

Abstract
Epidemiological evidence has suggested that some pediatric leukemias may be initiated in utero and, for some pairs of identical twins with concordant leukemia, this possibility has been strongly endorsed by molecular studies of clonality. Direct evidence for a prenatal origin can only be derived by prospective or retrospective detection of leukemia-specific molecular abnormalities in fetal or newborn samples. We report a PCR-based method that has been developed to scrutinize neonatal blood spots (Guthrie cards) for the presence of numerically infrequent leukemic cells at birth in individuals who subsequently developed leukemia. We demonstrate that unique or clonotypic MLL-AF4 genomic fusion sequences are present and detectable in neonatal blood spots from individuals who were diagnosed with acute lymphoblastic leukemia at ages 5 months to 2 years and, therefore, have arisen during fetal hematopoiesis in utero. This result provides unequivocal evidence for a prenatal initiation of acute leukemia in young patients. The method should be applicable to other fusion genes in children with common subtypes of leukemia and will be of value in attempts to unravel the natural history and etiology of this major subtype of pediatric cancer.
Epidemiological evidence suggests that exposures or events that occur prenatally or in infancy might play a role in the etiology of pediatric acute leukemia, the most common type of childhood cancer in developed countries (1–3). Being able to backtrack leukemic clones to the time of such events would have a considerable impact on our understanding of the natural history of the disease and on the design and interpretation of epidemiological studies. This requires access to both a leukemia-specific marker and, retrospectively, appropriate biological material. The only biological markers that can provide definitive identification of a leukemic clone are clonotypic alterations in DNA that are present in the leukemic cells at diagnosis, e.g., unique nonconstitutive mutations or rearrangements of genes (4–6). These provide specific and sensitive molecular markers for tracking the disease clone with the significant caveat that the mutant DNA sequence identified is not necessarily the initiating or first mutation in the leukemia and, therefore, might be absent at early stages of clonal evolution. The one readily available retrospective source of DNA from leukemic children is the Guthrie card or blood spot taken routinely by heel prick on most newborns (7, 8). Normally used to detect evidence of inborn errors of metabolism, DNA from these spots has been used to detect constitutive mutations (9–11) and exogenous viral sequences (12, 13) using PCR but not, as far as we are aware, for acquired molecular abnormalities in leukemia or other cancers. We reasoned that blood spot DNA from individuals who developed leukemia at a young age would enable us to test the idea that the leukemic clone with its acquired molecular marker could have an in utero fetal origin and therefore be present, albeit at a low level and in a clinically covert form, at birth. To test this hypothesis, we chose the leukemia for which there is both an appropriate marker and the best indirect evidence for initiation in utero. This is infant acute lymphoblastic leukemia with chromosome 11q23 translocations involving the MLL gene (14). In these leukemias, the MLL (or ALL-1, HRX, and HTRX) gene at 11q23 fuses in-frame with any 1 of more than 20 alternative partner genes (15). The resultant product is a hybrid protein with proposed altered transcriptional regulation properties (reviewed in refs. 16–18). The most frequent chromosomal rearrangement in infant acute lymphoblastic leukemia is the t(4;11) (q21;q23); this generates a MLL-AF4 gene fusion, which is believed to be the functional oncogene, as well as a reciprocal AF4-MLL fusion (19). Studies on pairs of infant identical twins with concordant leukemia indicated that in these cases, the unique MLL gene rearrangement was nonconstitutive and acquired during fetal hemopoiesis (20, 21). We now show directly by retrospective testing that the clonotypic MLL-AF4 fusion gene sequences of leukemic cells from young patients (5 months to 2 years of age) are present in their neonatal blood spots.
MATERIALS AND METHODS
DNA, Cell Samples, and Tissue Culture.
Leukemic DNA with the t(4;11) chromosome translocation (= MLL-AF4 fusion), confirmed by reverse transcription (RT)-PCR were obtained from three patients at diagnosis, 5, 6, and 24 months of age. RS4;11 (22) cells were grown at 37°C, 5% CO2/95% air in RPMI 1640 medium supplemented with 10% fetal calf serum (GIBCO), penicillin/streptomycin, and 4 mM l-glutamine, and used in preliminary experiments to determine the detection limits of the t(4;11) rearrangement. Guthrie cards (S&S 2992 standard) were obtained by means of the repositories in regional laboratories and were used for the study with informed consent. Other than these three cases, no other Guthrie cards from infants with MLL-AF4 fusions have been tested. At the time of their use as a source of genomic DNA in these experiments, the Guthrie cards had been stored for a period of 3 years for patient 1, 4 years for patient 2, and 5 years and 2 months for patient 3.
