0 of the donor–acceptor FRET pair used in this study (39 Å), the contribution of the noninteracting acceptors (outside the annular lipid shell) to the energy transfer efficiencies dominates considerably over the contribution of protein bound lipids. This leads to decreased sensitivity of the donor quenching profile for different selectivity constants (Fig. 5), and as such nearly error-free measurements are required in order to recover accurate selectivity constants. This shortcoming can be minimized by selecting a donor–acceptor pair with a smaller Förster radius, which will result in a larger contribution of annular shell acceptors to the donor quenching profiles and a larger tolerance of the model to uncertainty in the data (Fig. 4).
Fig. 5.
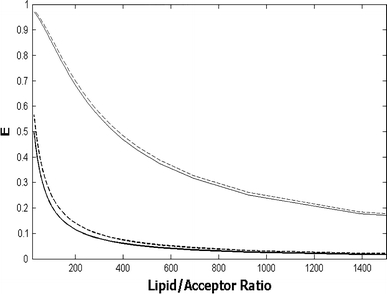
Simulations for energy transfer efficiencies between donors and acceptors in lipid bilayers using the model from Fernandes et al. (2004) (Eqs. 3–6). Donors and acceptors are assumed to be located in different planes in the bilayers and energy transfer to the two lipid bilayer leaflets at the same distance (l = 18.9 Å) is considered. A value of 16 Å was used for R e and the distance from the donor in the protein to acceptors in the annular lipid shell was assumed to be 9.5 Å. Dark and light curves correspond to results obtained with Förster radii of 20 and 40 Å, respectively. Full lines correspond to results from simulations assuming no preferential interaction with the acceptor (K S = 1) and dashed lines correspond to K S = 2
More recently, Capeta et al. (2006) proposed a model for FRET with acceptor enrichment surrounding donors, inspired by the distribution function used by Rotman and Hartmann (1988) in three-dimensional crystals, in that, around each donor, three regions are considered: (1) an exclusion region closest to the donor (R < R 1), reflecting the radius of the protein; (2) the annular region (R 1 < R < R 2), for which there is an increased probability of finding acceptors, characterized by a parameter B; and (3) a region for which the acceptor concentration is equal to the overall value (R > R 2). The resulting local acceptor concentration is a step function of the donor–acceptor distance (Fig. 6).
Fig. 6.
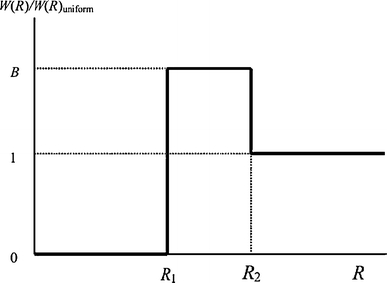
Plot of the ratio between the acceptor distribution function considered in the model of Capeta et al. (2006) and that for uniform distribution, showing the parameters R 1, R 2, and B. Reprinted with permission from Capeta et al. (2006); copyright 2006, Springer
The analytical law for the donor decay in the presence of acceptor is given as a function of five dimensionless variables: a dimensionless average acceptor concentration γ, the reduced time λ, the reduced interplanar spacing β w, the reduced exclusion distance β 1, and the relative enrichment factor for the acceptor in the annular region, B (see original reference for derivation). Numerical integration of the decay equation over time was carried out, in order to calculate numerical FRET efficiency curves (γ, E) for chosen (B, β w, β 1) triads. Empirical five-parameter functions of the form
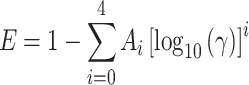 |
7 |
could fit well to the numerical results, and best-fit parameter values are given for multiple (B, β w, β 1) sets. As an illustration, the AChR/trans-parinaric acid FRET data of Poveda et al. (2002) (see “Absence of noninteracting species in FRET analysis”) are analyzed with this formalism, and moderate enrichment of the acceptor (≈25% relative to uniform distribution) is inferred (Fig. 7).
Fig. 7.
