

Abstract
A live, cold-passaged (cp) candidate vaccine virus, designated respiratory syncytial virus (RSV) B1 cp-52/2B5 (cp-52), replicated efficiently in Vero cells, but was found to be overattenuated for RSV-seronegative infants and children. Sequence analysis of reverse-transcription–PCR-amplified fragments of this mutant revealed a large deletion spanning most of the coding sequences for the small hydrophobic (SH) and attachment (G) proteins. Northern blot analysis of cp-52 detected multiple unique read-through mRNAs containing SH and G sequences, consistent with a deletion mutation spanning the SH:G gene junction. Immunological studies confirmed that an intact G glycoprotein was not produced by the cp-52 virus. Nonetheless, cp-52 was infectious and replicated to high titer in tissue culture despite the absence of the viral surface SH and G glycoproteins. Thus, our characterization of this negative-strand RNA virus identified a novel replication-competent deletion mutant lacking two of its three surface glycoproteins. The requirement of SH and G for efficient replication in vivo suggests that selective deletion of one or both of these RSV genes may provide an alternative or additive strategy for developing an optimally attenuated vaccine candidate.
Respiratory syncytial virus (RSV), the leading cause of severe viral respiratory illness in pediatric populations throughout the world (reviewed in ref. 1), accounts for approximately 90,000 hospitalizations in infants and children in the United States each year (2). The importance of RSV as a respiratory pathogen makes development of a safe and effective RSV vaccine a public health priority (3). Although a number of approaches to RSV vaccine development have been taken, live RSV vaccines may provide the best alternative for immunizing young infants, because a live vaccine would mimic natural infection, induce a balanced cellular and humoral immune response, and be unlikely to produce enhanced disease (4).
RSV exists as two antigenically distinct subgroups, A and B, and both RSV A and RSV B infections are capable of inducing severe lower respiratory tract disease (5–7). For this reason, a bivalent live RSV vaccine containing attenuated RSV A and RSV B components would be most desirable. Recently, a live attenuated RSV A candidate vaccine has been identified that appears to be safe and immunogenic in infants and children over 6 months of age (8). In addition, a cold-passaged (cp) RSV B candidate vaccine, designated RSV B1 cp-52/2B5 (cp-52), was derived by passage of the RSV B1 wild-type (wt) virus 52 times at low temperature (21–32°C) (9). Cp-52 was shown to be restricted in replication in vivo but still able to induce RSV serum-neutralizing antibody responses in cotton rats, African green monkeys, and chimpanzees (9). Also, it was found to be phenotypically stable after prolonged replication in cotton rats (9). Here, we describe the phase I evaluation of the cp-52 candidate vaccine in adults, children, and infants. Although this virus mutant grew to high titer (>107.0 plaque-forming units (pfu)/ml) in Vero cell culture, it was poorly infectious and overattenuated for humans. When we sought to elucidate the genetic basis for its overattenuation, we made an unexpected discovery that this cp-52 virus, which is replication competent in vitro, contains a large deletion that ablates the synthesis of two of its three virion glycoproteins, namely the small hydrophobic (SH) and attachment (G) glycoproteins.
MATERIALS AND METHODS
Clinical Studies.
The isolation and characterization of RSV B1 wt and cp-52 have been described elsewhere (9). Virus suspensions of the wt (lot RSV B1) and cp-52 mutant (lot RSV B-10) were grown in Vero cell culture and were found to be free of adventitious agents by Louis Potash (Dyncorp/PRI, Rockville, MD). The titers of the wt RSV B1 strain and RSV B1 cp-52 were 105.0 and 105.5 pfu/ml, respectively. When necessary, the viruses were diluted in L-15 medium (BioWhittaker) immediately before use.
Guidelines for human experimentation of the Joint Committee for Clinical Investigation of the Johns Hopkins University School of Medicine were followed in the conduct of clinical studies in adults, infants, and children. The RSV B1 wt virus and cp-52 each were evaluated in open-label, nonrandomized trials in healthy adults between 18 and 45 years of age. Evaluation of the wt RSV B1 virus was performed in the Johns Hopkins University Center for Immunization Research (CIR) isolation unit, and evaluation of the vaccine strain was performed in outpatient studies at the CIR, both as previously described (8). Nineteen volunteers in the inpatient study received 104.7 pfu of RSV B1 wt, and 17 volunteers in the outpatient study received 105 pfu of RSV B1 cp-52. Both viruses were administered intranasally in a 0.5-ml inoculum.
