

Abstract
Homology modelling is normally the technique of choice when experimental structure data are not available but three-dimensional coordinates are needed, for example, to aid with detailed interpretation of results of spectroscopic studies. Herein, the state of the art of homology modelling will be described in the light of a series of recent developments, and an overview will be given of the problems and opportunities encountered in this field. The major topic, the accuracy and precision of homology models, will be discussed extensively due to its influence on the reliability of conclusions drawn from the combination of homology models and spectroscopic data. Three real-world examples will illustrate how both homology modelling and spectroscopy can be beneficial for (bio)medical research.
Keywords: Homology modelling, CASP, Spectroscopy
Introduction
Knowledge of the three-dimensional structure of proteins is a prerequisite for much research in fields as diverse as protein engineering, human genetics and drug design. Only two spectroscopic techniques, nuclear magnetic resonance (NMR) and X-ray, can produce high-resolution three-dimensional coordinates of macromolecules. Most other spectroscopic techniques either add information to such three-dimensional coordinates, or require these coordinates for detailed interpretation of their results. NMR and X-ray are very elaborate techniques, and worldwide only about 30 protein structures are solved per day. In the time needed to read the above abstract, on the other hand, about 50 sequences were determined (worldwide) and deposited in international, freely and easily accessible sequence databases. Consequently, the necessity for homology modelling is only increasing.
In its most elementary form, homology modelling involves calculating the structure of a protein for which only the sequence is known using its alignment with a homologous protein for which the structure is known.
The first homology modelling articles were published as early as the late 1970s (Greer 1980), and since then we have kept using and improving the same concepts described in those ground-breaking articles. The process starts with the detection of a suitable template; an alignment is produced; insertions, deletions and residue substitutions are performed; the model is optimized; and since the late 1990s there is consensus that structure validation is needed to detect the unavoidable errors in the final model.
In the early 1990s many homology models were built (and unfortunately also published; also in EBJ) just for the sake of modelling, but since the mid 1990s homology models are considered tools that can aid with the design of experiments and with the interpretation of their results, although occasionally things can still go very wrong in the literature.
G-protein-coupled receptors (GPCRs) are by far the most important target for the pharmaceutical industry, and due to the scarcity of GPCR structure data these are also the most frequently modelled molecules. Until August 2000 GPCR models were either built ab initio, or using the non-homologous bacteriorhodopsin as a homology modelling template. In August 2000 the structure of bovine opsin became available (Palczewski et al. 2000), and since then GPCR homology modelling has been a serious possibility. GPCR models built before that historical moment are better forgotten (Oliveira et al. 2004). Unfortunately, people kept building, using and publishing ab initio models even after the bovine opsin structure became available (Orry and Wallace 2000). Today, four GPCR structures are available that can be used as template; Fig. 1 shows the principal differences between the bovine opsin structure and the structure of the beta-2 adrenergic receptor (Cherezov et al. 2007), one of the recently resolved GPCR crystal structures. Long before August 2000 the importance of GPCR structure models for drug design triggered a very large number of spectroscopic experiments, focussed on a variety of aspects.
Fig. 1.
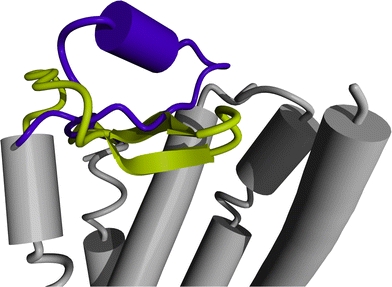
The most striking difference between the crystal structures of rhodopsin (PDBid 1f88, Palczewski et al. 2006) and the beta-2 adrenergic receptor (PDBid 2rh1, Cherezov et al. 2007) concerns the structure and location of the extracellular loop between helix IV and V. The loop IV–V in rhodopsin forms a β-sheet that folds into the binding pocket (yellow), whereas loop IV–V in the beta-2 adrenergic receptor forms an α-helix and extends towards the extracellular environment (purple)
The following series of short paragraphs illustrate the mutual relation between spectroscopy and homology modelling. Most examples are drawn from the GPCR research field. These examples are just illustrations and neither imply a judgment nor pretend completeness.
Exposed residue labelling
In two studies Davison and Findlay (1986a, b) identified residues that were exposed to the membrane environment or the retinal binding site, respectively, by labeling opsin with photoactivated l-azido-4-[125]iodobenzene. This study was carried out long before the first crystal structure of any GPCR became available, and determining which residues were labelled allowed Davison and Findlay to get a more complete picture of the three-dimensional organisation of the opsin molecule.
Site-directed spin labelling
The group of Khorana used site-directed spin labelling to analyse the structure and light-dependent changes of part of GPCRs. The electron paramagnetic resonance (EPR) spectrum of each spin-labelled mutant was analysed in terms of residue accessibility and mobility. In the article where they describe the region extending from helix VII to the palmitoylation sites in the rhodopsin molecule (Altenbach et al. 1999; Cai et al. 1999) they concluded that this region had extensive tertiary interactions. After Oliveira et al. (1999)—correctly—modelled this region as a helix that runs parallel to the membrane, the interpretation of the EPR data could be extended significantly, indicating how modelling can help interpret spectroscopic measurements.
Fluorescence resonance energy transfer
Turcatti et al. (1996) studied ligand–receptor interactions in the neurokinin-2 receptor (NK2) using fluorescence resonance energy transfer (FRET). A fluorescent unnatural amino acid was introduced at known sites into NK2. Inter-molecular distances were determined by measuring the fluorescence resonance energy transfer between the fluorescent unnatural amino acids and a fluorescently labelled NK2 heptapeptide antagonist. A similar approach was used to measure distances between the cholecystokinin receptor and a natural agonist (Harikumar et al. 2002) and between the secretin receptor and secretin analogues (Harikumar et al. 2007). Distances obtained were used as constraints to improve models for ligand–receptor interactions. The NK2 results were interpreted in terms of an obviously very poor bacteriorhodopsin structure. Looking at their results more than 10 years later, and with four GPCR structures at hand, we can see that they located the NK2 ligand largely at the correct place. This tells us how to trust or distrust the secretin FRET results and illustrates how spectroscopy can help improve modelling.
Fourier-transform infrared spectroscopy
Molecular models of rhodopsin based upon electron density projection maps that were constructed before the first crystal structure became available proposed a specific interaction between transmembrane (TM) helices III and V, which appeared to be mediated by amino acid residues Glu122 and His211 on TM helices III and V, respectively. Beck et al. (1998) used a combination of site-directed mutagenesis and Fourier-transform infrared spectroscopy (FTIR) to validate this hypothesis.
