

Abstract
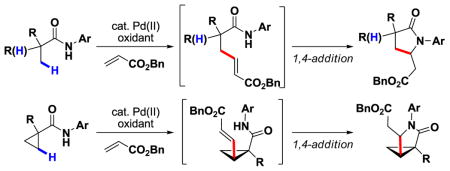
The first Pd(II)-catalyzed sp3 C–H olefination reaction has been developed using N-arylamide directing groups. Following olefination, the resulting intermediates were found to undergo rapid 1,4-addition to give the corresponding γ lactams. Notably, this method was effective with substrates containing α-hydrogen atoms and could be applied to effect methylene C–H olefination of cyclopropane substrates.
The Pd-catalyzed Mizoroki–Heck reaction, which couples olefins with aryl and vinyl halides and sulfonates is a uniquely powerful transformation for forging C–C bonds and plays a pivotal role in modern organic synthesis.1 Given its widespread use, the development of complementary methods to couple unactivated C–H bonds with olefins is highly attractive. Indeed, as early as 1967, Fujiwara and Moritani reported the first example of Pd-mediated C–H olefination of excess benzene (eq 1).2 Since then, several research groups have sought to expand the synthetic utility of arene C–H olefination by improving the reactivity, controlling the positional selectivity, and using the arene as the limiting reagent; this has been achieved by exploiting electron-rich heterocycles,3 proximate directing groups4 and ligand assistance.3b,4d At the same time, Pd(II)-mediated aryl C–H olefination has emerged as an efficient tool for constructing diverse carbon skeletons of natural products.4d,5
While aryl C–H olefination via either electrophilic palladation or other C–H activation pathways has been extensively explored in recent years, Pd-catalyzed olefination of unactivated sp3 C–H bonds remains undeveloped, with no examples reported to date. Because sp3 C–H olefination is mechanistically distinct from aryl C–H olefination, the conceptual and practical challenges in developing this technology are numerous.6 Firstly, Pd(II)-mediated sp3 C–H cleavage is far less facile than sp2 C–H cleavage, and as a consequence, devising reaction conditions for directed sp3 C–H activation to occur in the presence of competitively coordinating olefins is a tremendous challenge. Additionally, following sp3 C–H activation, carbopalladation of the resulting [Pd(II)–R] intermediate across a double bond is less well established.7 For instance, undesired β-hydride elimination is often a competitive pathway, particularly in the case of intermolecular olefination. We were encouraged with respect to this latter potential barrier by the pioneering work of Fu in their investigation of intramolecular olefination of unactivated, β-hydrogen-containing alkyl halides catalyzed by a Pd/N-heterocyclic carbene complex.8 Moreover, based on our recent discovery of highly efficient Pd(II)-catalyzed sp3 C–H activation using an N-arylamide (CONHAr) directing group,9 we conjectured that the former problem could also be overcome.
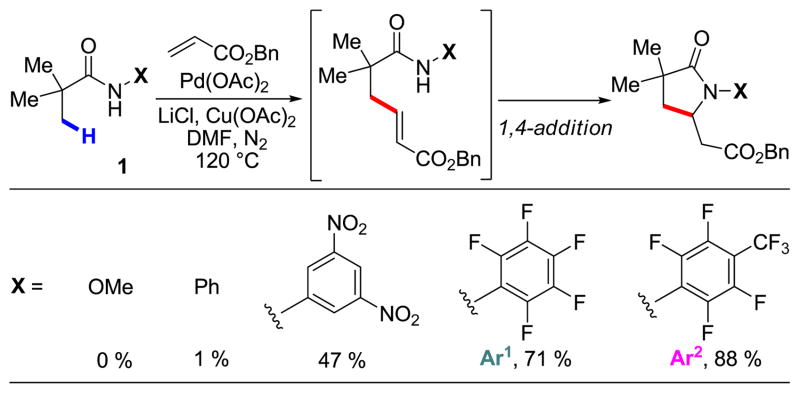
With these considerations in mind, we initiated our study on β-olefination of N-arylpivalamide 1 by carrying out a systematic screening of reaction conditions (Table 1). Gratifyingly, we found that olefination of 1 with benzyl acrylate occurred using Pd(OAc)2 (10 mol%) as the catalyst and Cu(OAc)2 (2 equiv) as the terminal oxidant in DMF. The products were isolated as the corresponding γ-lactams formed via a tandem intramolecular 1,4-addition of the amide to the newly installed acrylate. The yield with CONHAr1 directing group was increased from 45% to 71% by adding LiCl (2 equiv) to the reaction mixture. Two potential roles for LiCl are (1) serving as a source of Cl– which can act as a ligand to prevent the deactivation of Pd(0) to Pd black,10 and (2) inducing the in situ formation of a chloro-bridging complex which is more prone to carbopalladation.7a
Table 1.
 |
Conditions: 0.2 mmol of substrate, 0.1 mL of benzyl acrylate, 10 mol % Pd(OAc)2, 2.0 equiv of LiCl, 2.0 equiv of Cu(OAc)2, 1 mL of DMF, 120 °C, N2, 12 h.