Determination of Clonotypic Patient-Specific MLL-AF4 and AF4-MLL Genomic-Fusion Sequences.
Localization of the MLL gene breakpoint region was carried out as described (23). Essentially, high molecular weight genomic DNA was subject to nested long-range PCR by using primers that span the fusion site between MLL and the partner AF4 gene. Purified PCR products were digested with restriction enzymes, end-labeled with fluorescent dUTP, repurified, and subjected to automatic fluorescence-based analysis as described (24). After localization, breakpoint regions were sequenced by direct Taq cycle-sequencing by using primers selected on the basis of high resolution restriction maps.
PCR.
Reagents for PCR, including equipment and samples, were kept physically isolated from potential sources of contamination by the use of separate laboratories and laminar flow hoods, filtered pipette tips, and Gilson pipettes previously unexposed to sample material. PCR was performed in a Perkin–Elmer Gene Amp 9600 automated PCR processor.
Dilution Experiments.
Cellular dilution experiments to determine the PCR detection limit of the t(4;11) rearrangement was performed by using the cell line RS4;11 diluted into blood mononuclear cells from a healthy adult volunteer. The dilutions contained cell mixtures of 10−1 to 10−6 RS4;11 cells. DNA was extracted from each mixture by standard methods and long-range PCR reactions were performed.
Amplification of the MLL-AF4 translocation region in the RS4;11 cell line was performed by using a long-range PCR system (Expand Long Template PCR system, Boehringer Mannheim) under the following conditions: 1× final buffer (50 mM Tris⋅HCl, pH 9.2/16 mM (NH4)2SO4/2.25 mM MgCl2), 12 pmol each of 27-mer oligonucleotide primers (see below), 500 μM final concentration dNTPS, and 2.5 units of Taq and Pwo enzyme mix, for 10 min at 92°C, 30 sec at 65°C, and 2 min at 68°C × 30 cycles, followed by 7 min of extension at 68°C. Primers were designed from published sequence data of the MLL gene and t(4;11)/MLL-AF4 fusion region deposited in GenBank (25): (forward) 5′-TACACTTGTGTGAACTGTACTGAGCGG-3′ and (reverse) 5′-GCTGTCCTCCATCTGAATTAATGGCTG-3′.
“Artificial” Guthrie Cards.
Blood from a healthy volunteer was taken and immediately placed on ice. No anticoagulating agents were added. Serial dilutions of RS4;11 cells carrying the MLL-AF4 rearrangement were added in PBSA for a final dilution range of 10−1 to 10−5 RS4;11 cells. Thirty-three microliters of each mixture was spotted onto filter paper to fill a 1-cm “Guthrie” circle representing approximately 3.5 × 104 lymphocytes per Guthrie spot. The cards were left to dry at room temperature and away from light for 4 weeks prior to DNA amplification of the genomic MLL-AF4 fusion sequence by long-range PCR.
PCR Analysis.
To enhance the amplification efficiency of genomic DNA from Guthrie card and artificial Guthrie card specimens by eluting potential PCR contaminants, a procedure developed by Makowski et al. was followed (26). Guthrie pieces to be amplified were incubated in 0.5 ml ddH2O twice for 30 min each, then dried in a vacuum dessicator prior to PCR amplification reaction.
PCR amplification on diagnostic DNA and a segment or tranche equivalent to approximately one-eighth of the blood spot from Guthrie card samples was conducted in 0.6-ml thin-walled PCR tubes at 50 μl final volume of the following: 1× buffer (60 mM KCL/15 mM Tris, pH 8.8/1.5 mM MgCl2), 200 μM dNTPs, 12 pmol each of 5′ and 3′ oligonucleotide primers, and 2.5 units of Amplitaq DNA polymerase (Perkin–Elmer). Reactions were covered with 30 μl of mineral oil.
For the amplification of patient-specific t(4;11) fusion sequences, oligonucleotides were designed from 300–400 bp of patient-specific sequence data of each patient’s MLL-AF4 genomic fusion region or the reciprocal product AF4-MLL genomic fusion region: patient 1, (forward) 5′-CCTCACCCAAATTCCCTAAGTG-3′ and (reverse) 5′-CACAAGCAGGAACTAGGGTTTAG-3′; patient 2, (forward) 5′-AGCAGACGACTTCCAAACCGC-3′ and (reverse) 5′-TCCTGACCTTGTGATCCGCCT-3′; patient 3 (forward) 5′-GCATCTCCATTGCTCAAAGACA-3′ and (reverse) 5′-ATGCCCACTACTGGCACAGAGAA-3′.