Small-angle scattering
Small-angle scattering has occasionally been used to assist with homology modelling of water-soluble proteins. Mascarenhas et al. (1992) studied crotoxin. The sequence identity with a template structure was generally high enough to build a good homology model, but the structure of one large loop remained highly ambiguous. Small-angle neutron scattering data corresponded much better with one of the two models made, thus solving this problem. Comoletti et al. (2007) studied the structure of the neuroligins and their complex with neurexin using small-angle neutron scattering and small-angle X-ray scattering. A high-resolution structure of the neurexin was available but no structure was available for the neuroligins. However, the neuroligins have significant sequence similarity with acetylcholine esterase, making it possible to build a homology model. Scattering from neuroligin constructs was similar to that previously obtained from acetylcholine esterase structures (Marchot et al. 1996), indicating that the homology model was valid.
Fluorescent ligands
Turcatti et al. (1995) studied the NK2 receptor using a number of fluorescent ligands, differing only in the length of the spacer between the fluorescent probe and the peptide ligand. By analyzing the different levels of fluorescence related to the spacer lengths they found that the binding pocket of the NK2 receptor was buried at a depth of 5–10 Å.
Modelling and spectroscopy of the M13 protein
The final example to illustrate the never-ending love story of modelling and spectroscopy was recently reviewed by the Hemminga group. In a beautiful review (Vos et al. 2009) entitled “From ‘I’ to ‘L’ and back again: the odyssey of membrane-bound M13 protein” they illustrate how a large series of spectroscopic techniques have been employed worldwide over a period of more than 20 years to continuously update the structure model of the M13 protein in its membrane-bound form. The (mainly spectroscopic) techniques used during this whole odyssey include NMR, site-specific and other solid-state NMR, X-ray fibre diffraction, cryo-electron microscopy, site-directed spin labelling and site-directed introduction of fluorescence probes, similar to that described above for the GPCR studies, fluorescence energy transfer, site-specific infrared dichroism etc. Throughout this odyssey of Hemminga and others, homology modelling and spectroscopy were both applied. This example nicely illustrates the importance of homology modelling for spectroscopy, and vice versa. In some cases the spectroscopic results triggered the need to analyse the model, while in other cases the model suggested the spectroscopic experiments, and in some cases both went hand in hand. Throughout this odyssey, the model improved and the spectroscopy became more sophisticated.
The M13 odyssey also shows that spectroscopic techniques developed over the years, allowing for continuously more precise and more accurate measurements. At this same time homology modelling improved too. In the next paragraphs we will discuss the latest developments in homology modelling and its remaining unsolved problems. We will also illustrate the usefulness of homology models, sometimes in combination with spectroscopic techniques, with a few real-world examples.
Method overview: the eight steps of homology modelling
Homology modelling is usually described as a multi-step process in which the number of steps typically varies from X to Y. Here we use an eight-step plan (Fig. 2). Over the years each of these eight steps has undergone extensive scrutiny and has been the topic of much research. Consequently, models built today with a fully automatic web server are considerably more accurate than the first modelling approach used four decades ago by Browne et al. using wire and plastic models of bonds and atoms (Browne et al. 1969).
Fig. 2.
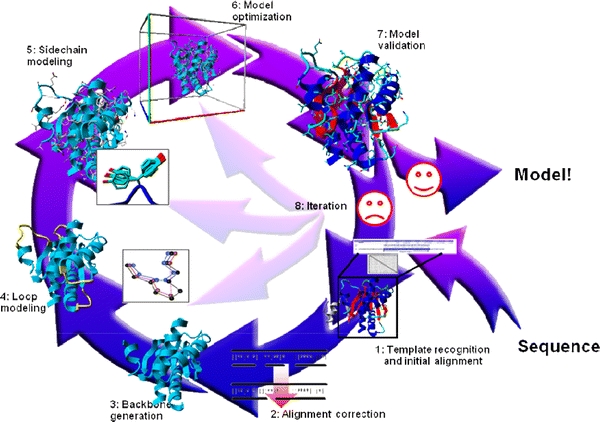
The eight steps of homology modelling. The first step involves finding a suitable homologous protein whose structure can be used as a modelling template, and generating the initial alignment between that template and the model sequence. In step 2 this alignment is refined using, for example, knowledge obtained from the template structure. In step 3 the backbone is generated and deletions are performed so that, temporarily forgetting insertions, the backbone of the template looks like that of the model as much as possible. In step 4 gaps in the model are closed, and optionally loops are constructed ab initio. In step 5, the side-chains are added using rotamer libraries to find the best rotamer for that local backbone conformation. Step 6 consists of molecular-dynamics simulation of the complete model in order to remove (the majority of) the introduced errors. In step 7 the model is checked for remaining errors using validation software. Depending on the outcome of the validation step we either approve the model or iterate the modelling process (step 8) starting from steps 1–6
Here, we will discuss the latest innovations and developments in each of the eight homology modelling steps.
Step 1: Template recognition and initial alignment
Traditionally, the modelling template is found by performing a BLAST (Altschul et al. 1990) search against the Protein Data Bank (PDB) (Berman et al. 2003). This approach is often successful when the query is highly similar to structures in the database. In contrast, templates that are close to the possible homology modelling threshold are harder to find or may even remain undetected (Sander and Schneider 1991). The development of PSI-BLAST (Altschul et al. 1997) and fold-recognition methods (e.g. Jones 1999) improved the detection of these difficult-to-find templates because these methods use a profile instead of a single sequence to search the database. Furthermore, the growing number of structures collected in the PDB makes it every year easier to find a homologous one.
Finding the best possible template is not limited to searching the PDB with (PSI-)BLAST. There may be several candidate templates with similar sequence identities to the query. In that case, the optimal template must be selected based on other criteria. The X-ray resolution is, although frequently used as such, only a limited measure of structure quality because it says something about the experimental data, not about the quality of the structure model. I.e. with 1.8-Å data one can typically make a better structure model than with 2.2-Å data, but whether or not this better structure model is actually made depends on the crystallographer and the software used.
The crystallographic residual, the so-called R-factor, says something about the correlation of the structure model with the experimental data and therefore seems more indicative than the X-ray resolution alone. Unfortunately, acceptable and even seemingly encouraging R-factors can be attained by adding more parameters to the structure models, effectively over-fitting/over-refining the model (Brändén and Jones 1990). This problem was solved by the introduction of the free R-factor (Brünger 1992), which is much more robust against over-fitting because the value is calculated only with the fraction of the X-ray data that was not used to build the structure model. Therefore, R-free can be seen as a description of how well the structure model predicts an ‘independent’ measurement.
While very robust, the free R-factor is a global indicator: it describes the structure model as a whole, but not a particular part of the protein that may interest us. A local measure of fit with the experimental data is needed, for instance, the real-space R-factor (Jones et al. 1991). For PDB entries these values can be obtained from the electron density server (EDS) (Kleywegt et al. 2004).
So, with the X-ray resolution, the (free) R-factor and the real-space R-factor, one can select a proper template from the (PSI-)BLAST results. That is, one can select a template that corresponds well with the X-ray experiment. A more in-depth analysis of the template structure is needed to see whether it also corresponds to our current knowledge of protein structures. Structure validation scores such as the Ramachandran Z-score (Hooft et al. 1997), the fraction of Ramachandran plot outliers (Laskowski et al. 1993), side-chain rotamer normality scores (Hooft et al. 1996a), residue packing scores (Vriend and Sander 1993), hydrogen-bond network quality (Hooft et al. 1996b) and many others are used to obtain insight into the geometric quality of the template structure. Most of these validation scores, both global and local, can be obtained via the PDB and the linked databanks.