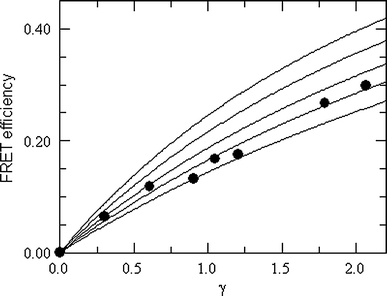
Approximate theoretical FRET efficiency curves for reduced donor–acceptor transverse distance β w = 0.375 and reduced exclusion distance β 1 = 1.25 (relative enrichment factor B, from bottom to top: B = 1.05, B = 1.25, B = 1.5, B = 2, B = 3; see original paper for approximation details) and experimental results of FRET between AChR tryptophan and trans-parinaric acid in egg-PC/DMPA/cholesterol (2:1:1) vesicles (Poveda et al. 2002). The best fit is obtained for B = 1.25. Reprinted with permission from Capeta et al. (2006); copyright 2006, Springer
This formalism presents several advantages and disadvantages. An advantage, relative to the formalism of Fernandes et al. (2004) as originally derived, is that it is directly applicable to proteins of any given size (as long as the protein can be approximated by a cylinder, with the donor located on its axis). The most important disadvantage probably resides in the complex analytical theory underlying the model, which forces most users to use the user-friendly empirical fitting functions given in the paper. Inevitably, the fitting functions’ parameters can only be presented for discrete values of the model parameters. Therefore, approximations have to be made in most cases (the only alternative would be to avoid the fitting functions and to calculate the FRET efficiency by numerical computation of the exact solution).
The model described in the previous section is reminiscent of an older treatment derived by Polozova and Litman (2000) for analysis of FRET between phospholipids labeled with pyrene and the rhodopsin retinal group in model membranes with different lipid compositions. To account for selectivity of rhodopsin for pyrene-labeled lipids, the authors developed a cluster model in which the protein (acceptor) is surrounded by a homogeneous cluster of lipids presenting a different donor lipid concentration than the bulk membrane. In the model, these clusters are circular and characterized by a cluster radius (R c). An important difference from the FRET viewpoint, as will be commented below, is that the donor is now the lipid species whereas the acceptor is the protein.
The equations presented by the authors for the time-resolved donor fluorescence emission are the following:
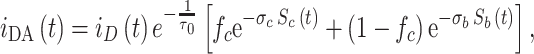 |
8 |
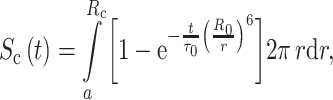 |
9 |
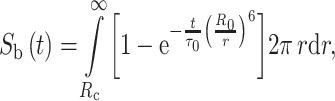 |
10 |
where f c is the fraction of donor lipid in clusters, and σ c and σ b are the superficial concentrations of the acceptor protein inside and outside of the cluster. σ c is a constant that depends only on the size of the cluster as it is assumed that only one protein molecule resides in each protein–lipid cluster, independently of the protein–lipid ratio. This model was fitted to the experimental FRET efficiencies. Due to correlation between parameters (acceptor concentration and R c), it was necessary to measure with two different lipid probes expected to present different protein affinities (di22:6-PE-Pyr and di16:0-PE-Pyr), and fit the model simultaneously to the two sets of data. When using proteoliposomes with a lipid composition of di22:6-PC/di16:0-PC/cholesterol (7:3:3) or di22:6-PC/16:0,22:6-PC/cholesterol (3:7:3), no protein–lipid selectivity was required to fit the model to the data. However, when the concentration of di:16:0-PC was increased at the expense of di22:6-PC (di22:6-PC/di16:0-PC/cholesterol (3:7:3)), FRET efficiencies from di22:6-PE-Pyr increased while those from di16:0-PE-Pyr decreased, and best fits were achieved using R c = 35 Å and a concentration of unsaturated lipids inside the clusters three times higher than that of saturated lipids. Since the authors assume the distance of closest protein–lipid approach (a in Eq. 9) to be 18 Å, the recovery of such a large R c implies enrichment of lipid around the protein beyond the first shell of lipids. Cholesterol was crucial in the enrichment of unsaturated lipids around the protein and no lipid segregation was observed in the absence of protein, indicating that segregation of unsaturated lipids is due to mutual affinity with the protein, rather than partition of the protein into preformed lipid domains.