Optimal thermocycling conditions were established for each patient-specific pair of MLL-AF4 primers by using diagnostic DNA.
The N-RAS DNA sequence was amplified as an internal control for Guthrie card DNA with N-RAS-specific primers: (forward) 5′-CTGGTGTGAAATGACTGAGT-3′ and (reverse) 5′-GGTGGGATCATATTCATCTA-3′, and the following thermocycling conditions: denaturing, annealing, and extension steps were performed for 1 min at 95°C, 1 min at 55°C, 1 min at 72°C for 30 cycles.
PCR products were routinely size-fractionated on agarose gels with molecular weight DNA markers (Boehringer Mannheim) and visualized by ethidium bromide staining. To sequence Guthrie specimen MLL-AF4 or AF4-MLL PCR products, each 10 μl asymmetric PCR reaction contained 3 μl of low-melt gel-purified PCR product, 4 μl of Dye terminator Cycle Sequencing Ready reaction mix (Perkin–Elmer), 3.2 μM of upstream or downstream primer, and was run through 30 cycles of 30 sec at 95°C, 30 sec at 55°C, and 1 min at 70°C. Sequence reactions were carried out by using an Applied Biosystems 373A automated DNA sequencer and further analyzed by geneworks 2.1 (IntelliGenetics).
RESULTS
Dilution Experiments with Leukemic Cells in Blood Spots.
The cell line RS4;11 carries a t(4;11) translocation in which the MLL gene is fused with a partner gene, AF4, located at 4q21 (27). The break in MLL occurs in the small intron 7 and genomic rearrangements can be detected in DNA by PCR by using primers as previously defined (ref. 25, and see Materials and Methods). Dilution experiments on the RS4;11 cell line were used to establish the detection sensitivity of the t(4;11) rearrangement by PCR. Analysis on DNA extracted from serial dilutions of RS4;11 cells indicated a detection limit of 0.01 ng (≈10 cells) (data not shown). To establish the detection limit of the t(4;11) rearrangement in Guthrie card DNA, serial dilutions of RS4;11 cells were artifically mixed with normal blood, without coagulant, for a final dilution range of 10−1 to 10−5, whereby 33 μl drops were immediately spotted onto Guthrie filter paper. This volume filled the standard 1-cm diameter circle used in clinics and hospitals and represented approximately 3 × 104 cells. PCR amplification on these artificial Guthrie specimens indicated a detection limit of the MLL-AF4 breakpoint sequence at 10−4 (1 leukemic cell in 10,000 normal) (Fig. 1). These preliminary data obtained in two repeat experiments indicate that MLL-AF4 fusion sequences were preserved after drying of cells on Guthrie cards and could be detected at low copy number.
Figure 1.
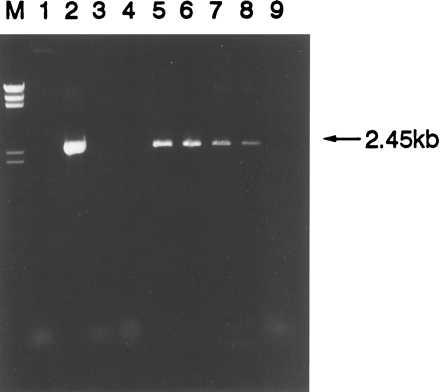
Amplification of 2.45 kb product of MLL-AF4 rearrangement from RS4;11 cell line DNA and RS4;11 “imitation” Guthrie specimens by PCR. Lanes: M, DNA marker II; 1, no DNA control; 2, 100 ng positive control RS4;11 DNA; 3, 100 ng human placenta negative control DNA; 4, 100 ng normal genomic negative control DNA; 5–9, 10−1 through 10−5 dilutions of RS4;11 cells on imitation Guthrie specimens.
PCR on Diagnostic DNA.
