

Abstract
Two new monocyclic analogs of the natural AChE inhibitor cyclophostin and two exocyclic enol phosphates were synthesized. The potencies and mechanisms of inhibition of the bicyclic and monocyclic enol phosphonates and the exocyclic enol phosphates toward human AChE are examined. One diastereoisomer of the bicyclic phosphonate exhibits an IC50 of 3 μM. Potency is only preserved when the cyclic enol phosphonate is intact and conjugated to an ester. Kinetic analysis indicates both a binding and a slow inactivation step for all active compounds. Mass spectrometric analysis indicates that the active site Ser is indeed phosphorylated by the bicyclic phosphonate.
Acetylcholinesterase (AChE) catalyzes the hydrolysis of the neurotransmitter acetylcholine (ACh) to choline and acetic acid. This enzyme belongs to the very large serine hydrolase class of enzymes1 which are characterized by the classic α/β hydrolase fold structure and the famous catalytic triad composed of an acidic residue, His, and Ser. This latter residue serves as the nucleophile in the attack on ester or amide substrates, and as such is the site of modification by irreversible inhibitors.
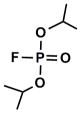
Among the most potent and well studied irreversible AChE inhibitors are the organophosphates (OP). The chemical warfare agents Sarin and VX, insecticides such as malathion, and the well known hydrolase inhibitor DIFP belong to this class of compounds (Fig. 1). These potent inhibitors are highly electrophilic and possess excellent leaving groups, leading to strong reactivity and phosphorylation of the active site Ser residue.
Fig. 1.

Structures of Some Organophosphate Inhibitors of AChE.
Once this Ser has been modified by an OP, the serine-OP inhibitor adduct can undergo a number of subsequent reactions.2 These include: i) Simple hydrolysis, restoring the Ser residue. This is generally not efficient among OPs. ii) Aging, in which ester hydrolysis occurs, generating monoester at Ser. This is typically a slow reaction, with half lives of hours. iii) Reactivation by treatment with an oxime (HON=R), which attacks the adduct and liberates Ser and a new oxime adduct. Indeed, oximes are used as antidotes for chemical warfare agents which target AChE.2 This reaction is faster against VX, Sarin, and paraoxon than other OPs.
AChE is also of interest due to its central role in memory and Alzheimer’s disease (AD). The memory loss in AD patients is aggravated by reduced levels of ACh.3 Boosting ACh levels by inhibiting AChE has proven to be an effective means of managing early stages of the disease. While three of the AD drugs currently on the market are AChE inhibitors,3 the increase in elderly population and incidence of the disease has spurred continued interest in new inhibitors of this enzyme.4, 5
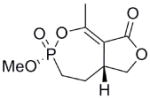
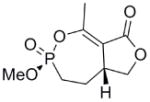
This clinical interest has intensified the search for natural product inhibitors of AChE. Polycyclic structures are a common characteristic of these compounds.6 To our knowledge, only two phosphorus-containing natural product AChE inhibitors have been reported.6 The better characterized of these is cyclophostin (1), a compound which has been isolated from the NK901093 strain of the bacterium Streptomyces lavendulae (Table 1).7 Cyclophostin was reported to exhibit an IC50 of 0.8 nM toward insect AChE.8 We recently reported the synthesis of two diastereoisomeric bicyclic phosphonate analogs (2a) and (2b) of cyclophostin which exhibit low μM IC50s toward human AChE. Furthermore, the activity was not compromised in the synthetic precursors (3a) and (3b) lacking the lactone ring (Figure 2).9
Table 1.
Inhibition of AChE by Cyclic Phosphonatesa
| Compound | Structure | IC50, uM | Ki, uMb | k2, min−1b | k−2, min−1b |
|---|---|---|---|---|---|
| 1 | 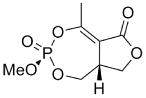 |
8e-4c | n/a | n/a | n/a |
| 2a | 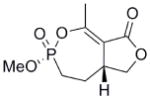 |
3d | 24 ± 6.0 | 0.3 ± 0.06 | 0.02 |
| 2b | 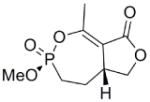 |
30d | 140 ± 58 | 0.4 ± 0.1 | 4e-3 |
| 3a |  |
≈ 35 | n/a | n/a | n/a |
| 3b | 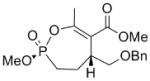 |
≈ 6 | n/a | n/a | n/a |
| 9 | 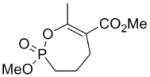 |
7 | 76 ± 20 | 0.2 ± 0.03 | 0.01 |
| 10 | 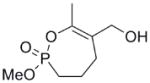 |
600 | n/a | n/a | n/a |
| 15 | 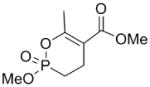 |
5 | 310 ± 130 | 4.2 ± 1.5 | 0.13 |
| 17a | 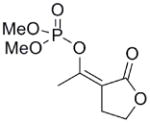 |
>1000 | n/a | n/a | n/a |
| 17b | 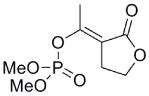 |
70 | n/a | n/a | n/a |
| DIFP | 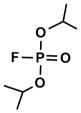 |
0.12 | 6.0±1.0 | 1.0 ± 0.1 | 0.13 |
Fig. 2.

Structures of Cyclophostin and Selected Phosphonate Analogs.
Cyclophostin is an unusual AChE inhibitor in that it combines the polycyclic structure common to natural product inhibitors6 with a phosphate center, the central feature of the well known organophosphorus inhibitors like Sarin.10 The cyclic phosphate provides opportunities to explore the relationship between structural strain and inhibitor reactivity, as well as great potential in exploring structure-activity relationships. Described here is the synthesis and first mechanistic study of a new class of cyclic enol phosphonate inhibitors of AChE derived by modification of the basic cyclophostin structure.