After cp-52 was shown to be well tolerated in adults, it was evaluated in randomized, double-blind, placebo-controlled phase I trials in infants and children 6–59 months of age at the Johns Hopkins University Center for Immunization Research (CIR). The candidate vaccine was evaluated at a dose of 104 or 105 pfu in 22 RSV-seropositive children and 26 RSV-seronegative children, who were screened for level of RSV serum-neutralizing antibody by a 60% complement-enhanced plaque reduction assay as previously described (8). Each subject received 0.5 ml of vaccine or placebo intranasally. In the pediatric studies, the ratio of vaccinees to placebo recipients was approximately 2:1. Seropositive study participants and seronegative study participants were evaluated at the CIR for respiratory and febrile illnesses as previously described (8, 10).
Nasal wash specimens for virus isolation were obtained on each day of observation from all subjects who participated in these studies. Fresh undiluted nasal wash specimens were titered by plaque assay on Vero cell monolayer cultures maintained under a semisolid overlay at 32°C, and results were expressed as log10 pfu/ml (9). Nasal wash samples also were inoculated into tubes containing Vero cell monolayers and were identified as RSV-positive by using an indirect immunofluorescence assay (Bartels Microscan, Baxter Healthcare, Bellevue, WA). For purposes of calculation, samples in which virus was not detected or did not produce plaques were assigned an infectivity titer of 100.6 pfu/ml.
Sera for measurement of RSV-specific antibodies were obtained from adults and RSV-seropositive children before and 4 weeks after inoculation of virus, and from RSV-seronegative children before and 8 weeks after inoculation. Sera were tested for antibodies to RSV by the plaque-reduction neutralization assay (11, 12), and the RSV antibody titers were expressed as reciprocal mean log2. Laboratory evidence of infection with RSV wt or vaccine strain was defined as isolation of RSV and/or a 4-fold or greater rise in serum RSV neutralizing antibody titer. The Fisher’s exact test (two-tailed) was used to compare the percent of adults shedding wt and candidate vaccine virus.
Sequence Analysis.
Vero cell monolayer cultures were infected with either the RSV B1 wt parent or cp-52 mutant virus at a multiplicity of infection (moi) of 0.2. After development of cytopathic effect at 3–5 days postinfection, infected cultures were frozen and thawed, and genomic RNA was extracted from clarified supernatants by using Trizol-LS reagent (Life Technologies, Grand Island, NY). Reverse transcription–PCR amplifications spanning the RSV genome were performed by using the GeneAmp XL RNA PCR Kit (Perkin–Elmer) and primer pairs specific to the RSV subgroup B strain 2B, which is highly related to B1 (unpublished observations). Briefly, reverse transcription was performed for 1 hr each at 55°C and 60°C, followed by hot start PCR with initial denaturation at 94°C for 3 min and 40 cycles of 94°C for 1 min, 55°C for 0.5 min, and 70°C for 5 min, followed by extension at 70°C for 10 min. A consensus sequence for the PCR amplified products was generated by using the Applied Biosystems-PRISM fluorescent dye terminator cycle sequencing kit with AmpliTaq DNA polymerase, FS and the Applied Biosystems 377 DNA sequencer (Perkin–Elmer). Sequences were analyzed by using the MacVector gene analysis program (Oxford Molecular, Oxford, UK).
Analysis of Gene Transcription Products.
Total cell-associated RNA was isolated from Vero cells 48 hr after infection with either RSV B1 or cp-52 virus at a moi of 2. RNA was extracted with Trizol-LS reagent and analyzed by Northern blotting by using RSV B1-specific M, SH, G, and F gene probes (see Fig. 1A) as described in the Fig. 2 legend. Two G gene-specific probes designated G and Gsm were used: the G gene probe contains ≈380 nucleotides from the central portion of the G gene transcription unit, and the Gsm probe contains ≈300 nucleotides derived from the 3′ end of the mRNA (Fig. 1A).
Figure 1.