Unlike that in RS4;11 cells, breaks in the majority of patients with MLL gene fusions occur in the relatively large intron 6 (1.4 kb) or 8 (3.5 kb), as well as in intronic regions of the partner gene (28). A consequence is that the resultant genomic fusions are likely to fall within very large regions with respect to flanking coding sequences and will be difficult to detect even by long-range PCR, especially if present at low copy number. This difficulty is circumvented in the analysis of low leukemic cell numbers in treated patients (i.e., minimal residual disease) by RT-PCR analysis of cDNA in which the large intronic sequences are deleted (29). We were unable to detect MLL-AF4 fusion sequences by RT-PCR in cDNA prepared from artificial Guthrie cards containing RS4;11 cells (data not shown), although β-hemoglobin cDNA was detectable as previously reported (30). We therefore had to obtain genomic sequence information on the MLL-AF4 fusion in diagnostic leukemic cell DNA prior to PCR analysis of the same patients’ Guthrie card. For direct sequencing of diagnostic leukemic DNA across the fusion site, we have used a rapid new technique (23) that incorporates long-range PCR, fluorescent end-labeling of restriction digest fragments, and automatic fluorescence-based analysis. Appropriate intron sequences relatively close to and on either side of the MLL-AF4 junction or the reciprocal AF4-MLL junction were then identified and used to design clonotypic or patient-specific primers for short-range genomic amplification by PCR of Guthrie card DNA.
Dilution experiments on diagnostic DNA with clonotypic primers of patients 1 and 2 resulted in a reproducible PCR detection limit of the MLL-AF4 fusion sequence at 10 and 100 pg of DNA, respectively (Fig. 2 A–C), and detection for patient 3 of 1–10 pg (Fig. 2D). PCR amplification of diagnostic DNA from an individual patient by using specific primers from the other patients gave no amplification product (Fig. 2).
Figure 2.
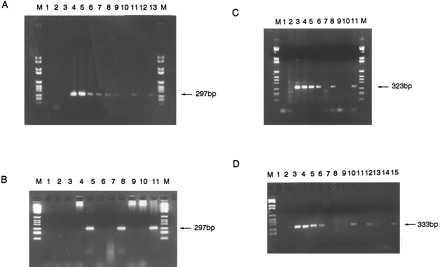
(A) PCR amplification of 297-bp-specific MLL-AF4 breakpoint region from diagnostic patient 1 leukemic DNA dilutions and two one-eighth segments of Guthrie spot. Lanes: M, DNA marker VI; 1, no DNA control; 2, 100 ng human placenta; 3, 100 ng patient 1 DNA with nonspecific primers; 4–10, patient 1 diagnostic DNA dilutions 100 ng–10 pg; 11, one-eighth segment patient 1 Guthrie; 12, “normal” Guthrie negative control; 13, one-eighth segment patient 1 Guthrie; M, DNA marker. (B) PCR on diagnostic DNA and third and fourth pieces of Guthrie spot from patient 1. Lanes: M, DNA marker VI; 1, no DNA control; 2, 100 ng human placenta negative control; 3 and 4, negative control one-eighth segments of normal Guthries 1 and 2; 5, third one-eighth segment of patient 1 Guthrie; 6 and 7, negative control normal Guthries 3 and 4; 8, fourth one-eighth segment of patient 1 Guthrie; 9 and 10, negative control normal Guthries 5 and 6; 11, patient 1 diagnostic DNA; M, DNA marker VI. (C) PCR amplification of specific 323-bp product of the AF4-MLL breakpoint region from patient 2 diagnostic DNA and two segments of Guthrie spot. Lanes: M, DNA marker VI; 1, no DNA control; 2, 100 ng patient 2 DNA with nonspecific primers; 3–6, patient 2 DNA 100 ng–0.1 ng; 7, 100 ng human placenta negative control; 8, one-eighth segment patient 2 Guthrie; 9 and 10, one-eighth segments of normal, negative control Guthries; 11, one-eighth segment of patient 2 Guthrie; M, DNA marker VI. (D) PCR amplification of 333-bp product specific to patient 3 AF4-MLL breakpoint region from diagnostic DNA and four one-eighth segments of Guthrie spot. Lanes: M, DNA marker VI; 1, no DNA control; 2, 100 ng human placenta negative control; 3–6, patient 3 diagnostic DNA 10 ng–10 pg; 7, patient 3 DNA with nonspecific primers; 8 and 9, one-eighth segments of negative control normal Guthries; 10–15, six one-eighth segments of patient 3 Guthrie spot.
PCR on Guthrie Specimens.