RESULTS
Synthesis of Monocyclic Phosphonate Analogs
Following a similar synthetic sequence to that used in the synthesis of the bicyclic phosphonates,8 the phosphono allylic carbonate (4) was reacted with methyl acetoacetate using a palladium (0) catalyst (Scheme 1). The unsubstituted carbonate was more reactive than the previously studied substituted carbonates8 and the reaction was complicated by formation both the mono- and the di-allyl products (5 & 6).
Scheme 1.

Hydrogenation of the vinyl phosphonate (5) with hydrogen gas over Pd on carbon in methanol gave the saturated phosphonate (7) in quantitative yield. Selective mono demethylation was achieved by reaction with NaI in refluxing acetonitrile to give the sodium salt, which was treated directly with Amberlite IR120 resin to give the corresponding phosphonic acid (8). Cyclization of the crude phosphonic acid (8) with a combination EDC and HOBt gave the 7-membered ring cyclic enol phosphonate (9) (Scheme 1). The methyl ester was reduced with an excess of diisobutyl aluminum hydride (DIBAL-H) to give the corresponding allylic alcohol (10).
For the synthesis of the 6-membered cyclic phosphonate (Scheme 2), methyl acetoacetate (10) was added to dimethyl vinyl phosphonate (12) under basic conditions (t-BuOK) to give the phosphonate (13) in 62% yield. Demethylation, protonation and cyclization as described above gave the 6-membered cyclic enol phosphonate (15) in 40% yield.
Scheme 2.

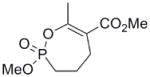
The exocyclic enol phosphates were prepared selectively by phosphorylation of acetyl butyrolactone (16) with dimethyl chlorophosphate under the appropriate conditions (Scheme 3). The Z isomer (17a) was produced when sodium hydride was used as base in THF solution, whereas the E isomer (17b) was the predominant product when Hunig’s base in CH2Cl2 was used.
Scheme 3.

Inhibition of AChE by Bicyclic Phosphonate Analogs of Cyclophostin
To understand what features of cyclophostin are important for inhibition of human AChE, the new compounds were tested for activity (Table 1). As reported previously,9 one diastereoisomer of the bicyclic phosphonate analog exhibits an IC50 of 3 μM, the best of the series. The other bicyclic diastereomer has an IC50 of 30 μM. This stereospecificity is more apparent in Fig. 3, which illustrates the inhibitor concentration dependence of the relative activity. We note that these IC50’s are substantially poorer than the value reported for the phosphate natural product from insect.8
Fig. 3. Inhibition AChE by (2a) and (2b).

34 nM human AChE was incubated with inhibitor for 30 min, then assayed. See Experimental section for additional details. (2a) Circles and solid line and (2b) squares and dashed line. The lines are not fits, but rather meant to guide the eye
Interestingly, the monocyclic 6-membered ring analog (15) and the 7-membered ring analog (9) have IC50’s comparable to the bicyclic analog (2a). However, reduction of ester carbon of (9) to an alcohol (10) dramatically increases the IC50 (100-fold), indicating either the importance of the electron withdrawing ester in enhancing the leaving group characteristics of the enol from the phosphonate center and/or important hydrogen bonding accepting contacts for the carbonyl carbon (see Discussion). Moreover, the E isomer of the compound which has an intact lactone ring but is open at the phosphorus center (17b) is reasonably active, but the corresponding Z isomer (17a) is less so (Table 1). Collectively, these results indicate that there is a requirement for structural alignment of the phosphorus center with the carboxy ester/lactone for inhibitory activity.
Mechanism of Inhibition
As stated in the introduction, no description of the type of AChE inhibition exhibited by cyclic OPs has been previously reported. To establish the mode(s) of action for these compounds, a more detailed analysis was conducted. Since most organophosphates irreversibly inhibit serine hydrolases, we began by assessing the general irreversibility of cyclic OP inhibition of AChE. This was accomplished by incubating the enzyme with (2a) for 30 min, removing the inhibitor using a desalting column, and then assessing residual activity. As shown in Fig. 4, residual AChE activity after incubation and removal of the inhibitor is barely detectable. This is consistent with irreversible inhibition on the timescale of this experiment, most likely by covalent modification (see below).
Fig. 4. Irreversibility of Inhibition by (2a).

E, activity of enzyme incubated in buffer for 30 min and subsequently desalted. E + I, activity of enzyme treated with inhibitor for 30 min but not desalted. The average activity for three trials was 0.2 % of initial activity. E + I + desalt, activity of enzyme incubated with inhibitor for 30 min and subsequently purified by desalting gel. See Experimental section for additional details.
To dissect this behavior, residual enzyme activities were obtained as a function of inhibitor concentration and incubation time for all compounds with low micromolar IC50’s (Fig. 5).
Fig. 5. Kinetics of AChE Inactivation by Cyclic Organophosphonates.

Left column: plots of percent residual activity vs. incubation time for the indicated inhibitors. Right column: kobs derived from global analysis vs. [I]. Data were collected and processed as described in Experimental. For (2a), [I] = 0.5, 1, 2, 4, 8, 16 uM; (2b), [I] = 5, 10, 20, 40, 80 uM; (9) [I] = 5, 10, 20, 40, 80 uM; (15) [I] = 5, 10, 20, 40, 80, 160 uM; DIFP [I] = 2, 4, 8, 16 uM.
The mechanism in Scheme 4, rather than the resolutely irreversible inactivation mechanism of Kitz and Wilson,11 was utilized because it permits the exploration of reversible inactivation. kobs values derived from these data were plotted vs. inhibitor concentration to obtain KI, which represents the dissociation equilibrium constant or affinity for the inhibitor, and k2 and k−2, the forward and reverse rate constants for the modifying inactivation step (see Experimental). These data are summarized in Table 1. Here the basis of the difference in IC50’s for (2a) and (2b) can be more easily appreciated. Affinity for (2a) is about 6-fold higher than for (2b). The rate constants for inactivation are more comparable, differing by no more than two-fold. Interestingly, (15) binds AChE more weakly than the larger compounds, but inactivates the enzyme at a much faster rate (10–20 fold).