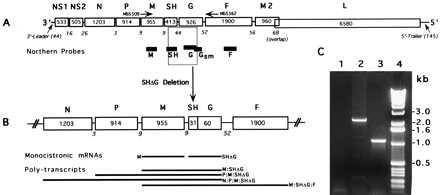
Genetic map of the RSV B1 parental strain (15,225 nts) and its deletion mutant, cp-52 (13,933 nts). Genes are listed on top according to encoded proteins: NS1 and NS2, nonstructural proteins; N, nucleocapsid protein; P, phosphoprotein; M, matrix protein; SH, small hydrophobic protein; G, attachment protein; F, fusion protein; M2, second matrix protein; L, large polymerase protein. The numbers in boxes are gene lengths and numbers below are the length of the intergenic regions with the exception of M2:L, which has a 68-nt overlap rather than an intergenic region. Map is not to scale. (A) Genetic map of wt B1. Noncoding 3′-leader (44 nt) and 5′-trailer (145 nt) are the potential genomic and antigenomic promoters. A primer pair (MSS509/MSS562) used for amplification of fragment IIa across the deleted region of cp-52 virus is depicted by arrows. Genomic location of the RNA probes used for Northern analysis is depicted by solid bars under the RSV B1 genome. (B) Genetic map of cp-52 with deleted SH and G gene regions. Two monocistronic gene products, M and SHΔG, detectable by M and Gsm gene probes, respectively, and several polycistronic transcription products identically detectable by both of these probes are depicted at the bottom. (C) Ethidium bromide-stained 1% agarose gel showing reverse transcription–PCR amplification products generated by primers MSS509 and MSS562 that used RSV B1 or cp-52 genomic RNAs. PCR product amplified from cp-52 RNA (lane 3) was found to be ≈1.3 kb smaller than that from B1 RNA (lane 2). Lane 1 is a reagent control and lane 4 shows size markers (1-kb ladder, Life Technologies).
Figure 2.
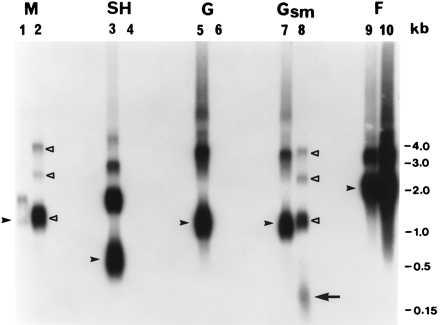
Northern blot hybridization of total intracellular RNA extracted from B1 and cp-52 virus-infected Vero cells. Replicate RNA samples (5 μg) were fractionated by electrophoresis for 3.5 hr at 90 V in a 1.2% agarose-2.2 M formaldehyde gel in 1× Mops buffer (pH 7.0). RNA was transferred in 20× SSC (1× SSC is 0.15 M NaCl and 0.015 M sodium citrate) to 0.2 μm Nytran nylon membrane with a TurboBlotter system (Schleicher & Schuell), and then was fixed by UV crosslinking. Negative-sense riboprobes (≈300 to 400 nt) labeled with [α-32P]UTP were prepared by in vitro transcription of B1 virus-specific PCR products (containing T7 promoter sequence) by using a MAXIscript T7 kit (Ambion, Austin, TX). Probe map positions on the RSV B1 genome are shown in Fig. 1A; 1.25 × 107 cpm of each probe were used for hybridization at 65°C in Rapid-hyb buffer (Amersham). Stringency washes of 15 min each were done twice in 2× SSC, 0.1% SDS at room temperature and twice in 0.2× SSC, 0.1% SDS at 65°C. The 65°C washes were separated by room temperature treatment with 1 μg/ml RNase A in 2× SSC for 15 min to remove nonspecifically bound probe (Promega). The blot was exposed to x-ray film for 6.5 hr. B1, lanes 1, 3, 5, 7, and 9; cp-52, lanes 2, 4, 6, 8, and 10. RSV B1-specific monocistronic mRNA transcripts corresponding to M, SH, G, and F genes are indicated by filled arrowheads. Identical polytranscripts unique for cp-52 virus that were detected independently by the M and Gsm probes are marked by open triangles in lanes 2 and 8 (see Fig. 1B for identification). The predicted SHΔG transcript is identified by the long arrow in lane 8. It should be noted that a short exposure revealed that the cp-52 M-specific signal in lane 2 (marked by the open triangle closest to the bottom) consists of two RNA species of similar size: a monocistronic M mRNA that is identical to the RNA identified by the filled arrowhead in B1 lane 1, and an M:SHΔG read-through transcript. The weak monocistronic M signal for B1 virus (lane 1) that was consistently observed in independent experiments indicates inefficient transcription termination and/or mRNA instability in RSV B1 virus.
Identification of G Glycoprotein by Western Blot.
Vero cell monolayer cultures were infected with either B1 or cp-52 virus at an moi of 1, or were mock-infected. Cells were harvested at 30 hr postinfection into lysing buffer (1% Nonidet P-40/0.4% deoxycholic acid/66 mM EDTA/10 mM Tris⋅HCl, pH 7.4), and cell nuclei were removed by centrifugation (1,000 × g). Proteins from crude cell lysates were separated by electrophoresis on 8–16% gradient polyacrylamide-SDS gels under denaturing, but nonreducing conditions and analyzed by Western blotting with RSV G protein-specific mAb K6 purified from murine ascites fluid (13). A biotinylated horse anti-mouse IgG was used with an avidin DH and biotinylated horseradish peroxidase H detection system.
Identification of F or G Glycoproteins in Viral Plaques by Immunostaining.