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as an internal standard.
As established in our previous report, electron-withdrawing substituents (CF3, F, and NO2) on the N-aryl group dramatically enhance the reaction.9 Indeed, the more electron-withdrawing CONHAr2 group improved the yield further to 88% (Table 1). Notably, the directing groups for sp3 C–H activation previously developed in our laboratory, such as carboxylic acid, hydroxamic acid, oxazoline and pyridine,11 were not reactive under these conditions. The choice of solvent was also critical, with polar and strongly-coordinating amide solvents such as DMF, DMA and NMP giving the best reactivity. Our preliminary screening was carried out using Cu(OAc)2 (2 equiv) for reoxidation of Pd(0); however, further optimization revealed that a mixture of Cu(OAc)2 and AgOAc (1.1 equiv each) gave the highest yield (90% by 1H NMR) even when the less reactive CONHAr1 directing group was used. We also found that using N2 in place of air gave consistently higher yields.
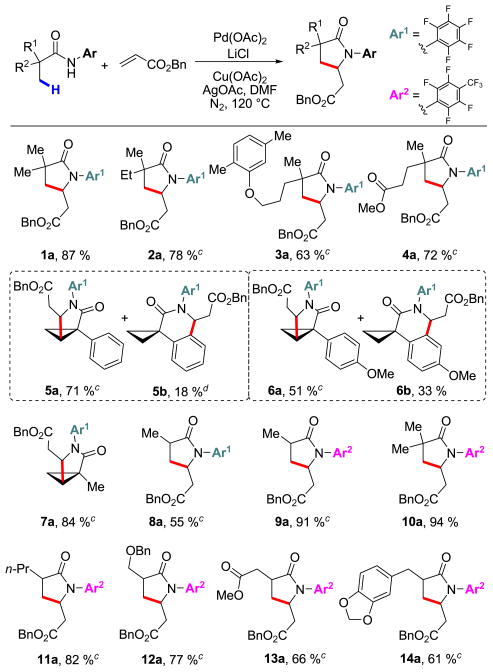
With the optimized conditions in hand, we converted a wide variety of commercial carboxylic acids into the corresponding CONHAr1 amides to examine the scope of the olefination protocol. Products containing ether (3a, 6a, 6b, 12a, and 14a) and ester (4a and 13a) groups could be obtained in good yields. Remarkably, this method was also found to be effective in activating methylene C–H bonds in cyclopropane substrates (5a, 6a, and 7a). For substrates 5 and 6, ortho-olefination of the aryl groups also took place, affording δ-lactam side products (5b and 6b) in 18% and 33 %, respectively. For substrate 7, the cyclopropyl C–H bond was selectively olefinated in the presence of an α-methyl group to give 7a as the only product in 84% yield. Lowering the temperature to 90 °C, we found that the cyclopropane substrates could still be olefinated effectively without a major decline in yield (7a, 77%).
In our previous reports concerning sp3 C–H activation reactions using carboxylic acids and their derivatives,11 substrates that contained α-hydrogen atoms were unreactive. Thus, the substrate scope was limited to compounds with quaternary α-carbon atoms. Nevertheless, we recently established that the facile conversion of the carboxylic acids to the corresponding CONHAr1 amides provides a remedy to this problem, allowing access to a broad range of substrates containing α-hydrogen atoms.9 We thus subjected substrate 8 to the olefination conditions described above; however, 8a was obtained in only 55% yield. More complex substrates with α-hydrogen atoms gave virtually no product. In an effort to overcome this issue, we hypothesized that the reactivity could be improved by exploiting the CONHAr2 directing group. Hence, we attempted C–H olefination of substrate 9, and to our delight we observed a considerable increase in the product yield, giving 9a in 91%. Using the CONHAr2 group, we successfully olefinated an array of α-hydrogen-containing substrates (9, 11, 12, 13, and 14) in good to excellent yields.
A plausible mechanism for this process involves initial amide-directed C–H insertion by Pd(II) to generate an alkylpalladium intermediate which reacts with the olefin via a carbopalladation, followed by β-hydride elimination to give the olefination product. Pd(0) is reoxidized to Pd(II) by Ag(I)/Cu(II) to close the catalytic cycle.
In summary, we have developed a Pd(II)-catalyzed reaction protocol for the direct olefination of sp3 C–H bonds. After β-C–H olefination, the amide products underwent 1,4-conjugate addition to give the corresponding lactam compounds. The reaction conditions could also be applied to effect olefination of cyclopropyl methylene C–H bonds and substrates containing α-hydrogen atoms. Studies to expand the scope to simple carboxylic acid substrates and to develop enantioselective sp3 C–H olefination reactions of gem-dimethyl- and cyclopropyl groups are currently underway in our laboratory.