While there is no doubt that the results fully support the authors’ conclusions, the quantitative validity of the formalism seems doubtful. Equations 8–10 are based on the derivation by Fung and Stryer (1978) of the kinetics of FRET in planar geometry. The latter authors originally derived the expression of the FRET S term (used here in Eqs. 9–10) by accounting for the interactions between one given donor molecule (all donors being equivalent) and all acceptors surrounding it. This was done by introducing the distribution function of donor–acceptor distances for uniform two-dimensional geometry. Now consider a circular cluster with an acceptor molecule in the center and donors surrounding it. It is clear that the donors within the cluster are not equivalent (unless they are all located at the same distance to the acceptor, which is generally not the case, and obviously not valid for the multiple annular lipid layers reported), and there is no circular symmetry or uniform distribution of acceptors surrounding each of them. Therefore, the FRET term for donors belonging to a cluster (Eq. 9) is not correct, and the effect of this inaccuracy in the results presented for R c and the ratio of concentration of lipid species in the clusters is not clear.
Concluding remarks
The sensitivity of FRET over distances of typically 10–100 Å is particularly suitable for studies of protein interactions, as protein dimensions are often in this range. The extraordinary sensitivity of fluorescent techniques was responsible for the widespread use of FRET for detection of protein interactions, a trend that was highly accentuated with the use of fluorescent proteins and genetic engineering (Tsien 1998). However, the application of FRET methodologies to the study of association between membrane components has been generally limited to high-affinity interactions, such as protein–protein interactions or those typically observed for membrane proteins and specifically bound lipids. For FRET studies of lower-affinity interactions between membrane proteins and lipids it is very likely that noninteracting species contribute to the FRET process, increasing the complexity of FRET data analysis. Quantitative studies in these conditions will either rely on meeting specific conditions that allow for disregard of energy transfer to noninteracting acceptors or on an accurate description of this FRET contribution.
The different examples described in this review illustrate the usefulness of FRET in the quantification of protein–lipid selectivity. FRET’s lack of dependence on direct contact also allows for probing of specific lipid enrichment away from the first shell of lipids around the protein, a property absent from other techniques used for protein–lipid selectivity studies which depend on protein–lipid contact. The extension of lipid enrichment around the protein to several lipid shells is likely to occur for large proteins and has the potential to induce significant heterogeneity in membrane composition and to originate membrane compartmentalization, a phenomenon that is likely to be of high biological relevance. These factors, together with the general advantages of fluorescence measurements, reinforce the value of FRET studies in the characterization of protein–lipid interactions.
Acknowledgments
Financial support for this work was provided by Fundação para a Ciência e Tecnologia (Portugal).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
The more you see: spectroscopy in molecular biophysics.