Viral plaques that developed on Vero cell monolayer cultures were immunostained by using a mouse anti-RSV F or G mAb-immunoperoxidase system as described previously (14). mAbs used to identify the RSV F and G glycoproteins in the plaques formed by B1 wt or the cp-52 mutant were kindly provided by Larry Anderson, Centers for Disease Control and Prevention, Atlanta, GA (mAbs 131–2 g, 130–5f, 92–11C, and 102–10B) and Edward Walsh, University of Rochester School of Medicine, Rochester, NY (mAb L9).
Analysis of Viral Growth at Low Temperature.
To generate multicycle growth curves for RSV B1 wt and cp-52 viruses, Vero cell monolayers were infected with either virus at a moi of 0.01, and growth was assessed at 25°C. Aliquots of the supernatant were removed daily for 14 days postinfection, and virus was quantitated by plaque titration on Vero cell monolayer cultures incubated at 32°C.
RESULTS
Response of Adults and Children to wt RSV B1 and RSV B1 cp-52.
The RSV B1 wt virus infected 53% of the adult volunteers and caused upper respiratory tract illness in 5 of the 10 infected adults. This degree of virulence of the wt virus in adults allowed us to assess the effect of the cp-52 mutations on attenuation. In contrast to individuals who received wt virus, only 6% of adults who received cp-52 shed virus [P = .003, Fisher’s (two-tailed) exact test, Table 1]. This indication of attenuation of the cp-52 virus in adults suggested that it was safe to evaluate this candidate vaccine mutant in seropositive children, and subsequently in seronegative children. The cp-52 vaccine candidate infected seropositive and seronegative children, but the frequency and magnitude of virus shedding were low, especially compared with RSV subgroup A vaccines that had been evaluated similarly (8). In adults and children, vaccine virus was shed between days 3 and 10 after inoculation, likely the result of viral replication rather than recovery of the inocula. The limited shedding of cp-52, coupled with the absence of a serum antibody response by infected vaccinees (Table 1), indicated that cp-52 was infectious but overattenuated for susceptible humans. The cp-52 virus therefore had sustained one or more host-range mutations that did not restrict replication in Vero cells, but nonetheless were attenuating for humans.
Table 1.
Response of adults to RSV wild-type or to RSV B1 cp-52 mutant virus and of infants and children to RSV B1 cp-52 or placebo
| Subjects | RSV B1 administered | Dose (log10 pfu) | No. of subjects | % Infected | Virus isolation, nasal wash
|
% with indicated illness
|
Serum neutralizing antibody titer, reciprocal mean (SD) log2
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Shedding virus | Peak titer, mean (SD) log10 pfu/ml | Febrile | URI | LRI | OM | Any RSV-like | Pre | Post | % with rise | |||||
| Adults | wt | 4.7 | 19 | 53 | 53 | 3.1 (1.3) | 5 | 21 | 0 | 0 | 26 | 10.2 (1.2) | 10.6 (0.9) | 6 |
| cp-52 | 5.0 | 17 | 6 | 6 | 1.9† | 6 | 0 | 0 | 0 | 6 | 9.3 (0.8) | 9.4 (0.9) | 0 | |
| Sero + | cp-52 | 4.0 | 4 | 25 | 25 | ≤0.6* | 75 | 50 | 0 | 0 | 75 | 9.1 (0.8) | 8.5 (0.7) | 0 |
| children | cp-52 | 5.0 | 11 | 45 | 45 | 0.9† (0.6) | 9 | 18 | 0 | 0 | 27 | 9.9 (1.6) | 9.7 (1.4) | 0 |
| Placebo | 0.0 | 7 | 0 | 0 | ≤0.6 | 14 | 0 | 0 | 0 | 14 | 10.2 (1.9) | 9.7 (1.8) | 0 | |
| Sero − | cp-52 | 4.0 | 7 | 14 | 14 | 1.3† | 28 | 14 | 0 | 14 | 28 | 4.3 (0.1) | 4.3 (0.1) | 0 |
| children | cp-52 | 5.0 | 9 | 11 | 11 | 1.9† | 22 | 56 | 0 | 22 | 67 | 4.4 (0.3) | 4.4 (0.3) | 0 |
| Placebo | 0.0 | 10 | 0 | 0 | ≤0.6 | 20 | 30 | 0 | 10 | 50 | 4.3 (0.1) | 4.4 (0.2) | 0 | |
Healthy adults, 15- to 59-month-old RSV seropositive and 6- to 24-month-old RSV seronegative children were enrolled in these studies. For the purposes of this study, seropositive children were those with an RSV serum plaque reduction neutralizing antibody titer >1:40. URI, upper respiratory tract illness; LRI, lower respiratory tract illness; OM, otitis media.