Another possible step in template selection is optimisation of the template before the actual modelling. We have recently shown that validation scores such as the Ramachandran Z-score and the number of atomic clashes (bumps) can be improved by a fully automated re-refinement of the PDB entry with its original experimental data (Joosten et al. 2009). In addition, the crystallographic R-factor, or rather the free R-factor, is also improved by this re-refinement. This optimisation is particularly useful for templates that will be used for drug docking studies because their success often depends critically on the quality of the atomic model. The benefit of re-refinement is tightly correlated with sequence identity between the template and the model sequence. That is, any improvement of the atomic coordinates of a residue is lost when this residue (or just its side-chain) has to be rebuilt. Fortunately, even with low sequence identity, there may be regions of the template that are not changed in the modelling process and thus can be improved by re-refinement.
Of course, when sufficient central processing unit (CPU) time is available to the modeller, it may be beneficial to use a number of (re-refined) PDB entries as templates, instead of a single one.
Step 2: Alignment correction
Having identified one or more possible modelling templates using the initial screening described above, more sophisticated methods are needed to arrive at better alignment. Molecular class-specific information systems (MCSISs) can be a great asset in the homology modelling process. MCSISs contain a large amount of heterogeneous data on one particular class of proteins. The GPCRDB (http://www.gpcr.org/7tm/; Horn et al. 2003) is a good example of such a system. It contains sequences, structures, mutation data, ligand binding data and much more. All of this information is used (directly or indirectly) for creating sequence profiles for GPCR (sub-)families. Profile-based alignments (Oliveira et al. 1993) are used to generate alignments that are of significantly higher quality than alignments generated by automatic methods based solely on sequence data. Currently, MCSISs are available for only a small number of protein families and therefore in most cases other sequence alignment tools are needed.
Many programs are available to align a number of related sequences, for example, CLUSTALW (Thompson et al. 1994). The resulting multiple sequence alignment implicitly contains a lot of additional information. For example, if at a certain position only exchanges between hydrophobic residues are observed, it is highly likely that this residue is buried. Multiple sequence alignments are also useful to place deletions or insertions in areas where the sequences are strongly divergent. To consider this knowledge during the alignment, one uses the multiple sequence alignment to derive position-specific scoring matrices, which are also called profiles (e.g. Taylor 1986; Dodge et al. 1998). In recent years, new programs such as MUSCLE and T-Coffee have been developed that use these profiles to generate and refine the multiple sequence alignments (Edgar 2004; Notredame et al. 2000).
When building a homology model, we are in the fortunate situation of having an almost perfect profile: the known structure of the template. We simply know that a certain alanine sits in the protein core and must therefore not be aligned with a glutamate. Alignment techniques such as SSALN make use of this structural knowledge found in the template (Qiu and Elber 2006).
Step 3: Backbone generation
Aligned residues occupy the same position in the template and model. Coordinates can thus simply be copied over to create the initial model backbone. In practice, there are many ways to improve this crude recipe. First, the template is likely to be present more than once in the PDB (e.g. a bundle of NMR structures, multiple copies in the crystal, or solved multiple times under different conditions). Here, one can use structure validation tools (Hooft et al. 1996a, Laskowski et al. 1993), such as the PDBREPORT databank (http://www.cmbi.ru.nl/gv/pdbreport/) to pick the best one, correcting errors where possible. Second, one can combine multiple templates, because residues missing in one template can sometimes be found in the other, or because the alignment covers more than one template, which is common for multi-domain targets. The well-known SwissModel server (http://swissmodel.expasy.org/; Arnold et al. 2006) selects fragments from different PDB files that locally are most similar to the corresponding model fragment. The Zhang (http://zhang.bioinformatics.ku.edu/I-TASSER/; Pandit et al. 2006) and Robetta (http://robetta.bakerlab.org/; Chivian et al. 2003) modelling servers have successfully extended this concept and are today among the best homology modelling servers available online. Some methods do not even create a single backbone at all; instead they use the alignment to derive restraints (hydrogen bonds, backbone torsion angles etc.), and only later build the model, while trying to satisfy the restraints (Sali and Blundell 1993).
Step 4: Loop modelling
Any insertion or deletion in the alignment implies a structural change of the backbone, and can thus not be modelled in the previous step. Since these changes usually take place outside regular secondary structure elements, their prediction is referred to as loop modelling. There are two major approaches to the problem: first knowledge-based methods (Michalsky et al. 2003), which search the PDB for known loops with high sequence similarity to the target and endpoints that match the anchor residues between which the loop has to be inserted, and second, energy-based methods, which sample random loop conformations while minimizing an energy function (Xiang and Honig 2002). Since loops never fit the anchor points exactly, they have to be closed, using for example an algorithm borrowed from robotics (Canutescu and Dunbrack 2003). In practice, a combination of both methods is common.
Step 5: Side-chain modelling
The most successful approaches to side-chain prediction are knowledge based. They use libraries of common side-chain rotamers extracted from high-resolution X-ray structures (Dunbrack and Karplus 1993; Chinea et al. 1995; Lovell et al. 2000). An essential feature of these libraries is backbone dependence, hence they store the distribution of the side-chain dihedral angles (χ 1, χ 2 etc.) as a function of the backbone dihedrals φ and ψ. This not only increases the accuracy, but also helps to shrink the search space (i.e. the possible combinations of interacting side-chain rotamers) to a size that can be handled, for example using dead-end elimination (Canutescu et al. 2003). The prediction accuracy is usually highest for residues in the hydrophobic core, where more than 90% of all predicted χ 1 angles fall within ±20° of the experimental values, but significantly lower for residues on the surface where the percentage drops to 70%, and further down to 50% for the combined χ 1/χ 2 accuracy (Canutescu et al. 2003). This is mainly caused by the electrostatic and hydrogen-bonding interactions, which are partly solvent mediated and much more difficult to get right than the simple repulsive van der Waals interactions in the core, but also partly due to the fact that flexible side-chains on the surface tend to adopt multiple conformations, which are additionally influenced by crystal contacts, so there simply may not be a single correct conformation at all. Nevertheless, the surface residues are among the most important ones to get right; they mediate all the interactions, and applications such as drug design or protein docking thus critically depend on them.