PCR was performed on Guthrie spot slices as described. For all three patients tested, an amplified product of the same base-pair size as in the diagnostic DNA sample was detectable in Guthrie card blood spot DNA (Fig. 2). These data were confirmed in two or three independent experiments for each patient sample with separate slices of the blood spot. In total, 4 of 4 slices were positive for patient 1, 3 of 3 were positive for patient 2, and 4 of 6 were positive for patient 3. The absence of any product in the negative controls—no DNA, 100 ng human placenta DNA, and normal (nonleukemic) Guthrie specimens, indicated that no cross contamination occurred (Fig. 2). N-RAS was amplified as a control for intact DNA and successful PCR (data not shown). To verify that the breakpoint fusion sequence amplified from the patients’ blood spot was identical to that of their diagnostic leukemic DNA, PCR products amplified from blood spot DNA were purified and sequenced as described. The MLL-AF4 or AF4-MLL fusion sequence from each patients’ Guthrie spot DNA was verified by both strands as identical to that of their respective diagnostic leukemic DNA (Fig. 3).
Figure 3.

Sequence of three patients (labeled 1, 2, and 3) diagnostic leukemic DNA (unshaded upper) and the (identical) MLL-AF4 or AF4-MLL fusion sequence from Guthrie DNA (shaded lower). Patient-specific oligonucleotide primers were derived from underlined sequence. (▾), MLL-AF4 or AF4-MLL breakpoint fusion for each patient.
DISCUSSION
The detection of leukemic gene fusion sequences in DNA by retrospective screening of neonatal blood spots is a novel finding anticipated from earlier studies with four pairs of identical twin infants (2–10 months of age) with leukemia, whose malignant cells had the same acquired rearrangement of the MLL gene (20, 21). It was argued that the sharing of a unique nonconstitutive gene rearrangement by the leukemic cells of two identical twins was only possible if the abnormality arose in a single cell in one fetus, the clonal progeny of which then colonized the other twin in utero via intraplacental anastomoses. If this interpretation is correct, then fetal blood and presumably neonatal blood would be expected to contain leukemic cells. It could be argued, however, that the selection of concordant leukemias in twins biased analysis of MLL gene fusions to those that must have originated before birth in utero. It was therefore theoretically possible that a prenatal origin did not hold true for discordant twin leukemias (i.e., leukemia in one twin only of an identical pair) or in nontwinned infants, i.e., most patients. The present study removes this doubt by providing direct molecular evidence that the patient and clone-specific MLL-AF4 (or AF4-MLL) gene fusion sequence in each of three patients tested was already present at birth.
In two of the selected cases the leukemia required 5 to 6 postnatal months for clinical manifestation, which is the average age at presentation for infant ALL with MLL-AF4 gene fusions. A small proportion of patients with ALL 1–2 years of age have MLL-AF4 and an even smaller number with MLL-AF4 are between 2 and 15 years of age (31). Our third case (patient 3) therefore represents the tail end of the age distribution of incidence for this biological subset of leukemias and is particularly instructive, because it illustrates the fact that leukemias with a fetal origin are not necessarily only those that develop and are diagnosed in the first year of life. The later age of diagnosis in this case might be associated with later onset in the fetus and/or a lower clone size at birth. We have not attempted to accurately quantify the numbers of cells in each blood spot that have the leukemic molecular marker but we can make some approximate estimates. The sensitivity of the assay varied with each patient-specific fusion gene sequence and primer pair. Dilution experiments with diagnostic DNA indicated that 10 pg of DNA, or as few as 1–10 cells per segment (≈one-eighth of blood spot or ≈4 μl of blood) could be detected. The intensity of the PCR amplified bands in these cases, when compared with those in the limiting dilution analysis of diagnostic material, although at best only a crude quantitative estimate, suggests that the number of MLL-AF4 fusion gene copies retrievable from the Guthrie specimens was probably around 20 per slice (or 200 per spot) for patients 1 and 2, but only 1 or 2 (10–20 per spot) in patient 3. This estimate, if correct, would accord with the observation that some sections (2 of 6) of the blood spot from patient 3 scored negative for the MLL-AF4 sequence. It is of interest that in this small series of patient samples, the case with the longest latency (patient 3) had the fewest detectable leukemic cells in neonatal blood spot. If we assume blood counts were normal at birth in these three patients, then with approximately 4 × 104 mononuclear cells per blood spot, these estimated values of leukemic cell numbers translate into a leukemic burden at birth of between 1 in 4,000 and 1 in 200 cells. In future studies it will be of interest to evaluate more systematically the relationship between fusion gene frequency and the interval from birth to clinical diagnosis. However, there may not be a simple linear relationship between these two variables either as indicated by serial PCR monitoring of minimal residual disease in treated patients with ALL (32).