Scheme 4.
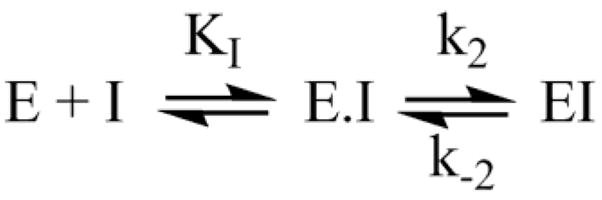
Rate constants for inactivation are slow, with it taking many minutes for inactivation. This is consistent with the irreversibility of inactivation exhibited by (2a) in the above desalting experiment. Also interesting is that k−2, which reflects the reversibility of inactivation, is small enough that little recovery of activity is likely to be observed in experiments lasting only a few hours.
To get some perspective, data are compared to the well known irreversible inhibitor DIFP analyzed similarly (Table 1). While none of the synthesized compounds exhibit a KI as tight as DIFP, k2 values for the active phosphonates are remarkably comparable to those obtained for DIFP.
Active Site Ser Modification
Oxime Reactivity
When organophosphates react with serine hydrolases, the active site Ser becomes phosphorylated (Scheme 5). This modification can sometimes be reversed with oximes (HON=R), strong nucleophiles which can displace the phosphorus center, and restore the Ser residue. In addition to serving as chemical antidotes to chemical warfare agents,2 oximes can be used to assess the nature of the phosphorylation reaction and the fate of the moiety. If enzyme activity is restored upon treatment with oxime, then the adduct is stable; if not, then either spontaneous hydrolysis or aging is suspected, although this is not always the case. 12 In the former reaction, the phosphate is released from the enzyme, restoring the Ser residue. In the latter ester, hydrolysis results in the release of alcohol and the retention of a monoester enzyme-phosphate adduct, which does not react with oximes (Scheme 5). A number of AChE-OPs adducts react reasonably well in this fashion.2
Scheme 5.

To understand the reactivity of cyclic phosphonate-AChE adduct, oxime reactivation was examined for human AChE reacted with (2a). Pralidoxime was added to a preincubated mixture of enzyme and inhibitor, and activity was monitored as a function of time. As shown in Fig. 6, at 35 mM, pralidoxime alone inhibits the enzyme to almost 50% (E+P), but sufficient activity remains to analyze the effect of the oxime on an enzyme-inhibitor adduct. Interestingly, extended incubation of the enzyme with inhibitor spontaneously results in a very small restoration of activity (compare E+I at 30 min vs. 120 min). This is consistent with the above experiments which indicate that enzyme-inhibitor adduct formation is very slowly reversible. Indeed, a kobs of approx. 0.04 min−1 can be calculated from 1% activity at 120 min, a value consistent with the data in Table 1. The addition of pralidoxime has only a very small effect on this recovery, just outside experimental error (E+I+P).
Fig. 6. Oxime Reactivation of AChE.

After 30 min incubation of human AChE with (2), pralidoxime was introduced and enzyme activity monitored. E+P, enzyme and pralidoxime control. E+I, enzyme and inhibitor control. E+I+P, enzyme and inhibitor and pralidoxime. Conditions are given in the text. All rates are expressed relative to the enzyme control at the same time point in the experiment.
Mass Spectrometry Analysis of Inhibitor-Modified Enzyme
Since treatment with pralidoxime did not result in significant reactivation, the enzyme-inhibitor complex was subjected to proteolysis, Zip-tip purification13 and MALDI-TOF mass spectrometry to determine if a Ser adduct could be detected.
Trypsin digestion of the enzyme yields the fragment L178-R219 with an expected mass of 4269.8 Da (Table 2; see Supplemental Material for mass spectra). Indeed, a fragment with an m/z of 4267.9 Da was detected. Treatment of the enzyme with (2a) results in a fragment with a mass of 4500 Da, also detected where expected. This corresponds to a change in mass of 235 Da, which is within experimental error of the expected mass for the adduct minus one proton. This confirms that (2a) modifies the active site Ser.
Table 2.
Mass Spectrometry Analysis of AChE-OP Adductsa
| Species | m/z Calculated | m/z Measured |
|---|---|---|
| L178-R219 | 4269.8a | 4267.9 |
| OP-L178-R219 | 4500.8 | 4503.8 |
| OP moiety | 234.2 | 235.9 |
Calculated using PeptideMass.
DISCUSSION
Structure-Activity Relationships in Cyclic Phosphonate AChE Inhibitors
As stated above, this is the first inhibition study of cyclic OPs against AChE. Comparisons to the properties of the acyclic fluoroinhibitor DIFP are instructive: This well known inhibitor is notoriously unstable in aqueous solution; these compounds are easily handled in aqueous solutions for enzyme assays and indefinitely stable in isopropanol solution and either 10 equiv. acetic acid or 10 equiv. imidazole over at least 24 h. (data not shown). A number of the cyclic derivatives of cyclophostin show good potency, with IC50’s in the lower micromolar range, only about an order of magnitude weaker than DIFP. This suggests that a cyclic phosphonate can exhibit a reactivity that is similar to that observed with fluorophosphates. There is evidence that ring strain plays a role in reactivity: (15) displays a k2 (inactivation constant) that is 20-fold higher than the less strained, seven-membered ring analog (9). However, the KI for (15) reflects a poorer fit in the active site (4-fold weaker binding than (9)), a feature that tempers the effectiveness of the inhibitor.