References
- Abrams FS, London E. Extension of the parallax analysis of membrane penetration depth to the polar region of model membranes: use of fluorescence quenching by a spin-label attached to the phospholipids polar headgroup. Biochemistry. 1993;32:10826–10831. doi: 10.1021/bi00091a038. [DOI] [PubMed] [Google Scholar]
- Albert AD, Young JE, Yeagle PL. Rhodopsin–cholesterol interactions in bovine rod outer segment disk membranes. Biochim Biophys Acta. 1996;1285:47–55. doi: 10.1016/S0005-2736(96)00145-9. [DOI] [PubMed] [Google Scholar]
- Antollini SS, Barrantes FJ. Disclosure of discrete sites for phospholipid and sterols at the protein–lipid interface in native acetylcholine receptor-rich membrane. Biochemistry. 1998;37:16653–16662. doi: 10.1021/bi9808215. [DOI] [PubMed] [Google Scholar]
- Antollini SS, Barrantes FJ. Unique effects of different fatty acid species on the physical properties of the Torpedo acetylcholine receptor membrane. J Biol Chem. 2002;277:1249–1254. doi: 10.1074/jbc.M106618200. [DOI] [PubMed] [Google Scholar]
- Antollini SS, Soto MA, Bonini de Romanelli I, Gutiérrez-Merino C, Sotomayor P, Barrantes FJ. Physical state of bulk and protein-associated lipid in nicotinic acetylcholine receptor-rich membrane studied by laurdan generalized polarization and fluorescence energy transfer. Biophys J. 1996;70:1275–1284. doi: 10.1016/S0006-3495(96)79684-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini IC, Antollini SS, Gutiérrez-Merino C, Barrantes FJ. Sphingomyelin composition and physical asymmetries in native acetylcholine receptor-rich membranes. Eur Biophys J. 2002;31:417–427. doi: 10.1007/s00249-002-0230-6. [DOI] [PubMed] [Google Scholar]
- Capeta RC, Poveda JA, Loura LMS. Non-uniform membrane probe distribution in resonance energy transfer: application to protein–lipid selectivity. J Fluoresc. 2006;16:161–172. doi: 10.1007/s10895-005-0036-x. [DOI] [PubMed] [Google Scholar]
- Corbin JA, Dirkx RA, Falke JJ. GRP1 Pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid. Biochemistry. 2004;43:16161–16173. doi: 10.1021/bi049017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornea RL, Jones LR, Autry JM, Thomas DD. Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry. 1997;36:2960–2967. doi: 10.1021/bi961955q. [DOI] [PubMed] [Google Scholar]
- Coutinho A, Loura LMS, Fedorov A, Prieto M. Pinched multilamellar structure of aggregates of lysozyme and phosphatidylserine-containing membranes revealed by FRET. Biophys J. 2008;95:4726–4736. doi: 10.1529/biophysj.108.134379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–606. doi: 10.1016/S0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- Dansen TB, Westerman J, Wouters FS, Wanders RJA, Van Hoek A, Gadella TWJ, Jr, Wirtz KWA. High-affinity binding of very-long-chain fatty acyl-CoA esters to the peroxisomal non-specific lipid-transfer protein (sterol carrier protein-2) Biochem J. 1999;339:193–199. doi: 10.1042/0264-6021:3390193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport L, Dale RE, Bisby RH, Cundall RB. Transverse location of the fluorescent probe 1, 6-diphenyl-1, 3, 5-hexatriene in model lipid bilayer membrane systems by resonance energy transfer. Biochemistry. 1985;24:4097–4108. doi: 10.1021/bi00336a044. [DOI] [PubMed] [Google Scholar]
- Everett J, Zlotnick A, Tennyson J, Holloway PW. Fluorescence quenching of cytochrome b5 in vesicles with an asymmetric transbilayer distribution of brominated phosphatidylcholine. J Biol Chem. 1986;261:6725–6729. [PubMed] [Google Scholar]
- Fairclough RH, Cantor CC. The use of singlet–singlet energy transfer to study macromolecular assemblies. Meth Enzymol. 1978;48:347–379. doi: 10.1016/S0076-6879(78)48019-X. [DOI] [PubMed] [Google Scholar]