Step 6: Model optimisation
Once all these steps are completed, one obtains the initial homology model, which hopefully looks broadly similar to the (usually unknown) target structure. The minor details, however, such as the precise backbone conformation, hydrogen-bonding networks or certain side-chain rotamers, are often wrong. While this deficiency keeps scientists working on experimental structure determination busy, predictors strive to bridge the gap between model and target (the ‘last mile’ of the protein folding problem) using various optimisation and refinement techniques, the most prominent ones being molecular dynamics (Krieger et al. 2004) and Monte Carlo simulations (Misura et al. 2006). For a given model, there are unfortunately many more paths leading away from the target than towards it, and combined with the limited accuracy of empirical force fields, this makes it very easy to reduce the model accuracy during the refinement. Consequently, the best optimisation was often no optimisation. We did well in the early Critical Assessment of Structure Prediction (CASP) homology modelling competitions simply by not performing MD simulations on the models [except for 25 energy-minimisation steps with CNS (Brünger et al. 1998) to introduce the same local geometric features that CNS put into the real structure against which our prediction would be compared]. Nevertheless, steady progress over the past years has changed this rule of thumb (see “Results”).
Step 7: Model validation
Protein structures were error free until the landmark article on Procheck by the Thornton group (Laskowski et al. 1993). This article can be seen as the beginning of the realisation that crystallographers and NMR spectroscopists actually use experimental techniques to determine their coordinates. With the release of the first WHAT_CHECK (Hooft et al. 1996a), structure validation became a common household technique for most scientists and, although not at the speed we hoped, errors in protein structures are becoming less frequent. The two main bottlenecks are the introduction of improved technologies in all structure solution software used all over the world, and the fact that the detection of an error does not implicitly mean that the error can be removed.
Step 8: Iteration
If the model is not good enough, (part of) the modelling process has to be repeated. For instance, wrong side-chain conformations can be improved by iterating the process from step 5 onwards. Sometimes, this iteration step means that one has to start the modelling process all over again using another template or alignment. Alternatively, one can start several modelling processes using different templates. The resulting models can be combined in the end to produce a hybrid model that consists of the strongest points of each separate model.
Unsolved problems and future directions in homology modelling
While many scientific disciplines face huge difficulties when trying to experimentally validate theoretical predictions, protein modelling is in a fortunate situation: since 1994, the biennial Critical Assessment of Structure Prediction (CASP) contests (Moult et al. 2007) have provided an ideal opportunity to evaluate the accuracy of today’s many protein structure prediction methods. During each CASP season (lasting about 4 months once every 2 years), about 200 research groups try to predict the structures of ~100 proteins, the CASP targets. The target sequences are provided to CASP by structural biology laboratories just before the corresponding structures are solved. The predictions are thus true blind predictions, allowing performance to be measured on realistic test cases, locating areas of progress as well as still unsolved problems.
CASP regularly shows that the eight homology modelling steps summarized above allow reliable models to be built in many cases, from which a lot of structural and functional insights can be derived. However, these eight steps are unfortunately not sufficient to actually solve the protein structure prediction problem via homology modelling as soon as enough templates become available. Figure 3 shows CASP8 targets T0498 and T0499: both proteins are 56 amino acids long, 53 of which are conserved (95% sequence identity). Still, the two structures are entirely different; just three point mutations completely change the fold. While this is an extreme example of human protein engineering art (He et al. 2008), also naturally occurring proteins with similar sequences often show surprising structural diversity (Kosloff and Kolodny 2008), leading classic homology modelling to fail miserably. The prion protein (Prusiner 1998) and other amyloid-forming proteins provide an even more dramatic case; here 100% identical sequences can exist in two totally different structures. Obviously, the homology modelling problem is tightly intertwined with the more general protein folding problem itself. Even if a close template is available, there can always be structurally divergent regions, which are either expected from the poor local alignment, or unexpectedly caused by critical point mutations, or widely differing crystal packing contacts.
Fig. 3.
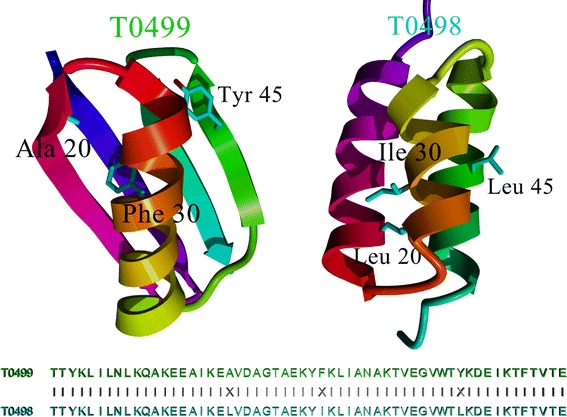
Comparison of CASP8 targets T0498 and T0499. The sequences of both proteins are 95% (53 of 56) identical (only residues 20, 30 and 45 differ), yet the structures are totally different. Classic homology modelling predicted T0499 correctly (which looks like the related homology modelling templates in the PDB), but failed completely for T0498. Since the structures of T0498 and T0499 have not been released yet, this figure is based on a closely related pair with PDB IDs 2jws and 2jwu from the same authors, who showed by NMR spectroscopy that these two structures look essentially the same as T0498 and T0499 (He et al. 2008)
The only way to handle these difficult cases is to apply more general ab initio folding algorithms, which do not depend on template structures, but try to simulate the complete folding process from the stretched-out conformation. As it turns out, this one-algorithm-fits-all approach is currently the most successful one at CASP (Chivian et al. 2003; Pandit et al. 2006): if available, it uses known templates (or fragments thereof) only to guide the search, but does not depend on them. As a side-effect, this allows hybrid models to be built, combining the best parts from multiple templates.
Despite these encouraging developments, the protein folding problem is far from solved. The best models are still built by those who got the alignment right in the first place, which unfortunately implies that structural diversity is often missed: one cannot yet ignore the difficult-to-align regions and simply predict them with ab initio folding instead. The sequence alignment problem will thus remain an active research field for years to come.
Noteworthy progress has been made with model optimisation to bridge the structural gap between initial model and target. While in the early days of CASP, predictors were well advised to keep the backbone of their model fixed (the frozen-core approach), simply because the danger of messing up the model was too large, the situation is quite different today: force field accuracy (Krieger et al. 2004) and sampling efficiency (Misura et al. 2006) have improved to a level that allows well-performing methods such as Modeller-CSA (Joo et al. 2008), Rosetta (Chivian et al. 2003), undertaker (Vriend 1990) and YASARA (Krieger et al. 2009) to free all atoms during the refinement, often moving models considerably closer to the target.
While homology modelling currently focusses on the protein in a model, other entities, i.e. carbohydrates, small molecules and ions, also make up important parts of certain proteins and protein complexes; for instance, zinc atoms in so-called zinc fingers are important for the stability of the protein, and the common protein haemoglobin would be useless without its heme groups and the iron atoms therein. Carbohydrates in glycoproteins perform numerous functions, ranging from providing stability to signalling and labelling for intracellular transport (Lütteke 2009). The many roles of non-protein entities make it obvious that homology modelling should look beyond the protein. A complete model should thus be more than a three-dimensional (3D) representation of an amino acid sequence. One major challenge for homology modelling is recognising binding sites for non-protein entities.