With the exception of (15), the rate constants for inactivation among the active cyclic phosphonates ((2a), (2b), and (9)) are comparable to that of DIFP. This effect is illustrated when comparing (9) with (10). These compounds only differ at the exocyclic functionality: The alcohol is a poorer leaving group and also a poor inhibitor, while the ester is more potent. However, other effects could also contribute, as these functionalities participate in noncovalent interactions differently. The ester could easily be making more complementary contacts in the active site. The poor IC50 of (10) makes assessing these possibilities technically difficult, however.
In contrast to the inactivation constants (k2 and k−2), the binding affinities (KI) for the synthesized phosphonates differ more substantially and thus are responsible for the differing IC50’s. The crystal structure for (2b)9 shows that the hydrogen and methoxy are on the same side, whereas in (2a) they are on opposite sides. The data presented here are sufficient to argue that (2a) makes more advantageous contacts in the active site than (2b). The active site architecture of AChE has been examined extensively.14 In addition to the catalytic triad, (H447, S203, Glu334), Ser229 and Glu202 and a few water molecules lie within a few angstroms of Ser203, providing opportunities for polar contacts. It seems unlikely that these compounds have the structural features necessary to take advantage of contacts to the well characterized choline binding site and the peripheral binding site, the latter of which is some 20 Å removed from the catalytic triad.14 However, through modification at the methyl groups, these opportunities could be exploited to attenuate inhibitor affinity in the manner of the structurally similar cyclipostins, which inhibit lipases.15
To put the potencies of these phosphonates into perspective, some comparison to IC50’s and kinetic parameters is in order. The closest OP structural relative of these compounds is monocrotophos, an acyclic OP pesticide with a conjugated enolate (Fig. 1). We are unaware of any kinetic analysis of these compound toward AChE, but an IC50 of about 10 μM has been reported recently against eel AChE.16 (2a) and (15) exhibited IC50’s of 70 and 20 μM, respectively, against this form of the enzyme (data not shown). Without dissecting the IC50’s into individual binding and rate constants, it would not be possible to ascribe the similarities to affinity vs. reactivity.
To understand the potency of these analogs of cyclophostin relative to a wider range of characterized OPs, it was necessary to fit the kinetic data to the more commonly used irreversible inactivation model.11 This yields KI and kinact, which are used to calculate ki, a first order rate constant for inactivation. Table 3 summarizes the results of this analysis, as well as literature ki’s for a number of OPs for comparison. Our ability to reproduce the literature value for DIFP provides confidence in making comparisons. When using this ki value, which is a function of both binding and presumed irreversible inactivation steps, it is clear that the cyclic OPs described here are less potent than DIFP by about two orders of magnitude, making their activity relative to the very potent VX, Soman, and Tabun especially modest. By this measure, these OPs are likely to be less toxic. For the evolution of these compounds into potential Alzheimer’s drugs, this is an attractive characteristic.
Table 3.
Inhibition of Human AChE by Organophosphatesa
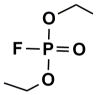
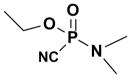
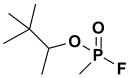
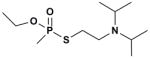
Oxime Reactivity
We have shown here that a cyclic phosphonate analog of cyclophostin indeed modifies the active site Ser residue of AChE, but is not subject to significant reactivation upon treatment with pralidoxime. Oximes are used as antidotes to the chemical warfare OPs, but they are not particularly effective. In a recent study, the kinetics of oxime reactivation was found to vary substantially among well known OPs, ranging from 1-0.01 min−1.2, 12 A detailed study was not conducted here, but reactivation of the cyclic OP-AChE adduct by pralidoxime in two hours is residual at best (about 0.04 min−1). The value of this estimate is shared by much smaller OPs like DIFP. Thus one cannot conclude that steric bulk is the sole determinant of oxime reactivity of the OP-enzyme adduct. Indeed, this is further supported by our studies of eel AChE. For this form of the enzyme, oxime reactivation of AChE-OP adduct is more pronounced (15–20%, data not shown). This indicates that details in the active site architecture are important, implying a role for other functional groups in the active site in attenuating both binding specificity and reactivity. Clearly there are opportunities for clarifying and exploiting these details in subsequent studies.
In summary, phosphonate analogs of the bicyclic phosphate natural product cyclophostin are, like simpler OPs, principally irreversible inhibitors of acetylcholinesterase. They bind AChE with slightly less affinity than the well known serine hydrolase inhibitor DIFP but modify the enzyme with similar rate constants. The catalytic Ser residue is modified by the cyclic OP, but treatment with an oxime does not result in reactivation. The less potent behavior of these cyclic OPs provide opportunities for further SAR studies.
EXPERIMENTAL
Materials and Methods
All reactions were carried out in oven dried glassware under an atmosphere of argon unless otherwise noted. 1H, 13C, and 31P NMR spectra were recorded in CDCl3 at 300, 75 and 121 MHz, respectively. 1H NMR spectra were referenced to residual CHCl3 (7.27 ppm). 13C NMR spectra were referenced to the center line of CDCl3 (77.23 ppm) and 31P NMR spectra were referenced to external 85% H3PO4 (0 ppm). Coupling constants, J, are reported in Hz. Human acetylcholinesterase, acetylthiocholine iodide, 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), pralidoxime iodide, and diisopropylfluorophosphate (DIFP) were purchased from Sigma (Saint Louis, MO). P6-DG desalting resin was obtained from Biorad (Hercules, CA). Trypsin was obtained from Promega (Madison, WI).