- Fernandes F, Loura LM, Koehorst R, Spruijt RB, Hemminga MA, Fedorov A, Prieto M. Quantification of protein–lipid selectivity using FRET: application to the M13 major coat protein. Biophys J. 2004;87:344–352. doi: 10.1529/biophysj.104.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes F, Loura LMS, Koehorst R, Dixon N, Kee TP, Prieto M, Hemminga M. Binding assays of inhibitors towards selected V-ATPase domains. Biochim Biophys Acta. 2006;1758:1777–1786. doi: 10.1016/j.bbamem.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Fernandes F, Loura LMS, Fedorov A, Prieto M. Absence of clustering of phosphatidylinositol-(4,5)-bisphosphate in fluid phosphatidylcholine. J Lipid Res. 2006;47:1521–1525. doi: 10.1194/jlr.M600121-JLR200. [DOI] [PubMed] [Google Scholar]
- Fernandes F, Neves P, Gameiro P, Loura LMS, Prieto M. Ciprofloxacin interactions with bacterial protein OmpF: modelling of FRET from a multi-tryptophan protein trimer. Biochim Biophys Acta. 2007;1768:2822–2830. doi: 10.1016/j.bbamem.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Fernandes F, Loura LMS, Chichón FJ, Carrascosa JL, Fedorov A, Prieto M. Role of helix 0 of the N-BAR domain in membrane curvature generation. Biophys J. 2008;94:3065–3073. doi: 10.1529/biophysj.107.113118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes F, Prieto M, Loura LMS (2009) Membrane microheterogeneity: Förster resonance energy transfer characterization of lateral membrane domains (companion paper) [DOI] [PubMed]
- Fung BK, Stryer L. Surface density determination in membranes by fluorescence energy transfer. Biochemistry. 1978;17:5241–5248. doi: 10.1021/bi00617a025. [DOI] [PubMed] [Google Scholar]
- Gadella TWJ, Jr, Wirtz KWA. The low-affinity lipid binding site of the non-specific lipid transfer protein. Implications for its mode of action. Biochim Biophys Acta. 1991;1070:237–245. doi: 10.1016/0005-2736(91)90170-D. [DOI] [PubMed] [Google Scholar]
- Gambhir A, Hangyás-Mihályné G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J. 2004;86:2188–2207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Merino C. Quantitation of the Förster energy transfer for two-dimensional systems. I. Lateral phase separation in unilamellar vesicles formed by binary phospholipid mixtures. Biophys Chem. 1981;14:247–257. doi: 10.1016/0301-4622(81)85025-9. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Merino C. Quantitation of the Förster energy transfer for two-dimensional systems. II. Protein distribution and aggregation state in biological membranes. Biophys Chem. 1981;14:259–266. doi: 10.1016/0301-4622(81)85026-0. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Merino C, Munkonge F, Mata AM, East JM, Levinson BL, Napier RM, Lee AG. The position of the ATP binding site on the (Ca2+ + Mg2+)-ATPase. Biochim Biophys Acta. 1987;897:207–216. doi: 10.1016/0005-2736(87)90417-2. [DOI] [PubMed] [Google Scholar]
- Holt A, de Almeida RFM, Nyholm TKM, Loura LMS, Daily AE, Staffhorst RWHM, Rijkers DTS, Koeppe RE, Prieto M, Killian JA. Is there a preferential interaction between cholesterol and tryptophan residues in membrane proteins? Biochemistry. 2008;47:2638–2649. doi: 10.1021/bi702235k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes WE, Larijani B, Parker PJ. Detecting protein–phospholipid interactions. J Biol Chem. 2002;277:22974–22979. doi: 10.1074/jbc.M201391200. [DOI] [PubMed] [Google Scholar]
- Hunte C. Specific protein–lipid interactions in membrane proteins. Biochem Soc Trans. 2005;33:938–942. doi: 10.1042/BST20050938. [DOI] [PubMed] [Google Scholar]
- Ilien B, Franchet C, Bernard P, Morisset S, Weill CO, Bourguignon J-J, Hibert M, Galzi JL. Fluorescence resonance energy transfer to probe human M1 muscarinic receptor structure and drug binding properties. J Neurochem. 2003;85:768–778. doi: 10.1046/j.1471-4159.2003.01717.x. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of fluorescence spectroscopy. New York: Kluwer; 2006. [Google Scholar]