Leukemias associated with MLL gene fusions have some unusual features. Most pediatric leukemias with MLL gene fusions clearly have a remarkably short latency (less than 1 year in most cases, though clearly more than 2 years in the case of our patient 3). MLL gene fusions are also associated with secondary leukemias of pediatric and adult patients previously treated with the topoisomerase II inhibitors etoposide (VP-16), teniposide (VP-26), or anthracyclines (33, 34). In addition, in these cases latency is short (1–3 years) compared with that recorded for other types of leukemogenic exposures and leukemic subtypes (3, 35). There is also a very high rate of concordance of leukemia in infant identical twins (36), most of which will have MLL fusion genes. The actual concordance rate is unknown; it is usually quoted as around 25% (or 1 in 4), but might approximate to 100% for those (≈60%) monozygotic twins that share a single, monochorionic placenta (36). Collectively, these data suggest that the presence of an in-frame MLL fusion gene in an “appropriate” cell type may be sufficient for the development of clinical acute leukemia. However, pediatric patients with translocations involving the MLL gene at diagnosis often have additional genetic abnormalities (37, 38), and although these may represent diverse secondary events, they might contribute critically to the pathogenesis of disease. It is therefore possible that MLL gene fusions arise more frequently in fetal development than the subsequent incidence rate of pediatric leukemia would indicate. It follows that it will be important to evaluate the frequency with which MLL-AF4 or other MLL gene fusions are present in unselected newborn blood samples. This cannot be done by scrutinizing Guthrie card blood spots with clonotypic primer-based PCR as in this study, but is possible by RT-PCR screening of cDNA prepared from cord blood and by using generic primers. Such a study has been initiated.
An important aspect of this study is the potential it provides to evaluate the natural history of leukemia in children with the common form of B cell precursor or common ALL, which peaks at 2–4 years of age and is the major subtype of pediatric leukemia and cancer. It has been suggested that some of these cases could also be initiated in utero, but involve postnatal exposures to infection (3, 36); the evidence is, however, largely epidemiological and indirect. Molecular studies on identical twins who developed T cell malignancy at 9 or 10 years of age provides compelling evidence that a fetal origin coupled with protracted latency is possible (39). Clonotypic molecular markers now exist for the common forms of childhood acute leukemia (40, 41), and it should be possible to tackle this important question by a similar analysis of Guthrie card neonatal blood spots. It is important in this respect to note that the blood spots in which we have been able to identify leukemia fusion gene sequences had been stored for up to 5 years before use in this study.
Finally, our data reinforces the notion that exposures of pregnant mothers and their unborn fetuses are critical, time-constrained events in the etiology of acute leukemias in young children or infants (1, 3). Candidate exposures, particularly with natural or medicinal inhibitors of topoisomerase II (3, 42) are being assessed in ongoing case/control epidemiological studies.
Acknowledgments
We are grateful to Drs. M. Addison, R. Jones, S. Evans, A. Heeley, and A. D. J. Pearson, who identified and provided Guthrie cards. We thank Dr. J. Brown for technical advice; Dr. C. M. Price and Ms. R. Dhat for RT-PCR analysis of diagnostic samples in the United Kingdom Children’s Cancer Study (UKCCS); Prof. L. Secker-Walker for providing information from the Leukaemia Research Fund/UKCCG karyotype database in ALL; Drs. T. Enver, L. M. Wiedemann, T. Leis, and J. L. Wiemels for critical comments; Barbara Deverson for help in manuscript production; and Dr. Sally Davies for first suggesting the possible use of Guthrie cards for these experiments. This work was supported by the Leukaemia Research Fund (United Kingdom), the Kay Kendall Leukaemia Fund (United Kingdom), and the Cancer Research Campaign. We thank the UKCCCR, the UKCCS Management Committee, the UKCCG, and physicians who referred patient diagnostic samples, including Drs. A. Will, R. Stevens, J. M. Chessells, F. E. Cotter, V. Broadbent, A. D. J. Pearson, M. Reid, and P. Middleton.