Drug docking software (e.g. Rarey et al. 1996; Nabuurs et al. 2007) can be used to detect the binding sites of compounds such as heme groups or coenzymes. However, relevant biological information is needed to select compounds that may be bound to the protein. Copying the binding site from the template structure is the simplest method, but does not work for ab initio folding models. For such models, spectroscopic analysis of the protein can provide insight into which compounds are bound. This approach is not limited to homology modelling; X-ray crystallography can also benefit from spectroscopic analysis of a protein to identify a bound compound (Chen et al. 2002).
Incorporating ions can be an additional step of the modelling process. Nayal and Di Cera (1996) have suggested a method to detect sodium binding sites in protein structures which can be extended to detect various other ion binding sites. Of course, any additional experimental data can guide this ion-site detection process. Especially tightly bound functional ions that co-purify with the protein can be detected by means of spectroscopic analysis. A significant number of PDB files have bound ions or water molecules that were erroneously assigned. We have observed H20, and Na+, K+, Mg2+, Ca2+ and NH+4 ions that should actually be one of the others in this list. This is the result of X-ray spectroscopy having difficulties distinguishing between H2O, NH+4, Na+ and Mg2+ because these entities have equally many electrons, as do K+ and Ca2+.
The power of force-field-based model optimisation methods can be significantly reduced when such problems include a difference in the ionic charge. It is therefore very important to (experimentally) validate the ions in template structures when these are important for the final homology model.
Carbohydrates can be modelled at the final stage of the homology modelling process, but this does not always reflect the protein folding process. Carbohydrates are not only added in post-translational modification (that is, when the protein is ‘done’), but also during the protein expression by the ribosome. They are important in the protein folding process and the detection of misfolded proteins (Parodi 2002). It may therefore prove interesting to add the necessary carbohydrates to the unfolded protein before ab initio folding. Apart from their role in protein folding, carbohydrates are sometimes important in oligomerisation of proteins; for instance, the neuraminidase protein from influenza shows different glycosylation states in its monomeric, dimeric and tetrameric states. The carbohydrates in tetrameric state provide extra stability (Fig. 4) and, in the case of the Spanish flu influenza virus, resistance to trypsin digestion leads to increased virulence (Wu et al. 2009). This shows the vital (and sometimes lethal) importance of considering carbohydrates in homology models.
Fig. 4.
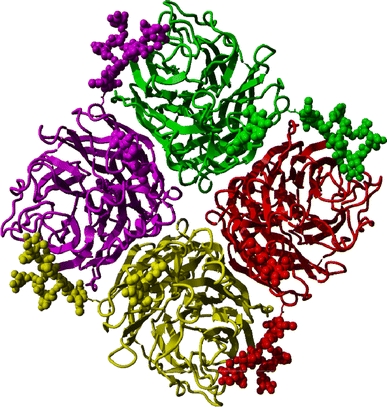
Tetrameric form of whale influenza neuraminidase (PDBid 2r8h, Smith et al. 2006), coloured by monomer. Protein chains are displayed in ribbon representation, carbohydrate atoms in ball representation. The carbohydrates of one monomer interact with the adjacent monomer, thus stabilising the tetramer
Results and discussion: other roles for homology modelling
In the 1990s most articles that included homology modelling described just how the model of one protein was constructed, and ended with the ominous sentence “…this model will help us perform our research on this intriguing protein”, after which the group would start working on something else. Here, we will illustrate the importance of homology modelling for the study of inheritable diseases, but the value of models has been amply illustrated in fields ranging from drug design to laundry powder enzyme engineering, from validating experimental structures to the design of humanized antibodies, and from mutation analysis to intelligent experimental design in many spectroscopic research projects.
In the following examples building the homology model was not the ultimate goal but one of the tools used to gain more information about a mutation, a disease or a process in the human body. These examples prove that homology models can be of great use in the (bio)medical field.
Modelling of the LRTOMT-COMT domain
In a study of non-syndromic deafness four pathogenic mutations were found in an as-yet uncharacterized gene which codes for two different proteins, called LRTOMT1 and LRTOMT2 (Ahmed et al. 2008). Three of these mutations were mis-sense mutations located in the catechol-O-methyltransferase (COMT, EC 2.1.1.6) domain of LRTOMT2; one introduced a stop codon causing the loss of a large fraction of the protein. The COMT domain catalyses the transfer of a methyl group from S-adenosyl-l-methionine (Ado-Met) to a hydroxyl group of catechol. No structure for LRTOMT2 was known, so we needed a homology model to study the mis-sense mutations in more detail.
Using the crystal structure of rat COMT [39% identity to LRTOMT2 over 212 amino acids (PDBid 1h1d, Bonifacio et al. 2002)] we were able to model the COMT domain of human LRTOMT2. The three mutated residues are located in helix 1 (p.R81Q), helix 2 (p.W105R) and the loop that follows helix 2 (p.E110K), and thus not in the hypothetical substrate-binding pockets. However, the loop is predicted to be important for the groove that binds the putative methyl acceptor. The Arg81 and Glu110 residues are predicted to form a hydrogen-bonded salt bridge between helix 1 and the loop, and Trp105 is predicted to make hydrophobic interactions in the core between these helices (Fig. 5). These residues may therefore be important for local protein stability and can affect the substrate binding region.
Fig. 5.

a Molecular model of the catechol-O-methyltransferase domain of LRTOMT2, residues 79–290. The affected residues are depicted in blue. The predicted ligands are coloured yellow, and the tyrosine residue (Tyr111) that lines the hydrophobic groove of the ligand binding site is shown in cyan. The boxed region containing residues affected by mis-sense mutations of LRTOMT2 is enlarged in panels b–d. The helices 1 (H1) and 2 (H2) are shown with wild-type residues Arg81, Trp105 and Glu110 depicted in blue and mutated residues in green. Hydrogen bonds are represented by yellow dotted lines. b The Arg81 and Glu110 residues form a salt bridge between helix 1 and the loop following helix 2. The Gln81 residue cannot form this salt bridge as it is not positively charged. Also, the formation of hydrogen bonds is impaired because of the smaller size of glutamine as compared with arginine. c The Trp105 residue is predicted to make hydrophobic interactions as a result of its large side-chain. Most of these interactions would be lost by the W105R substitution. d Substitution E110K is predicted to lead to the loss of hydrogen bonds and a salt bridge. There would likely be repulsion between the side-chains Lys110 and Arg81 as both are positively charged
Modelling of the ligand-binding domain of ESRRB
Sequence analysis revealed four mis-sense mutations of the estrogen-related receptor beta (ESRRB) gene leading to autosomal-recessive non-syndromic hearing impairment (Collin et al. 2008). Experimental results indicated that ESRRB is essential for inner-ear development and function. ESRRB encodes the estrogen-related receptor protein beta, a member of the nuclear hormone receptor (NHR) family. In general, members of this family share a zinc finger C4 DNA-binding domain at their N-terminus and a ligand-binding domain that is located near the C-terminus. The ligand-binding domain of nuclear hormone receptors is a well-conserved and highly organized structure containing 12 alpha-helices. Three mutations (p.L320P, p.V342L and p.L347P) were located within this ligand-binding domain. The fourth mutation was located in the DNA binding domain and will not be discussed here.