- Lee AG. Lipid–protein interactions in biological membranes: a structural perspective. Biochim Biophys Acta. 2003;1612:1–40. doi: 10.1016/S0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- Levi V, Rossi JPFC, Echarte MM, Castello PR, Flecha FLG. Thermal stability of the plasma membrane calcium pump. Quantitative analysis of its dependence on lipid–protein interactions. J Membrane Biol. 2000;173:215–225. doi: 10.1007/s002320001021. [DOI] [PubMed] [Google Scholar]
- Levi V, Rossi JPFC, Castello PR, Flecha FLG. Quantitative analysis of membrane protein–amphiphile interactions using resonance energy transfer. Anal Biochem. 2003;317:171–179. doi: 10.1016/S0003-2697(03)00132-5. [DOI] [PubMed] [Google Scholar]
- London E, Feigenson GW. Fluorescence quenching in model membranes. 1. Characterization of quenching by a spinlabeled phospholipid. Biochemistry. 1981;20:1932–1938. doi: 10.1021/bi00510a032. [DOI] [PubMed] [Google Scholar]
- Loura LMS, Fedorov A, Prieto M. Exclusion of a cholesterol analog from the cholesterol-rich phase in model membranes. Biochim Biophys Acta. 2001;1511:236–243. doi: 10.1016/S0005-2736(01)00269-3. [DOI] [PubMed] [Google Scholar]
- Maier O, Oberle V, Hoekstra D. Fluorescent lipid probes: some properties and applications (a review) Chem Phys Lipids. 2002;116:3–18. doi: 10.1016/S0009-3084(02)00017-8. [DOI] [PubMed] [Google Scholar]
- Marsh D. Protein modulation of lipids, and vice versa, in membranes. Biochim Biophys Acta. 2008;1778:1545–1575. doi: 10.1016/j.bbamem.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Marsh D, Horváth LI. Structure, dynamics and composition of the lipid–protein interface. Perspectives from spin-labelling. Biochim Biophys Acta. 1998;1376:267–296. doi: 10.1016/s0304-4157(98)00009-4. [DOI] [PubMed] [Google Scholar]
- Màzeres S, Schram V, Tocanne J-F, Lopez A. 7-Nitrobenz-2-oxa-1, 3-diazole-4-yl-labeled phospholipids in lipid membranes: differences in fluorescence behavior. Biophys J. 1996;71:327–335. doi: 10.1016/S0006-3495(96)79228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanaswami V, McNamee MG. Protein–lipid interactions and Torpedo californica nicotinic acetylcholine receptor function. 2. Membrane fluidity and ligand-mediated alteration in the accessibility of gamma subunit cysteine residues to cholesterol. Biochemistry. 1993;32:12420–12427. doi: 10.1021/bi00097a021. [DOI] [PubMed] [Google Scholar]
- Nemecz G, Jefferson JR, Schroederg F. Polyene fatty acid interactions with recombinant intestinal and liver fatty acid-binding proteins. J Biol Chem. 1991;266:17112–17123. [PubMed] [Google Scholar]
- Nomikos M, Mulgrew-Nesbitt A, Pallavi P, Mihalyne G, Zaitseva I, Swann K, Lai FA, Murray D, McLaughlin S. Binding of phosphoinositide-specific phospholipase C-ξ (PLC-ξ) to phospholipid membranes. J Biol Chem. 2007;282:16644–16653. doi: 10.1074/jbc.M701072200. [DOI] [PubMed] [Google Scholar]
- O’Keeffe AH, East JM, Lee AG. Selectivity in lipid binding to the bacterial outer membrane protein OmpF. Biophys J. 2000;79:2066–2074. doi: 10.1016/S0006-3495(00)76454-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap EHW, Bastiaens PIH, Borst JW, van den Berg PAW, van Hoek A, Snoek GT, Wirtz KWA, Visser AJWG. Quantitation of the interaction of protein kinase C with diacylglycerol and phosphoinositides by time-resolved detection of resonance energy transfer. Biochemistry. 1993;32:13310–13317. doi: 10.1021/bi00211a044. [DOI] [PubMed] [Google Scholar]
- Peelen SJCJ, Sanders JC, Hemminga MA, Marsh D. Stoichiometry, selectivity and exchange dynamics of lipid–protein interaction with bacteriophage M13 coat protein studied by spin label electron spin resonance. Effects of protein secondary structure. Biochemistry. 1992;31:2670–2677. doi: 10.1021/bi00125a006. [DOI] [PubMed] [Google Scholar]