This patient shed vaccine virus that did not plaque.
One adult, one seropositive child, and two seronegative children shed vaccine virus in titers ranging from 101.3 to 102.1 pfu/ml. Attempts to recover vaccine virus from snap-frozen nasal wash specimens by serial passage in Vero cell culture were unsuccessful, probably because low titers of virus were shed.
Genetic and Immunologic Analysis of wt RSV B1 and RSV B1 cp-52.
To understand the genetic basis of the host-range mutation(s), the nucleotide sequence of the B1 wt parent and cp-52 viruses was determined. The full RNA genome of B1 virus was amplified by reverse transcription–PCR as four overlapping fragments (I-IV) of ≈3.9-, 4.7-, 3.9-, and 4.7-kb length (data not shown). These amplified products were sequenced directly on both strands by using RSV 2B-specific primers. Consensus sequence of the full-length RSV B1 was determined and used for the amplification and sequence analysis of its cp-52 derivative. Reverse transcription–PCR of the cp-52 genomic RNA failed to amplify full-length fragment II (≈4.7 kb), which spans the M, SH, G, and F genes. Primer pairs were designed to amplify this region as two smaller fragments, from nucleotide 3,287 to 5,679 (IIa) and 5,465 to 7,707 (IIb). Fragment IIb that spanned the F gene was successfully amplified. Attempts to amplify fragment IIa that spanned the M, SH, and G genes (Fig. 1A) yielded a truncated product of ≈1.1 kb, which was ≈1.3 kb shorter than the full-length IIa fragment (Fig. 1C). Several other primer pair combinations spanning the IIa region also failed to produce a full-length product (data not shown), suggesting that a portion of this region was deleted in the cp-52 virus. Sequence analysis of the truncated IIa fragment revealed that most of the region spanning the SH and G genes of the cp-52 virus was deleted (Table 2, Fig. 1B), retaining only the first 31 nucleotides of the SH gene (including the gene-start signal) and the last 60 nucleotides of the G gene (including the gene-end signal). The remaining SH:G region could encode a chimeric transcript of ≈91 nucleotides that lacked a predicted ORF. In addition to the long deletion, cp-52 virus contains seven point mutations (Table 2), five of which code for amino acid changes (one in the F gene and four in the L gene), one that is silent (F gene), and one that is in the noncoding G:F intergenic region (Table 2).
Table 2.
Sequence comparison of RSV B1 and cp-52
| Gene | Genomic position | Nucleotide*
|
Amino acid change
|
||
|---|---|---|---|---|---|
| B1 | cp-52† | B1 → cp-52 | |||
| G:F | 5626 | C | A | Noncoding intergenic | |
| F | 6318 | A | G | Glu → Gly | 218‡ |
| 6460 | U | C | Silent | 265 | |
| L | 10973 | G | A | Arg → Lys | 822 |
| 13492 | A | C | Asn → His | 1662 | |
| 14164 | U | A | Leu → Ile | 1886 | |
| 14596 | U | C | Phe → Leu | 2030 | |
Positive (+) sense.
cp-52 also sustained a deletion of nucleotides 4249-5540 spanning the SH and G genes that is not shown in the table.
Number indicates position of amino acid in the indicated protein.
Northern blot analysis confirmed that the cp-52 virus lacked intact SH and G genes (Fig. 2). In contrast, identical monocistronic M and F gene products were produced, as expected, by the B1 and cp-52 viruses (Fig. 2, compare lanes 1 and 2 and 9 and 10). The patterns of RSV B1 RNA bands hybridizing with the G and Gsm probes (Fig. 2, lanes 5 and 7) were identical and were consistent with those predicted for the normal G gene transcription products. The M and Gsm probes detected unique and identical bands consistent with the predicted SHΔG-containing polytranscripts, namely, M:SHΔG, P:M:SHΔG, N:P:M:SHΔG, and/or M:SHΔG:F (Fig. 1B) in the cp-52 virus (Fig. 2, lanes 2 and 8). These bands were not seen with the wt B1 virus (Fig. 2, lanes 1 and 7). These polytranscripts could have been produced only as a consequence of the SHΔG chimeric gene structure that juxtaposes the M gene with the truncated G gene and removes the SH:G intergenic region, allowing read-through across the RSV SH:G gene junction. In addition, the Gsm probe identified the predicted SHΔG gene fusion transcript of ≈91 nucleotides (Fig. 2, lane 8), which also was authenticated by ribonuclease protection studies that used a cp-52 probe specific to the SH:G gene boundary (data not shown). Further evidence to support the G gene deletion in cp-52 virus was provided by Northern blot analysis of genomic RNA extracted from virions. A positive-sense B1-specific G gene probe that hybridized to full-length B1 RNA failed to react with cp-52 genomic RNA, whereas genomic RNA from both viruses hybridized with a control probe containing 3′-leader and NS1 gene sequences (data not shown).