To study the three mutations in the ligand binding domain in more detail we built a homology model of this domain using the structure of the estrogen-related receptor gamma (ESRRG) receptor (PDBid 1kv6, Greschik et al. 2002) as a template. It had 79% sequence identity over 229 amino acids. The molecular model showed that the three mis-sense mutations in the ligand binding domain are likely to affect the structure and stability (Fig. 6). Two of the mutations involved a leucine-to-proline mutation, L320P in helix 7 and L347P in helix 8. In general, the introduction of proline residues within helices reduces the stability of the helix, and therefore these mutations will disturb the structure of the helices and probably the complete ligand-binding domain. In addition, the loss of the leucine side-chain abrogates a number of hydrophobic interactions. The other mutation in this domain (V342L) substitutes a leucine for a valine residue, resulting in the occurrence of a somewhat larger side-chain that bumps into the molecular surface of helix one. This substitution is predicted to reduce the strength of the interaction between helix 1 and 8.
Fig. 6.
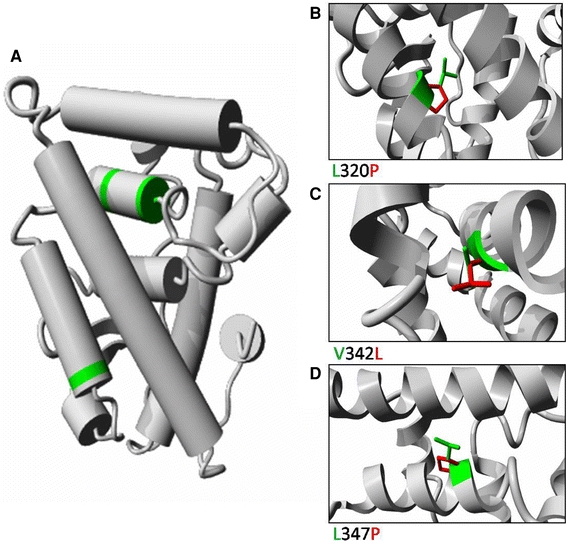
a Molecular modelling of the ligand-binding domain of the human ESRRB protein. The structure was deduced from the known ESRRB structure. The various helices are represented by cylinders. The three amino acids that are affected by the mis-sense mutations are indicated in green. Detailed views of the three mutations are shown in panels b, c and d. The wild-type residue is depicted in green, whereas the side-chain of mutant residue is presented in red. b L320 makes hydrophobic contacts in the core between two helices, the mutation to P causes loss of many hydrophobic interactions. Additionally, the P will disturb the structure of the helix. c V342 is tightly packed in the core between two helices. This core will be disturbed by the V342L mutation because L has a larger side-chain. d L347P will cause loss of hydrophobic interactions between the helices, and the introduction of P will disturb the structure of the helix
In summary, the molecular modelling data predicts that mutations of ESRRB will result in conformational changes near the substituted amino acids or decreased helix stability and are therefore likely affect the stability and function of the complete ligand-binding domain.
Functional states of alcohol oxidase
Van der Klei and co-workers studied four different conformational states of the flavoenzyme alcohol oxidase (AO) from the methylotrophic yeasts Hansenula polymorpha and Pichia pastoris, including assembly intermediates (Boteva et al. 1999). These proteins had to be homology modelled from the enzyme glucose oxidase that shows only 25% sequence identity with the AOs. With so little similarity, homology modelling is very difficult and large modelling errors are to be expected. An additional problem is that glucose oxidase is a dimer while AO is an octamer. The low quality of the model certainly precluded any form of protein–protein docking to construct an octamer from the dimer. The model fortunately revealed a series of hydrophobic surface patches, some of which have tryptophan residues at the surface. As there were also tryptophans observed in the model near the flavin adenine dinucleotide (FAD) group, the suggestion to apply spectroscopic techniques to extend the modelling study came shouting from Fig. 7. A series of spectroscopic techniques, including time-resolved fluorescence, fluorescence anisotropy decay, steady-state fluorescence, and visible and near-ultraviolet circular dichroism, was used to characterize native AO and several putative assembly intermediates. A good working hypothesis for the AO folding pathway could be derived. The study also triggered the search for chaperones that seemed necessary to allow FAD to bind to AO in vivo.
Fig. 7.
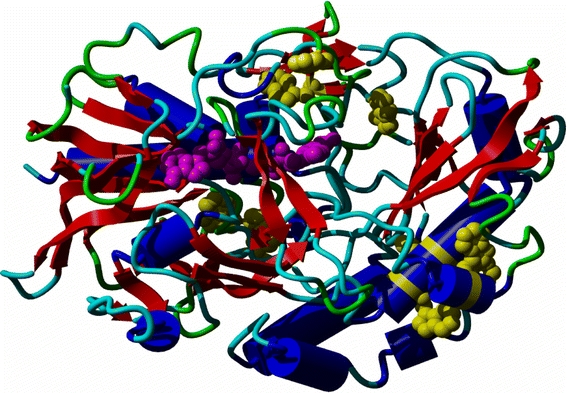
Model of one monomer of AO shown as ribbon; helix in blue, strand in red, loop and turn in shades of green. All tryptophan side-chains are shown in yellow; the FAD group is shown in purple
Conclusion
Homology modelling will always be needed because it is impossible to solve the three-dimensional structure for each determined sequence. An increasing number of scientists are now using protein structures for mutational analysis and experimental design. The process of homology modelling has improved dramatically over the years, but there are still many problems to solve. It is reassuring for homology modellers that modelling problems can often be solved using spectroscopic techniques. Spectroscopists, on the other hand, normally need homology models to fully harvest the results from their spectroscopic studies. This interplay brings us back to the title of this article: homology modelling and spectroscopy are indeed a never-ending love story.
Acknowledgments
This work was part of the BioRange programme of the Netherlands Bioinformatics Centre (NBIC), which is supported by a BSIK grant through the Netherlands Genomics Initiative (NGI). TIPharma is thanked for support. This work was funded in part by the EU project EMBRACE Grid which is funded by the European Commission within its FP6 Programme, under the thematic area ‘Life sciences, genomics and biotechnology for health’, contract number LUNG-CT-2004-512092. This work was also supported by the BioSapiens Network of Excellence project. The BioSapiens project is funded by the European Commission within its FP6 Programme, under the thematic area ‘Life sciences, genomics and biotechnology for health’, contract number LSHG-CT-2003-503265. G.V. wishes to thank M.A. Hemminga for being a warm-hearted friend and a great Ph.D. supervisor.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
The more you see: spectroscopy in molecular biophysics.
Contributor Information
Hanka Venselaar, Email: hvensela@cmbi.ru.nl.
Gert Vriend, Phone: +31-24-3619390, FAX: +31-24-3619395, Email: vriend@cmbi.ru.nl.