- Petrescu AD, Gallegos AM, Okamura Y, Strauss JF, Schroeder F. Steroidogenic acute regulatory protein binds cholesterol and modulates mitochondrial membrane sterol domain dynamics. J Biol Chem. 2001;276:36970–36982. doi: 10.1074/jbc.M101939200. [DOI] [PubMed] [Google Scholar]
- Polozova A, Litman BJ. Cholesterol dependent recruitment of di22:6-PC by a G protein-coupled receptor into lateral domains. Biophys J. 2000;79:2632–2643. doi: 10.1016/S0006-3495(00)76502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda JA, Encinar JA, Fernández AM, Mateo CR, Ferragut JA, González-Ros JM. Segregation of phosphatidic acid-rich domains in reconstituted acetylcholine receptor membranes. Biochemistry. 2002;41:12253–12262. doi: 10.1021/bi0200099. [DOI] [PubMed] [Google Scholar]
- Raghuraman H, Chattopadhyay A. Interaction of melittin with membrane cholesterol: a fluorescence approach. Biophys J. 2004;87:2419–2432. doi: 10.1529/biophysj.104.043596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romoser V, Ball R, Smrcka AV. Phospholipase Cβ2 association with phospholipid interfaces assessed by fluorescence resonance energy transfer. J Biol Chem. 1996;271:25071–25078. doi: 10.1074/jbc.271.41.25071. [DOI] [PubMed] [Google Scholar]
- Rotman SR, Hartmann FX. Non-radiative energy transfer in non-uniform codoped laser crystals. Chem Phys Lett. 1988;152:311–318. doi: 10.1016/0009-2614(88)80098-8. [DOI] [Google Scholar]
- Sanders JC, Ottaviani MF, van Hoek A, Visser AJWG, Hemminga MA. A small protein in model membranes: a time-resolved fluorescence and ESR study on the interaction of M13 coat protein with lipid bilayers. Eur Biophys J. 1992;21:305–311. doi: 10.1007/BF00188342. [DOI] [PubMed] [Google Scholar]
- Spruijt RB, Wolfs CJAM, Hemminga MA. Aggregation-related conformational change of membrane-associated coat protein of bacteriophage M13. Biochemistry. 1989;28:9158–9165. doi: 10.1021/bi00449a030. [DOI] [PubMed] [Google Scholar]
- Spruijt RB, Wolfs CJAM, Verver JWG, Hemminga MA. Accessibility and environmental probing using cysteine residues introduced along the putative transmembrane domain of the major coat protein of bacteriophage M13. Biochemistry. 1996;35:10383–10391. doi: 10.1021/bi960410t. [DOI] [PubMed] [Google Scholar]
- Stryer L, Haugland RP. Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam V, Hanley QS, Clayton AHA, Jovin TM (2003) In: Marriott G, Parker I (eds) Methods in enzymology: biophotonics part A: photophysics of green and red fluorescent proteins: implications for quantitative microscopy Academic Press, San Diego [DOI] [PubMed]
- Thomas DD, Carlsen WF, Stryer L. Fluorescence energy transfer in the rapid-diffusion limit. Proc Natl Acad Sci USA. 1978;75:5746–5750. doi: 10.1073/pnas.75.12.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Wang S, Martin E, Cimino J, Omann G, Glaser M. Distribution of phospholipids around gramicidin and D-β-hydroxybutyrate dehydrogenase as measured by resonance energy transfer. Biochemistry. 1988;27:2033–2039. doi: 10.1021/bi00406a033. [DOI] [PubMed] [Google Scholar]
- Williamson IM, Alvis SJ, East JM, Lee AG. Interactions of phospholipids with the potassium channel KcsA. Biophys J. 2002;83:2026–2038. doi: 10.1016/S0006-3495(02)73964-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfs CLAM, Horváth LI, Marsh D, Watts A, Hemminga MA. Spin label ESR of bacteriophage M13 coat protein in mixed lipid bilayers. Characterization of molecular selectivity of charged phospholipids for the bacteriophage M13 coat protein in lipid bilayers. Biochemistry. 1989;28:995–1001. doi: 10.1021/bi00452a018. [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]