Immunologic confirmation of the deletion of RSV G from cp-52 was provided when RSV-infected cell cultures were analyzed by Western blot (data not shown) and plaque immunostaining that used G protein-specific mAbs. As shown in Table 3, RSV-B cp-52 plaques were stained with mAbs specific for RSV F protein but not with those specific for RSV G protein. The failure of broadly reactive G protein-specific mAbs to detect G protein in cp-52 virus-infected cells by two different assays thus provides further evidence that an intact RSV G protein is not produced by this mutant virus.
Table 3.
Cells infected with the cp-52 mutant virus do not bind murine mAbs directed at epitopes shared by subgroup A and subgroup B RSV G glycoproteins
| mAb | Protein specificity of mAb | Subgroup specificity of mAb | Reactivity of indicated RSV with Ab
|
||
|---|---|---|---|---|---|
| A2 wt | B1 wt | cp52 | |||
| 131-2g | G | A,B | + | + | − |
| 130-5f | G | A,B | + | + | − |
| L9 | G | A,B | + | + | − |
| 92-11C | F | A | + | − | − |
| 102-10B | F | B | − | + | + |
Plaque immunostaining was performed as previously described (14). Vero cell monolayers in 24-well plates were inoculated with 50 pfu of indicated virus, incubated for 5 days at 37°C, fixed with methanol, and stained with RSV F- or G-specific mAbs. Each G-specific mAb was used at a dilution of 1:200. Each F-specific mAb was used at a dilution of 1:1,000.
Growth of RSV B1 wt and cp-52 at 25°C.
As shown in Fig. 3, titers of cp-52 in infected Vero cell culture supernatants were approximately 10- to 100-fold higher than RSV B1 throughout the course of replication. It is likely that cp-52 emerged as the dominant strain during cold passage because of this growth advantage.
Figure 3.
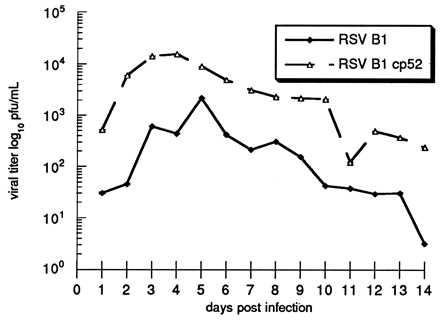
Multistep growth curves for RSV B1 wt and cp-52 viruses. Vero cell monolayer cultures were infected with either B1 or cp-52 at an moi of 0.01. Cultures were maintained at 25°C, and aliquots of supernatant were removed daily for 14 days postinfection, snap-frozen, and stored at −70°C. Thawed aliquots were titered by plaque assay on Vero cell monolayer cultures maintained under a semisolid overlay at 32°C as previously described, and results were expressed as log10 pfu/ml (8). Each point represent the mean ± SE of three experiments. The titers of the input viruses were 2.5 × 104 pfu/ml for RSV B1 wt and 2.3 × 104 pfu/ml for cp-52.
DISCUSSION
The serial passage of wt respiratory viruses at low temperature to select attenuated mutant viruses has been used to produce live attenuated influenza and human parainfluenza type 3 (PIV-3) candidate vaccines (15, 16), and most recently, a live attenuated RSV A candidate vaccine (8). Each of these candidate vaccines (cold-adapted influenza, cp-45 PIV-3, and RSV A 248/404) contain temperature-sensitive and non-temperature sensitive attenuating mutations that act in concert to restrict replication in rodents, primates, and humans (8, 17, 18), yet permit sufficient replication to induce virus-specific systemic and mucosal antibody responses. Although the genetic basis of attenuation of these candidate vaccines has not been fully defined, each possess a series of point mutations in the coding or regulatory regions of the genomes that specify the mutant phenotypes (19–21). In the present study, passage of RSV B1 at low temperature selected for a host-range mutant that was able to replicate efficiently in Vero cells, but was highly restricted in replication and poorly immunogenic in seronegative vaccinees. In contrast, seronegative vaccinees who received RSV A candidate vaccines in previous studies shed a moderate amount of virus and developed a high level of serum neutralizing antibodies (8). Thus, cp-52 appears to be overattenuated and is unlikely to prove useful as a vaccine strain.