Footnotes
References
- 1.Ross J A, Davies S M, Potter J D, Robison L L. Epidemiol Rev. 1994;16:243–272. doi: 10.1093/oxfordjournals.epirev.a036153. [DOI] [PubMed] [Google Scholar]
- 2.Shu X-O, Ross J A, Pendergrass T W, Reaman G H, Lampkin B, Robison L L. J Natl Cancer Inst. 1996;88:24–31. doi: 10.1093/jnci/88.1.24. [DOI] [PubMed] [Google Scholar]
- 3.Greaves M F. Lancet. 1997;349:344–349. doi: 10.1016/s0140-6736(96)09412-3. [DOI] [PubMed] [Google Scholar]
- 4.Rowley J D. Semin Hematol. 1990;27:122–136. [PubMed] [Google Scholar]
- 5.Rabbitts T H. Nature (London) 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 6.Sawyers C L. Lancet. 1997;349:196–200. doi: 10.1016/S0140-6736(96)07535-6. [DOI] [PubMed] [Google Scholar]
- 7.Guthrie R. In: Neonatal Screening for Inborn Errors of Metabolism. Bickel H, Guthrie R, Hammersen G, editors. Berlin: Springer; 1980. pp. 259–270. [Google Scholar]
- 8.McCabe E R B, Huang S-Z, Seltzer W K, Law M L. Hum Genet. 1987;75:213–216. doi: 10.1007/BF00281061. [DOI] [PubMed] [Google Scholar]
- 9.Jinks D C, Minter M, Tarver D A, Vanderford M, Hejtmancik J F, McCabe E R B. Hum Genet. 1989;81:363–366. doi: 10.1007/BF00283692. [DOI] [PubMed] [Google Scholar]
- 10.Gregersen N, Blakemore A I F, Winter V, Andresen B, Kolvraa S, Bolund L, Curtis D, Engel P C. Clin Chim Acta. 1991;203:23–34. doi: 10.1016/0009-8981(91)90153-4. [DOI] [PubMed] [Google Scholar]
- 11.Raskin S, Phillips J A, Kaplan G, McClure M, Vnencak-Jones C. PCR Methods Appl. 1992;2:154–156. doi: 10.1101/gr.2.2.154. [DOI] [PubMed] [Google Scholar]
- 12.Comeau A M, Hsu H-W, Schwerzler M, Mushinsky G, Walter E, Hofman L, Grady G F. J Pediatr. 1993;123:252–258. doi: 10.1016/s0022-3476(05)81697-x. [DOI] [PubMed] [Google Scholar]
- 13.Noda S, Eizuru Y, Minamishima Y, Ikenoue T, Mori N. J Virol Methods. 1993;43:111–122. doi: 10.1016/0166-0934(93)90094-8. [DOI] [PubMed] [Google Scholar]
- 14.Thirman M J, Gill H J, Burnett R C, Mbangkollo D, Mccabe N R, Kobayashi H, Ziemin van der Poel S, Kaneko Y, Morgan R, Sandberg A A, Chaganti R S K, Larson R A, Le Beau M M, Diaz M O, Rowley J D. N Engl J Med. 1993;329:909–914. doi: 10.1056/NEJM199309233291302. [DOI] [PubMed] [Google Scholar]
- 15.Pui C-H, Kane J R, Crist W M. Leukemia. 1995;9:762–769. [PubMed] [Google Scholar]
- 16.Bernard O A, Berger R. Genes Chromosomes Cancer. 1995;13:75–85. doi: 10.1002/gcc.2870130202. [DOI] [PubMed] [Google Scholar]
- 17.Canaani E, Nowell P C, Croce C M. Adv Cancer Res. 1995;66:213–234. doi: 10.1016/s0065-230x(08)60255-9. [DOI] [PubMed] [Google Scholar]
- 18.Downing J R, Look A T. In: Molecular Genetics and Therapy of Leukemia. Freireich E J, Kantarjian H, editors. Boston: Kluwer Academic; 1996. pp. 73–90. [Google Scholar]
- 19.Chen C-S, Sorensen P H B, Domer P H, Reaman G H, Korsmeyer S J, Heerema N A, Hammond G D, Kersey J H. Blood. 1993;81:2386–2393. [PubMed] [Google Scholar]
- 20.Ford A M, Ridge S A, Cabrera M E, Mahmoud H, Steel C M, Chan L C, Greaves M F. Nature (London) 1993;363:358–360. doi: 10.1038/363358a0. [DOI] [PubMed] [Google Scholar]
- 21.Gill Super H J, Rothberg P G, Kobayashi H, Freeman A I, Diaz M O, Rowley J D. Blood. 1994;83:641–644. [PubMed] [Google Scholar]