(E) Methyl 2-acetyl-5-(dimethoxyphosphoryl)pent-4-enoate (5)
To a solution of Pd2(dba)3 (0.055 g, 0.060 mmol) and dppe (0.073 g, 0.18 mmol) in anhydrous THF (5 mL) at room temperature, was added methyl acetoacetate (1.13 mL, 9.00 mmol), followed by the addition of a solution of phosphono allylic carbonate (4) (0.672 g, 3.00 mmol) in THF (20 mL). After the reaction was complete (TLC and 31P NMR analysis), the reaction mixture was partitioned between brine and Et2O. The layers were separated and the aqueous layer was re-extracted with Et2O. The combined organic layers were dried over anhydrous Na2SO4, filtered and the solvent was evaporated under reduced pressure to give the crude product. Purification using column chromatography (SiO2, 20–50% EtOAc in hexanes) gave the vinyl phosphonate (5) as pale yellow oil (0.32 g, 40 %). IR (neat, NaCl) 1743, 1717, 1635 cm−1; 1H NMR (CDCl3) δ 6.67 (1H, m), 5.69 (1H, m), 3.73 (6H, d, JHP =10.8 Hz), 3.67 (3H, s), 3.61 (1H, t, JHH = 7.3 Hz), 2.76 (2H, tt, JHH = 7.1 Hz, 1.7 Hz), 2.25 (3H, s); 13C NMR (CDCl3) δ 201.5, 169.4, 149.9 (d, JCP = 5.1 Hz), 119.0 (d, JCP =187 Hz), 58.1, 53.1, 52.7 (d, JCP = 7.4 Hz), 32.5 (d, JCP = 23.4 Hz), 29.1; 31P NMR (CDCl3) δ 20.6; HRMS (FAB, MH+) calcd. for C10H18O6P: 265.0891. Found 265.0829 and some dialkylation product (6) as a yellow oil (0.142 g, 23 %) IR (neat, NaCl) 1739, 1713, 1633 cm−1; 1H NMR (CDCl3) δ 6.52 (2H, m), 5.72 (2H, m), 3.77 (3H, s), 3.70 (12H, d, JHP =11.1 Hz ), 2.78 (2H, m, 2.17 (3H, s); 13C NMR (CDCl3) δ 202.2, 170.9, 146.9 (d, JCP = 5.3 Hz), 121.3 (d, JCP =186.2 Hz), 62.7, 53.1, 52.5 (d, JCP = 5.9 Hz), 37.0 (d, JCP = 23.3 Hz), 27.1 (d, JCP = 8.6 Hz); 31PNMR (CDCl3) δ 19.6; HRMS (FAB, MH+) calcd for C15H27O9P2 413.1130. Found 413.1143.
Methyl 2-acetyl-5-(dimethoxyphosphoryl)pentanoate (7)
To a solution of vinyl phosphonate (5) (0.175 g, 0.660 mmol) in methanol (2 mL), was added 10% palladium on charcoal (0.071 g, 0.066 mmol). The solution was first flushed with argon, followed by hydrogen gas. A reservoir of hydrogen was provided by a balloon. After the reaction was complete (31P NMR analysis), the reaction mixture was filtered through Celite® which was then further washed with CH2Cl2 (5 × 20 mL). The solvent was evaporated under reduced pressure to give the saturated phosphonate (7) as colorless oil (0.176 g, quant). IR (NaCl, neat) 1737, 1719 cm−1; 1HNMR (CDCl3) δ 3.66 (d, 6H, JHP = 10.8 Hz), 3.68 (3H, s), 3.38 (1H, t, JHH = 7.2 Hz), 2.17 (3H, s) 1.86 (2H, m), 1.76 (2H, m), 1.53 (2H, m); 13C NMR (CDCl3) δ 202.8, 170.3, 59.5, 52.9, 52.7, (d, JCP = 6.6 Hz), 29.4, 29.0 (d, JCP = 17.4 Hz) 24.9 (d, JCP = 141 Hz), 20.9 (d, JCP = 4.9 Hz); 31PNMR (CDCl3) δ 34.4; HRMS (FAB, MH+) calcd. for C10H20O6P: 267.0997. Found 267.0988.
7-Membered Cyclic Phosphonate (9)
To a solution of phosphonate (7) (0.115 g, 0.432 mmol) in acetonitrile (2 mL), was added NaI (0.072 g, 0.475 mmol) and the resulting mixture was heated at reflux overnight. The solvent was evaporated under reduced pressure to give the mono-sodium salt as a solid. The sodium salt was suspended in dry acetone and amberlite IR 120 resin was added. The resulting mixture was shaken until there was complete dissolution of the salt. The resin was removed by filtration and washed with additional acetone. The solvent was evaporated under reduced pressure to the phosphonic acid (0.116 g) as a red viscous liquid. The crude phosphonic acid (0.113 g, 0.448 mmol) was dissolved in dry CH2Cl2 (9 mL), and then EDC (0.112 g, 0.583 mmol) and HOBt (0.092 g, 0.673 mmol) were added followed by Hunig’s base (0.120 mL, 0.673 mmol). After 24 h, the solvent was evaporated in vacuo and the residue was dissolved in EtOAc. The solution was washed with 0.5N aq. HCl, saturated aq. NaHCO3, dried over MgSO4 and evaporated under reduced pressure. Purification by column chromatography (SiO2, 20%-40 % EtOAc in hexanes) gave the monocyclic phosphonate (9) (0.058 g, 55 %). IR (NaCl, neat) 1716, 1646 cm−1; 1H NMR (CDCl3) δ 3.81 (d, 3H, JHP = 11.2 Hz), 3.73 (3H, s), 2.65 (1H, m), 2.48 (1H, m), 2.32 (3H, s), 2.00 (4H, m); 13C NMR (CDCl3) δ 168.4, 159.4 (d, JCP = 7.9 Hz), 119.5 (d, JCP = 4.6 Hz), 52.6 (d, JCP = 7.1 Hz), 52.3, 26.6 (d, JCP = 2.6 Hz), 26.6 (d, JCP = 133 Hz), 21.4 (d, JCP = 7.5 Hz), 21.2; 31P NMR (CDCl3) δ 25.0; HRMS (EI+, M+) calcd. for C9H15O5P: 234.0657. Found 234.0659.