References
- Ahmed ZM, Masmoudi S, Kalay E, Belyantseva IA, Mosrati MA, Collin RW, Riazuddin S, Hmani-Aifa M, Venselaar H, Kawar MN, Tlili A, van der Zwaag B, Khan SY, Ayadi L, Riazuddin SA, Morell RJ, Griffith AJ, Charfedine I, Caylan R, Oostrik J, Karaguzel A, Ghorbel A, Riazuddin S, Friedman TB, Ayadi H, Kremer H. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat Genet. 2008;40(11):1335–1340. doi: 10.1038/ng.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Cai K, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 306–322 extending from helix VII to the palmitoylation sites in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999;38(25):7931–7937. doi: 10.1021/bi9900121. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Anang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Beck M, Sakmar TP, Siebert F. Spectroscopic evidence for interaction between transmembrane helices 3 and 5 in rhodopsin. Biochemistry. 1998;37(20):7630–7639. doi: 10.1021/bi9801560. [DOI] [PubMed] [Google Scholar]
- Berman HM, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10(12):980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Bonifacio MJ, Archer M, Rodrigues ML, Matias PM, Learmonth DA, Carrondo MA, Soares-Da-Silva P. Kinetics and crystal structure of catechol-o-methyltransferase complex with co-substrate and a novel inhibitor with potential therapeutic application. Mol Pharmacol. 2002;62:795–805. doi: 10.1124/mol.62.4.795. [DOI] [PubMed] [Google Scholar]
- Boteva R, Visser AJ, Flilippi B, Vriend G, Veenhuis M, van der Klei IJ. Conformational transitions accompanying oligomerization of yeast alcohol oxidase, a peroxisomal flavoenzyme. Biochemistry. 1999;38(16):5034–5044. doi: 10.1021/bi982266c. [DOI] [PubMed] [Google Scholar]
- Brändén C-I, Jones TA. Between objectivity and subjectivity. Nature. 1990;343:687–689. doi: 10.1038/343687a0. [DOI] [Google Scholar]
- Browne WJ, North AC, Philips DC, Brew K, Vanaman TC, Hill RL. A possible three-dimensional structure of bovine α-lactalbumin based on that of hen’s egg-white lysozyme. J Mol Biol. 1969;42:65–86. doi: 10.1016/0022-2836(69)90487-2. [DOI] [PubMed] [Google Scholar]
- Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, De Lano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Grystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cai K, Klein-Seetharaman J, Farrens D, Zhang C, Altenbach C, Hubbell WL, Khorana HG. Single-cysteine substitution mutants at amino acid positions 306–321 in rhodopsin, the sequence between the cytoplasmic end of helix VII and the palmitoylation sites: sulfhydryl reactivity and transducin activation reveal a tertiary structure. Biochemistry. 1999;38(25):7925–7930. doi: 10.1021/bi9900119. [DOI] [PubMed] [Google Scholar]
- Canutescu AA, Dunbrack RLJ. Cyclic coordinate descent: a robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canutescu AA, Shelenkov AA, Dunbrack RLJ. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415(6871):545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinea G, Padron G, Hooft RWW, Sander C, Vriend G. The use of position specific rotamers in model building by homology. Proteins. 1995;23:415–421. doi: 10.1002/prot.340230315. [DOI] [PubMed] [Google Scholar]
- Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CE, Bonneau R, Rohl CA, Baker D. Automated prediction of CASP-5 structures using the Robetta server. Proteins. 2003;53(suppl 6):524–533. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- Collin RW, Kalay E, Tariq M, Peters T, van der Zwaag B, Venselaar H, Oostrik J, Lee K, Ahmed ZM, Caylan R, Li Y, Spierenburg HA, Eyupoglu E, Heister A, Riazuddin S, Bahat E, Ansar M, Arslan S, Wollnik B, Brunner HG, Cremers CW, Karaguzel A, Ahmad W, Cremers FP, Vriend G, Friedman TB, Riazuddin S, Leal SM, Kremer H. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet. 2008;82(1):125–138. doi: 10.1016/j.ajhg.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoletti D, Grishaev A, Whitten AE, Tsigelny I, Taylor P, Trewhella J. Synaptic arrangement of the neuroligin/beta-neurexin complex revealed by X-ray and neutron scattering. Structure. 2007;15(6):693–705. doi: 10.1016/j.str.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison MD, Findlay JB. Modification of ovine opsin with the photosensitive hydrophobic probe 1-azido-4-[125I]iodobenzene. Labelling of the chromophore-attachment domain. Biochem J. 1986;234(2):413–420. doi: 10.1042/bj2340413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison MD, Findlay JB. Identification of the sites in opsin modified by photoactivated azido[125I]iodobenzene. Biochem J. 1986;236(2):389–395. doi: 10.1042/bj2360389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge C, Sander C, Schneider R. The HSSP database of protein structure-sequence alignments and family profiles. Nucleic Acids Res. 1998;26(1):313–315. doi: 10.1093/nar/26.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack RLJ, Karplus M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- Edgar R. MUSCLE: Multiple sequence alignment with high accuracy and high troughpout. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer J. Model for haptoglobin heavy chain based upon structural homology. Proc Natl Acad Sci USA. 1980;77(6):339–3397. doi: 10.1073/pnas.77.6.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greschik H, Wurtz JM, Sanglier S, Bourguet W, van Dorsselaer A, Moras D, Renaud JP. Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol Cell. 2002;9:303–313. doi: 10.1016/S1097-2765(02)00444-6. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Pinon DI, Wessels WS, Prendergast FG, Miller LJ. Environment and mobility of a series of fluorescent reporters at the amino terminus of structurally related peptide agonists and antagonists bound to the cholecystokinin receptor. J Biol Chem. 2002;277(21):18552–18560. doi: 10.1074/jbc.M201164200. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Lam PC, Dong M, Sexton PM, Abagyan R, Miller LJ. Fluorescence resonance energy transfer analysis of secretin docking to its receptor: mapping distances between residues distributed throughout the ligand pharmacophore and distinct receptor residues. J Biol Chem. 2007;282(45):32834–32843. doi: 10.1074/jbc.M704563200. [DOI] [PubMed] [Google Scholar]
- He Y, Chen Y, Alexander P, Bryan PN, Orban J. NMR structures of two designed proteins with high sequence identity but different fold and function. Proc Natl Acad Sci USA. 2008;105:14412–14417. doi: 10.1073/pnas.0805857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft RWW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- Hooft RWW, Sander C, Vriend G. Positioning hydrogen atoms by optimizing hydrogen bond networks in protein structures. Proteins. 1996;26:363–376. doi: 10.1002/(SICI)1097-0134(199612)26:4<363::AID-PROT1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, Vriend G (2003) GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res 1:31(1):294–297 [DOI] [PMC free article] [PubMed]
- Jones DT. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287(4):797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst A. 1991;47:110–119. doi: 10.1107/S0108767390010224. [DOI] [PubMed] [Google Scholar]