- 22.Stong R C, Korsmeyer S J, Parkin J L, Arthur D C, Kersey J H. Blood. 1985;65:21–31. [PubMed] [Google Scholar]
- 23.Leis, T., Repp, R., Metzler, M., Borkhardt, A., Schlager, F. & Lampert, F. (1997) Leukemia, in press. [DOI] [PubMed]
- 24.Repp R, Borkhardt A, Haupt E, Kreuder J, Brettreich S, Hammermann J, Nishida K, Harbott J, Lampert F. Leukemia. 1995;9:210–215. [PubMed] [Google Scholar]
- 25.Djabali M, Selleri L, Parry P, Bower M, Young B D, Evans G A. Nat Genet. 1992;2:113–118. doi: 10.1038/ng1092-113. [DOI] [PubMed] [Google Scholar]
- 26.Makowski G S, Davis E L, Aslanzadeh J, Hopfer S M. Nucleic Acids Res. 1995;23:3788–3789. doi: 10.1093/nar/23.18.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domer P H, Fakharzadeh S S, Chen C-S, Jockel J, Johansen L, Silverman G A, Kersey J H, Korsmeyer S J. Proc Natl Acad Sci USA. 1993;90:7884–7888. doi: 10.1073/pnas.90.16.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broeker P L S, Super H G, Thirman M J, Pomykala H, Yonebayashi Y, Tanabe S, Zeleznik-Le N, Rowley J D. Blood. 1996;87:1912–1922. [PubMed] [Google Scholar]
- 29.Biondi A, Rambaldi A, Rossi V, Elia L, Caslini C, Basso G, Battista R, Barbui T, Mandelli F, Masera G, Croce C, Canaani E, Cimino G. Blood. 1993;82:2943–2947. [PubMed] [Google Scholar]
- 30.Zhang Y H, McCabe E R B. Hum Genet. 1992;89:311–314. doi: 10.1007/BF00220548. [DOI] [PubMed] [Google Scholar]
- 31.Greaves M F. Leukemia. 1996;10:372–377. [PubMed] [Google Scholar]
- 32.Roberts W M, Estrov Z, Ouspenskaia M V, Johnston D A, McClain K L, Zipf T F. N Engl J Med. 1997;336:317–323. doi: 10.1056/NEJM199701303360501. [DOI] [PubMed] [Google Scholar]
- 33.Gill Super H J, McCabe N R, Thirman M J, Larson R A, Le Beau M M, Pedersen-Bjergaard J, P, P, Diaz M O, Rowley J D. Blood. 1993;82:3705–3711. [PubMed] [Google Scholar]
- 34.Pui C-H, Relling M V, Rivera G K, Hancock M L, Raimondi S C, Heslop H E, Santana V M, Ribeiro R C, Sandlund J T, Mahmoud H H, Evans W E, Crist W M, Krance R A. Leukemia. 1995;9:1990–1996. [PubMed] [Google Scholar]
- 35.Smith M A, McCaffrey R P, Karp J E. J Natl Cancer Inst. 1996;88:407–418. doi: 10.1093/jnci/88.7.407. [DOI] [PubMed] [Google Scholar]
- 36.Greaves M. Blood. 1993;82:1043–1051. [PubMed] [Google Scholar]
- 37.Heerema N A, Arthur D C, Sather H, Albo V, Feusner J, Lange B J, Steinherz P G, Zeltzer P, Hammond D, Reaman G H. Blood. 1994;83:2274–2284. [PubMed] [Google Scholar]
- 38.Cimino G, Lanza C, Elia L, Lo Coco F, Gaidano G, Biondi A, Pastore C, Serra A, Canaani E, Croce C M, Mandelli F, Saglio G. Br J Haematol. 1997;96:308–313. doi: 10.1046/j.1365-2141.1997.d01-2044.x. [DOI] [PubMed] [Google Scholar]
- 39.Ford A M, Pombo-de-Oliveira M S, McCarthy K P, MacLean J M, Carrico K C, Vincent R F, Greaves M. Blood. 1997;89:281–285. [PubMed] [Google Scholar]
- 40.Golub T R, Barker G F, Bohlander S K, Hiebert S W, Ward D C, Bray-Ward P, Morgan E, Raimondi S C, Rowley J D, Gilliland D G. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romana S P, Mauchauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, Bernard O A. Blood. 1995;85:3662–3670. [PubMed] [Google Scholar]
- 42.Ross J A, Potter J D, Robison L L. J Natl Cancer Inst. 1994;86:1678–1680. doi: 10.1093/jnci/86.22.1678. [DOI] [PubMed] [Google Scholar]