Reduced Monocyclic Phosphonate (10)
To a solution of monocyclic phosphonate (9) (0.065 g, 0.278 mmol) in CH2Cl2 (1 mL) at −78 °C was added dibal-H (1.1 mL, 1M solution in hexane) dropwise. The solution was allowed to warm slowly to room temperature. After the reaction was complete (TLC and 31P NMR analysis), the reaction mixture was partitioned between Et2O and saturated solution of Rochelle’s salt. The layers were separated and the aqueous layer was re-extracted with Et2O (2 × 4 mL). The combined Et2O layers were dried over Na2SO4, and the solvent was evaporated under reduced pressure. Purification by column chromatography (SiO2, 20–50% EtOAc in hexanes) gave the allylic alcohol (10) as colorless oil (0.046 g, 80 %). IR (neat, NaCl) 3361 cm−1; 1H NMR (CDCl3) δ 4.2 (1H, dd, JHP = 0.8 Hz, JHH = 12 Hz), 4.03 (1H, dd, JHP = 0.7 Hz, JHH = 11 Hz) 3.80 (d, 3H, JHP = 11 Hz), 2.36 (2H, t, JHH = 5.2 Hz), 2.14 (1H, m), 2.04 (1H, m), 2.0 (3H, s), 1.91 (2H, m). 13C NMR (CDCl3) δ 144.8 (d, JCP = 7.5 Hz), 124.2 (d, JCP = 5 Hz), 63.6, 52.2 (d, JCP = 7.0 Hz), 28.8 (d, JCP = 1.3 Hz), 27.0 (d, JCP = 134 Hz), 21.8 (d, JCP = 7.1 Hz), 18.0; 31PNMR (CDCl3) δ 27.1; HRMS (EI+, M+) calcd for C8H15O4P: 206.0708. Found 206.0711
Methyl 2-acetyl-4-(dimethoxyphosphoryl)butanoate (13)
To the suspension of potassium t-butoxide (0.56 g, 5.0 mmol) in THF (14 mL), was slowly added methyl acetoacetate (1.63 mL, 10 mmol), followed by dropwise addition of dimethyl vinyl phosphonate (0.60 mL, 5.0 mmol) over 5 minutes. The resulting reaction mixture was stirred at room temperature for 2 hours. After the reaction was complete (TLC and 31P NMR analysis), it was quenched by the addition saturated NH4Cl solution. The layers were separated and the aqueous layer was re-extracted with EtOAc (2 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure to give the crude product. Purification by column chromatography (SiO2, 40–60% EtOAc in hexanes) gave the phosphonate (13) (0.76 g, 60%) as an oil. IR (NaCl, neat) 1742, 1716 cm−1; 1H NMR (CDCl3) δ 3.72 (d, 6H, JHP = 10.5 Hz), 3.69 (3H, s), 3.60 (1H, t, JHH = 7.2 Hz), 2.23 (3H, s), 2.09 (2H, m), 1.74 (3H, m); 13C NMR (CDCl3) δ 202.5, 169.8, 59.1 (d, JCP = 13.9 Hz), 53.0, 52.9,(d, JCP = 6.6 Hz), 29.7, 22.4.(d, JCP = 141 Hz), 21.3 (d, JCP = 4.3 Hz); 31P NMR (CDCl3) δ 33.5; HRMS (FAB, MH+) calcd. for C9H18O6P: 253.0841. Found 253.0844.
6-Membered Cyclic Phosphonate (15)
To the solution of phosphonate (13) (0.1 g, 0.4 mmol) in dry CH3CN (0.5 mL) was added NaI (0.066 g, 0.48 mmol) and the resulting mixture was heated at reflux overnight. The solvent was evaporated under reduced pressure to give the mono-sodium salt as a solid. The sodium salt was suspended in dry acetone and amberlite IR 120 resin was added. The resulting mixture was shaken until there was complete dissolution of the salt. The resin was removed by filtration and washed with additional amounts of acetone. The solvent was evaporated under reduced pressure to give the phosphonic acid (0.095 g) as a viscous red liquid. The phosphonic acid (0.09 g, 0.38 mmol) was dissolved in dry CH2Cl2 (1.2 mL), EDC (0.094 g, 0.49 mmol) and HOBt (0.067 g, 0.57 mmol) were added followed by Hunig’s base (0.09 mL, 0.8 mmol). After 24 hours, the solvent was evaporated under reduced pressure and the residue was dissolved in EtOAc. The solution was washed with 0.5N aq. HCl, saturated aq. NaHCO3, dried over MgSO4 and evaporated under reduced pressure. Purification by column chromatography (SiO2, 10–30% EtOAc in hexanes) gave the cyclic phosphonate (0.033 g, 40%). IR (NaCl, neat) 1717, 1637 cm−1; 1H NMR (CDCl3) δ 3.83 (d, 3H, JHP = 11.2 Hz), 3.75 (3H, s), 2.89 (1H, m), 2.67 (1H, m), 2.31 (3H, d, JHP = 1.6 Hz), 2.01 (2H, m); 13C NMR (CDCl3) δ 167.6 (d, JCP = 0.9 Hz), 161.4 (d, JCP = 9.9 Hz), 108.2 (d, JCP = 11.5 Hz), 52.7 (d, JCP = 7.1 Hz), 52.1, 22.3 (d, JCP = 9.4 Hz), 21.0 (d, JCP = 5.2 Hz), 18.7 (d, JCP = 128 Hz); 31P NMR (CDCl3) δ 23.3; HRMS (EI+, M+) calcd. for C8H13O5P: 220.0505. Found 220.0497.