Supplementary Material
Scheme 1.

Pd-Catalyzed C–H Olefination Reactions
Table 2.
 |
Conditions: 0.2 mmol of substrate, 0.1 mL of benzyl acrylate, 10 mol % Pd(OAc)2, 2.0 equiv of LiCl, 1.1 equiv of Cu(OAc)2, 1.1 equiv of AgOAc, 1 mL of DMF, 120 °C, N2, 12 h.
Isolated yield,.
Obtained as a mixture of inseparable cis/trans diastereomers (see Supporting Information). Optically inactive starting materials were used.
Yield determined by 1H NMR.
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, the National Institutes of Health (NIGMS, 1 R01 GM084019-01A1) for financial support, Bristol Myers Squibb for a predoctoral fellowship (M. W.), and the National Science Foundation, the Department of Defense, and the Skaggs Oxford Scholarship program for predoctoral fellowships (K.M.E.).
Footnotes
Supporting Information Available: Experimental procedure and characterization of all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of the Mizoroki–Heck reaction, see: Heck RF. Org React. 1982;27:345.Tsuji J. Palladium Reagents and Catalysts. Wiley; Chichester, UK: 1995. Chapter 4.Bräse S, de Meijere A. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Wiley-VCH; New York: 2004. Chapter 5.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368.
- 2.(a) Moritani I, Fujiwara Y. Tetrahedron Lett. 1967:1119. [Google Scholar]; (b) Jia C, Kitamura T, Fujiwara Y. Acc Chem Res. 2001;34:633. doi: 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]
- 3.(a) Itahara T. Synthesis. 1979:151. [Google Scholar]; (b) Ferreira EM, Stoltz BM.J Am Chem Soc 2003125957812904010 [Google Scholar]; (c) Ma S, Yu S. Tetrahedron Lett. 2004;45:8419. [Google Scholar]; (d) Liu C, Widenhoefer RA. J Am Chem Soc. 2004;126:10250. doi: 10.1021/ja046810i. [DOI] [PubMed] [Google Scholar]; (e) Grimster NP, Gauntlett C, Godfrey CRA, Gaunt MJ. Angew Chem, Int Ed. 2005;44:3125. doi: 10.1002/anie.200500468. [DOI] [PubMed] [Google Scholar]; (f) Capito E, Brown JM, Ricci A. Chem Commun. 2005:1854. doi: 10.1039/b417035k. [DOI] [PubMed] [Google Scholar]
- 4.(a) Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. J Org Chem. 1998;63:5211. [Google Scholar]; (b) Boele MDK, van Strijdonck GTPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J Am Chem Soc. 2002;124:1586. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]; (c) Zaitsev VG, Daugulis O. J Am Chem Soc. 2005;127:4156. doi: 10.1021/ja050366h. [DOI] [PubMed] [Google Scholar]; (d) Wang DH, Engle KM, Shi BF, Yu JQ. Science. 2010;327:315. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Trost BM, Godleski SA, Genet JP. J Am Chem Soc. 1978;100:3930. [Google Scholar]; (b) Baran PS, Corey EJ. J Am Chem Soc. 2002;124:7904. doi: 10.1021/ja026663t. [DOI] [PubMed] [Google Scholar]; (c) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2004;126:9552. doi: 10.1021/ja046695b. [DOI] [PubMed] [Google Scholar]; (d) Beck EM, Hatley R, Gaunt MJ. Angew Chem Int Ed. 2008;47:3004. doi: 10.1002/anie.200705005. [DOI] [PubMed] [Google Scholar]
- 6.For a recent review on transition metal-mediated sp3 C–H functionalization reactions, see: Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem Eur J. doi: 10.1002/chem.200902374.
- 7.For carbopalladation of alkylpalladium complexes across olefins, see: Balavoine G, Clinet JC. J Organomet Chem. 1990;390:C84.For olefination of alkylpalladium complexes with vinylboronic acids, see: Dangel BD, Godula K, Youn SW, Sezen Bengü, Sames D. J Am Chem Soc. 2002;124:11856. doi: 10.1021/ja027311p.
- 8.Firmansjah L, Fu GC. J Am Chem Soc. 2007;129:11340. doi: 10.1021/ja075245r. [DOI] [PubMed] [Google Scholar]
- 9.Wasa M, Engle KM, Yu JQ. J Am Chem Soc. 2009;131:9886. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Amatore C, Jutand A. J Organomet Chem. 1999;576:254. [Google Scholar]; (b) Maehara A, Satoh T, Miura M. Tetrahedron. 2008;64:5982. [Google Scholar]
- 11.For a review, see: Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094. doi: 10.1002/anie.200806273.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.