Sequence analysis and in vitro studies indicated that the cp-52 virus sustained a large deletion that ablated synthesis of the SH and G surface glycoproteins. It is perhaps not completely surprising that an RSV lacking an SH gene can replicate effectively in vitro, because many paramyxoviruses lack this membrane glycoprotein and a recent report describes the absence of SH in the Enders strain of mumps virus despite its presence in other mumps strains (22). However, the mechanism by which an RSV lacking the attachment (G) glycoprotein can initiate infection remains to be determined. It is possible that naturally occurring cell surface lectins could serve as an alternate receptor for cp-52, and that the F protein might serve as a ligand for this receptor, as has been previously described for Sendai virus (23, 24). Whether the host range phenotype of cp-52 might result from a difference in lectins on the surface of Vero cells and human respiratory epithelium requires further study.
The mechanism by which this replication-competent deletion mutant arose was not clear initially, but because the cp-52 mutant was recovered after multiple cold passages, we considered the possibility that this mutant may have had a growth advantage over wt RSV in Vero cell culture at low temperature. The multicycle growth curve analysis indicated that cp-52 grew to significantly higher titer than wt virus in cell cultures incubated at low temperature, suggesting it may replicate more efficiently and/or be less cell-associated than wt virus. Hence, cp-52 is likely to have emerged during repeated cold passage of virus-infected culture fluids because of its growth advantage over wt virus. In addition, replication of cp-52 may have interfered with replication of the wt virus, as has been previously described for cold-adapted influenza and wt influenza viruses (25, 26). Recently, it has been shown that RSV with an engineered insertion exhibited decreased replicative capacity (27). Therefore, it also is possible that cp-52 may replicate more efficiently than wt virus because of its truncated genome. In addition, the cp-52 virus is clearly a host-range mutant, because its replication is highly restricted in rodents, nonhuman primates, and humans despite its efficient replication in Vero cell culture. Whether these host-range properties are the result of the five point mutations resulting in amino acid substitutions, the large SH:G deletion, or both awaits additional study.
Although the cp-52 mutant virus is not an appropriate RSV B vaccine candidate for RSV seronegative infants and children, we have learned that a large mutation involving the deletion of the RSV SH and G genes is compatible with efficient replication in cell culture. It is possible that deletion of a nonessential viral gene (such as SH) might contribute to the attenuation of future candidate vaccines. The use of cDNA technology (28) will allow the construction of a series of diverse recombinant viruses to assess the individual contribution of the point mutations and the SH and G deletions to the attenuation phenotype of cp-52. Once the critical mutations are identified, recombinant viruses containing these mutations can be produced and evaluated in preclinical and clinical trials for their usefulness in RSV vaccine development.
Acknowledgments
We thank Roberta Casey, Barbara Burns, Victoria Hodgins, Pamela Nehring, Anna Speaks, Ellen Davis, Bhagvanji Thumar, and Jean Froehlich for clinical and technical assistance, Martin Blair for assistance with manuscript preparation, and Robert Chanock for review of the manuscript. Special thanks go to Robert Lerch for helpful discussions and to Albert Cupo for assistance with Western blots. This work was supported by National Institutes of Health Contract AI-15095 and by Wyeth-Lederle Vaccines and Pediatrics.
ABBREVIATIONS
- cp
cold-passaged
- RSV
respiratory syncytial virus
- wt
wild type
- pfu
plaque-forming unit
- moi
multiplicity of infection
Footnotes
References
- 1.Collins P L, McIntosh K, Chanock R M. In: Fields Virology. Fields B N, editor. New York: Raven; 1996. pp. 1313–1351. [Google Scholar]
- 2.Anderson L J, Parker R A, Strikas R L. J Infect Dis. 1990;161:640–646. doi: 10.1093/infdis/161.4.640. [DOI] [PubMed] [Google Scholar]