(Z)-Dimethyl 1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl phosphate (17a)
To a suspension of sodium hydride (60% dispersion in mineral oil, 0.12 g, 3.0 mmol) in THF (14 mL) was added 2-acetyl butyrolactone (0.283 mL, 2.50 mmol) dropwise at room temperature over ~ 2 minutes. The mixture was stirred until effervescence ceased, then it was cooled to 0 °C, and dimethyl chlorophosphate (0.41 mL, 3.0 mmol) was added dropwise. After the reaction was complete (30 minutes. TLC and 31P NMR analysis), the reaction was quenched by the addition saturated aqueous NH4Cl (10 mL). The organic layer was separated and the aqueous layer was re-extracted with EtOAc (20 mL × 2). The combined organic layers were dried over anhydrous Na2SO4, filtered and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography (SiO2, 20–30 % EtOAc in hexanes) to give the enol phosphate (21a) (0.36 g, 62%). IR (NaCl, neat) 1754, 1685 cm−1; 1H NMR (CDCl3) δ 4.28 (t, 2H, JHH = 7.5 Hz), 3.88 (d, 6H, JHP = 11.5 Hz), 2.90 (2H, m), 2.16 (3H, dd, JHH = 1.6, 3.6 Hz); 13C NMR (CDCl3) δ 167.5, 152.8 (d, JCP = 7.0 Hz), 110.3 (d, JCP = 8.6 Hz), 64.5, 55.6 (d, JCP = 6.5 Hz), 26.5, 20.2; 31P NMR (CDCl3) δ −5.45; HRMS (FAB+, MH+) calcd. for C8H14O6P: 237.0528. Found 237.0534.
(E)-Dimethyl 1-(2-oxodihydrofuran-3(2H)-ylidene)ethyl phosphate (17b)
To the solution of 2-acetyl butyrolactone (0.28 mL, 2.5 mmol) in dry CH2Cl2 (7 mL) at room temperature was added Hunig’s base (2.2 mL, 12.5 mmol). The resulting solution was stirred for 5 minutes then the solution was cooled to −30 °C and dimethyl chlorophosphate (0.4 mL, 3 mmol) was added dropwise. The resulting solution was stirred at −30 °C for ~ 30 mins., then it was allowed to warm to room temperature. After the reaction was complete (TLC and 31P NMR analysis), the reaction mixture was diluted with CH2Cl2 (7 mL) and washed with saturated NH4Cl. The organic layer was separated and the aqueous phase was re-extracted with EtOAc (20 mL × 2). The combined organic layers were dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography (SiO2, 20–30% EtOAc in hexane) to give the enol phosphate (0.32 g, 54%). IR (NaCl, neat), 1751, 1688 cm−1; 1H NMR (CDCl3) δ 4.28 (t, 2H, JHH = 7.5 Hz), 3.84 (d, 6H, JHP = 11.5 Hz), 3.01 (2H, m), 2.49 (3H, d, JHH = 2.2 Hz); 13C NMR (CDCl3) δ 170.6, 157.1 (d, JCP = 7.0 Hz), 111.6 (d, JCP = 9.1 Hz), 64.7, 55.1 (d, JCP = 6.4 Hz), 25.9, 16.6; 31P NMR (CDCl3) δ −4.7; HRMS (FAB, MH+) calcd. For C8H14O6P: 237.0528. Found 237.0525.
Enzyme Assay
Recombinant AChE was quantitated using the extinction coefficient 119, 650 M−1cm−1 calculated from its amino acid sequence.17 34 nM AChE was assayed using Ellman’s reagent18 using 160 μM acetylthiocholine and 300 μM 5,5′-dithiobis-2-ntirobenzoic acid (DTNB) in 100 mM sodium phosphate, pH 8.0, in a cuvette thermostatted to 37°C. Thiocholine released by the enzyme reacts with excess DTNB, resulting in the formation of the thionitrobenzoate anion, which absorbs at 412 nm. Using an HP8453 diode array spectrophotometer, the reaction was monitored for 30 seconds and absorbance recorded every 2 sec to obtain the initial slope. This was then converted to μmol/min using an extinction coefficient of 14,150 M−1sec−1.
Determination of IC50s of Organophosphonates (OPs)
The inhibitors were solubilized in isopropanol and used in volumes of less than 5% of the total reaction volume. AChE (34 nM) was incubated for 30 min. with each compound at an array of concentrations in 20 mM Tris, 1% BSA, pH 7.5 at 25°C. The residual activity of the enzyme was assayed using 500 μM acetylthiocholine, a saturating concentration 10-fold higher than the Km of 46 μM for this substrate.19 The IC50, or compound concentration necessary to achieve 50% decrease in enzyme activity, was determined from a plot of the % residual activity vs. inhibitor concentration.
Assay for Irreversible Inhibition
34 nM AChE was incubated with 20 μM (2) at 25°C as described above. After 30 mins, the reaction mixture was passed through a P6-DG desalting gel filtration column equilibrated with 20 mM Tris, pH 7.5 to remove the inhibitor. The enzyme was subsequently requantitated and assayed for residual activity as described above.
Determination of Rate Constants for Binding and Inactivation
Rate constants for binding and inactivation steps were determined by assuming the data are well represented by Scheme 4, where KI is the equilibrium constant for binding (reversible), and k2 and k−2 are forward and reverse rate constants for inactivation.20, 21 Residual activity was measured as a function of incubation time and inhibitor concentration and globally fit using Scientist software to the equation:
| (eq. 1) |
where At equals the fraction of enzymatic activity remaining following pre-incubation time (t), A0 represents the enzymatic activity at time zero (i.e., the initial activity), and kobs is the observed rate of decay. The resulting constant k−2 and various kobs values were subsequently plotted vs. [I] and fit using Kaleidagraph software to the equation:
| (eq. 2) |
to yield KI and k2.
To facilitate comparison to literature data, the kinetic data were also globally fit to the irreversible inactivation model of Kitz and Wilson11 to yield KI and kinact. The first order rate constants for the compounds in Table 3 (ki) were obtained by dividing KI into kinact.