- Joo K, Lee J, Lee K, Kim BG, Lee J (2008) All-atom chain-building by optimizing MODELLER energy function using conformational space annealing. Proteins Nov [Ahead of print] [DOI] [PubMed]
- Joosten RP, Womack T, Vriend G, Bricogne G. Re-refinement from deposited X-ray data can deliver improved models for most PDB entries. Acta Cryst D. 2009;62:176–185. doi: 10.1107/S0907444908037591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wählby A, Jones TA. The Uppsala electron-density server. Acta Cryst D. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- Kosloff M, Kolodny R. Sequence-similar, structure-dissimilar protein pairs in the PDB. Proteins. 2008;71:891–902. doi: 10.1002/prot.21770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G. Making optimal use of empirical energy functions: force field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K (2009) Improving physical realism, stereochemistry and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77(suppl 9) [DOI] [PMC free article] [PubMed]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structure. J Appl Cryst. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. doi: 10.1002/1097-0134(20000815)40:3<389::AID-PROT50>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Lütteke T. Analysis and validation of carbohydrate three-dimensional structures. Acta Cryst D. 2009;65:156–168. doi: 10.1107/S0907444909001905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchot P, Ravelli RB, Raves ML, Bourne Y, Vellom DC, Kanter J, Camp S, Sussman JL, Taylor P. Soluble monomeric acetylcholinesterase from mouse: expression, purification, and crystallization in complex with fasciculin. Protein Sci. 1996;5:672–679. doi: 10.1002/pro.5560050411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas YP, Stouten PF, Beltran JR, Laure CJ, Vriend G. Structure–function relationship for the highly toxic crotoxin from Crotalus durissus terrificus. Eur Biophys J. 1992;21(3):199–205. doi: 10.1007/BF00196764. [DOI] [PubMed] [Google Scholar]
- Michalsky E, Goede A, Preissner R. Loops in proteins (LIP)—a comprehensive loop database for homology modelling. Protein Eng. 2003;16:979–985. doi: 10.1093/protein/gzg119. [DOI] [PubMed] [Google Scholar]
- Misura KM, Chivian D, Rohl CA, Kim DE, Baker D. Physically realistic homology models built with ROSETTA can be more accurate than their templates. Proc Natl Acad Sci USA. 2006;103:5361–5366. doi: 10.1073/pnas.0509355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult J, Fidelis K, Kryshtafovych A, Rost B, Hubbard T, Tramontano A. Critical assessment of methods of protein structure prediction—round VII. Proteins. 2007;69(suppl 8):3–9. doi: 10.1002/prot.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabuurs SB, Wagener M, de Vlieg J. Fleksy: a flexible approach to induced fit docking. J Med Chem. 2007;50(26):6507–6518. doi: 10.1021/jm070593p. [DOI] [PubMed] [Google Scholar]
- Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J Mol Biol. 1996;256(2):228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Paiva AC, Vriend G. A common motif in G protein-coupled seven transmembrane helix receptors. J Comp Aided Mol Des. 1993;7:649–658. doi: 10.1007/BF00125323. [DOI] [Google Scholar]
- Oliveira L, Paiva ACM, Vriend G. A low resolution model for the interaction of G proteins with G protein-coupled receptors. Prot Eng. 1999;12:1087–1095. doi: 10.1093/protein/12.12.1087. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Hulsen T, Lutje Hulsik D, Paiva AC, Vriend G. Heavier-than-air flying machines are impossible. FEBS Lett. 2004;564(3):269–273. doi: 10.1016/S0014-5793(04)00320-5. [DOI] [PubMed] [Google Scholar]
- Orry AJ, Wallace BA. Modeling and docking the endothelin G-protein-coupled receptor. Biophys J. 2000;79(6):3083–3094. doi: 10.1016/S0006-3495(00)76543-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Pandit SB, Zhang Y, Skolnick J. TASSER-Lite: an automated tool for protein comparative modeling. Biophys J. 2006;91:4180–4190. doi: 10.1529/biophysj.106.084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi AJ. Role of N-oligosaccharide endoplasmic reticulum processing reactions in glycoprotein folding and degradation. Biochem J. 2002;348:1–13. doi: 10.1042/0264-6021:3480001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95(23):13363–1383 [DOI] [PMC free article] [PubMed]
- Qiu J, Elber R. SSALN: an alignment algorithm using structure-dependent substitution matrices and gap penalties learned from structurally aligned protein pairs. Proteins. 2006;62:881–891. doi: 10.1002/prot.20854. [DOI] [PubMed] [Google Scholar]
- Rarey M, Kramer B, Lengauer T, Klebe G. FlexX: a fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261(3):470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sander C, Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins. 1991;9(1):56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- Smith BJ, Huyton T, Joosten RP, McKimm-Breschkin JL, Zhang JG, Luo CS, Lou MZ, Labrou NE, Garrett TP. Structure of a calcium-deficient form of influenza virus neuraminidase: implications for substrate binding. Acta Cryst D. 2006;62:947–952. doi: 10.1107/S0907444906020063. [DOI] [PubMed] [Google Scholar]
- Taylor W. Identification of protein sequence homology by consensus template alignment. J Mol Biol. 1986;188(2):233–258. doi: 10.1016/0022-2836(86)90308-6. [DOI] [PubMed] [Google Scholar]
- Thompson J, Higgins D, Gibson T. ClustalW: improving the sensitivity of progressive multiple sequence alignments through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcatti G, Vogel H, Chollet A. Probing the binding domain of the NK2 receptor with fluorescent ligands: evidence that heptapeptide agonists and antagonists bind differently. Biochemistry. 1995;34(12):3972–3980. doi: 10.1021/bi00012a015. [DOI] [PubMed] [Google Scholar]
- Turcatti G, Nemeth K, Edgerton MD, Meseth U, Talabot F, Peitsch M, Knowles J, Vogel H, Chollet A. Probing the structure and function of the tachykinin neurokinin-2 receptor through biosynthetic incorporation of fluorescent amino acids at specific sites. J Biol Chem. 1996;271(33):19991–19998. doi: 10.1074/jbc.271.33.19991. [DOI] [PubMed] [Google Scholar]
- Vos WL, Nazarov PV, Koehorst RB, Spruijt RB, Hemminga MA (2009) From ‘I’ to ‘L’ and back again: the odyssey of membrane-bound M13 protein. Trends Biochem Sci [Epub ahead of print] [DOI] [PubMed]
- Vriend G. WHAT IF: A molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-V. [DOI] [PubMed] [Google Scholar]
- Vriend G, Sander C. Quality control of protein models: directional atomic contact analysis. J Appl Cryst. 1993;26:47–60. doi: 10.1107/S0021889892008240. [DOI] [Google Scholar]
- Wu ZL, Ethen C, Hickey GE, Jiang W. Active 1918 pandemic flu viral neuraminidase has distinct N-glycan profile and is resistant to trypsin digestion. Biochem Biophys Res Commun. 2009;379(3):749–753. doi: 10.1016/j.bbrc.2008.12.139. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Honig B. Evaluating conformational free energies: the colony energy and its application to the problem of loop prediction. Proc Natl Acad Sci USA. 2002;99:7432–7437. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]