Oxime Reactivation Reaction
34 nM AChE was incubated with (2a) in 20 mM Tris, pH 7.5 for 30 mins at 25°C as described above. Subsequently, pralidoxime (Scheme 5) was added to the reaction mixture to a concentration of 35 mM. The enzyme was assayed at various timepoints in parallel with control samples.
Mass Spectrometric Analysis of Proteolytic Fragments
70 μM the enzyme was incubated with 1 mM (2) in for 30 min at 25°C in 20 mM Tris, pH 7.5. Excess compound was removed using a YM-3 Centricon (Millipore) to prevent inactivation of trypsin. The enzyme was then digested with 2 μg of trypsin at 37°C in 50 mM sodium phosphate, 0.1% SDS, 20% acetonitrile, pH 8.0. After two hours, the mixtures were evaporated to dryness.
Pellets were resuspended in 20 μL of 20% acetonitrile, 0.1 % TFA. By pumping a pipet several times, the sample was passed over a Zip-Tip (Millipore) column activated with 70% MeCN and preequilibrated with 20% acetonitrile, 0.1 % TFA. Stepwise elution was performed using 10 μL aliquots of MeCN/TFA mixture. 5 μL of each fraction was mixed with an equal volume of 10 mg/mL of trans-3,5-dimethoxy-4-hydroxycinnamic acid in 40% MeCN and 0.1 % TFA, spotted onto a plate, dried, and analyzed using an Applied Biosystems Voyager-DE STR MALDI instrument at the Danforth Plant Science Center.
Supplementary Material
Acknowledgments
The project described was supported by grant number R01-GM076192 from the National Institute of General Medicine. We thank National Science Foundation for grants to purchase the NMR spectrometer (CHE-9974801), and the mass spectrometer (CHE-9708640). We thank Mr. Joe Kramer and Prof. R.E.K. Winter for mass spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kidd D, Liu Y, Cravatt BF. Biochemistry. 2001;40:4005. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 2.Worek F, Thiermann H, Szinicz L, Eyer P. Biochem Pharmacol. 2004;68:2237. doi: 10.1016/j.bcp.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 3.Mount C, Downton C. Nat Med. 2006;12:780. doi: 10.1038/nm0706-780. [DOI] [PubMed] [Google Scholar]
- 4.Musial A, Bajda M, Malawska B. Curr Med Chem. 2007;14:2654. doi: 10.2174/092986707782023217. [DOI] [PubMed] [Google Scholar]
- 5.Zhou X, Wang XB, Wang T, Kong LY. Bioorg Med Chem. 2008;16:8011. doi: 10.1016/j.bmc.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 6.Hostettmann K, Borloz A, Urbain A, Marston A. Curr Org Chem. 2006;10:825. [Google Scholar]
- 7.This compound should not be confused with the carbohydrate cyclophostin in: de Kort M, Regenbogen A, Overkleeft H, Challiss R, Iwata Y, Miyamoto S, van der Marel G, van Boom J. Tetrahedron. 2000;56:5915.
- 8.Kurokawa T, Suzuki K, Hayaoka T, Nakagawa T. J Antibiotics. 1993;46:1315. doi: 10.7164/antibiotics.46.1315. [DOI] [PubMed] [Google Scholar]
- 9.Bandyopadhyay S, Dutta S, Spilling C, Dupureur C, Rath N. J Org Chem. 2008 doi: 10.1021/jo801453v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor P, Radic Z, Hosea N, Camp S, Marchot P, Berman H. Toxicology Letters. 1995;82–83:453. doi: 10.1016/0378-4274(95)03575-3. [DOI] [PubMed] [Google Scholar]
- 11.Kitz R, Wilson IB. J Biol Chem. 1962;237:3245. [PubMed] [Google Scholar]
- 12.Epstein TM, Samanta U, Kirby SD, Cerasoli DM, Bahnson BJ. Biochemistry. 2009;48:3425. doi: 10.1021/bi8023527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elhanany E, Ordentlich A, Dgany O, Kaplan D, Segall Y, Barak R, Velan B, Shafferman A. Chem Res Toxicol. 2001;14:912. doi: 10.1021/tx0100542. [DOI] [PubMed] [Google Scholar]
- 14.Szegletes T, Mallender WD, Rosenberry TL. Biochemistry. 1998;37:4206. doi: 10.1021/bi972158a. [DOI] [PubMed] [Google Scholar]
- 15.Vértesy L, Beck B, Brönstrup M, Ehrlich K, Kurz M, Müller G, Schummer D, Seibert G. Journal of Antibiotics. 2002;55:480. doi: 10.7164/antibiotics.55.480. [DOI] [PubMed] [Google Scholar]
- 16.Du D, Chen S, Song D, Li H, Chen X. Biosens Bioelectron. 2008;24:475. doi: 10.1016/j.bios.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. Protein Science. 1995;4:2411. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. Biochem Pharmacol. 1961;7:88. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 19.Radic Z, Pickering NA, Vellom DC, Camp S, Taylor P. Biochemistry. 1993;32:12074. doi: 10.1021/bi00096a018. [DOI] [PubMed] [Google Scholar]
- 20.Walker MC, Kurumbail RG, Kiefer JR, Moreland KT, Koboldt CM, Isakson PC, Seibert K, Gierse JK. Biochem J. 2001;357:709. doi: 10.1042/0264-6021:3570709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callan OH, So OY, Swinney DC. J Biol Chem. 1996;271:3548. doi: 10.1074/jbc.271.7.3548. [DOI] [PubMed] [Google Scholar]
- 22.Ordentlich A, Barak D, Kronman C, Ariel N, Segall Y, Velan B, Shafferman A. J Biol Chem. 1996;271:11953. doi: 10.1074/jbc.271.20.11953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.