- 3.Committee on Issues and Priorities for New Vaccine Development. New Vaccine Development: Establishing Priorities. National Academy Press, Washington, DC: Institute of Medicine; 1985. p. 342. [Google Scholar]
- 4.Murphy B R, Hall S L, Kulkarni A B, Crowe J E, Collins P L, Connors M, Karron R A, Chanock R M. Virus Res. 1994;32:13–36. doi: 10.1016/0168-1702(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 5.Taylor C E, Morrow S, Scott M, Young B, Toms G L. Lancet. 1989;1:777–778. doi: 10.1016/s0140-6736(89)92592-0. [DOI] [PubMed] [Google Scholar]
- 6.McConnochie K M, Hall C B, Walsh E E, Roghmann K J. J Pediatr. 1990;117:52–62. doi: 10.1016/s0022-3476(05)82443-6. [DOI] [PubMed] [Google Scholar]
- 7.Hall C B, Walsh E E, Schnabel K C, Long C E, McConnochie K M, Hildreth S W, Anderson L J. J Infect Dis. 1990;162:1283–1290. doi: 10.1093/infdis/162.6.1283. [DOI] [PubMed] [Google Scholar]
- 8.Karron R A, Wright P F, Crowe J E, Jr, Clements M L, Thompson J, Makhene M, Casey R, Murphy B R. J Infect Dis. 1997;176:1428–1436. doi: 10.1086/514138. [DOI] [PubMed] [Google Scholar]
- 9.Crowe J E, Jr, Bui P T, Firestone C-Y. J Infect Dis. 1996;173:829–839. doi: 10.1093/infdis/173.4.829. [DOI] [PubMed] [Google Scholar]
- 10.Karron R A, Wright P F, Hall S L, Makhene M, Thompson J, Burns B A, Tollefson S, Steinhoff M C, Wilson M H, Harris D O, Clements M L, Murphy B R. J Infect Dis. 1995;171:1107–1114. doi: 10.1093/infdis/171.5.1107. [DOI] [PubMed] [Google Scholar]
- 11.Connors M, Collins P L, Firestone C-Y, Sotnikov A V, Waitze A, Davis A R, Hung P P, Chanock R M, Murphy B R. Vaccine. 1992;10:475–484. doi: 10.1016/0264-410x(92)90397-3. [DOI] [PubMed] [Google Scholar]
- 12.Connors M, Kulkarni A B, Firestone C-Y, Holmes K L, Morse H C, III, Sotnikov A V, Murphy B R. J Virol. 1992;66:7444–7451. doi: 10.1128/jvi.66.12.7444-7451.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh E E, Hall C B, Schlesing J J, Brandriss M W, Hildreth S, Paradiso P. J Gen Virol. 1989;70:2953–2961. doi: 10.1099/0022-1317-70-11-2953. [DOI] [PubMed] [Google Scholar]
- 14.Murphy B R, Sotnikov A V, Lawrence L A, Banks S M, Prince G A. Vaccine. 1990;8:497–502. doi: 10.1016/0264-410x(90)90253-i. [DOI] [PubMed] [Google Scholar]
- 15.Maasab H F, DeBorde D C. Vaccine. 1985;3:355–369. doi: 10.1016/0264-410x(85)90124-0. [DOI] [PubMed] [Google Scholar]
- 16.Belshe R B, Hisson F K. J Med Virol. 1982;10:235–242. doi: 10.1002/jmv.1890100403. [DOI] [PubMed] [Google Scholar]
- 17.Wright P F, Karzon D T. Prog Med Virol. 1987;34:70–88. [PubMed] [Google Scholar]
- 18.Hall S L, Stokes A, Tierney E L, London W T, Belshe R B, Newman F C, Murphy B R. Virus Res. 1992;22:173–184. doi: 10.1016/0168-1702(92)90049-f. [DOI] [PubMed] [Google Scholar]
- 19.Subbarao E K, Kawaoka Y, Murphy B R. J Virol. 1993;67:7223–7228. doi: 10.1128/jvi.67.12.7223-7228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stokes A, Tierney E L, Sarris C M, Murphy B R, Hall S L. Virus Res. 1993;30:43–52. doi: 10.1016/0168-1702(93)90014-e. [DOI] [PubMed] [Google Scholar]
- 21.Firestone C-Y, Whitehead S S, Collins P L, Murphy B R, Crowe J E., Jr Virology. 1996;225:419–422. doi: 10.1006/viro.1996.0618. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi K, Tanabayashi K, Hishiyama M, Yamada A. Virology. 1996;225:156–162. doi: 10.1006/viro.1996.0583. [DOI] [PubMed] [Google Scholar]
- 23.Markwell M A K, Portner A, Schwartz A L. Proc Natl Acad Sci USA. 1985;82:978–982. doi: 10.1073/pnas.82.4.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bitzer M, Lauer U, Baumann C, Spiegel M, Gregor M, Neubert W J. J Virol. 1997;71:5481–5486. doi: 10.1128/jvi.71.7.5481-5486.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitaker-Dowling P, Lucas W, Youngner J S. Virology. 1990;175:358–364. doi: 10.1016/0042-6822(90)90420-v. [DOI] [PubMed] [Google Scholar]
- 26.Brown E G, Dimock C F, Hannah K. J Virol. 1992;66:6314–6321. doi: 10.1128/jvi.66.11.6314-6321.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bukreyev A, Camargo E, Collins P L. J Virol. 1996;70:6634–6641. doi: 10.1128/jvi.70.10.6634-6641.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins P L, Hill M G, Camargo E, Grosfeld H, Chanock R M, Murphy B M. Proc Natl Acad Sci USA. 1995;92:11563–11567. doi: 10.1073/pnas.92.25.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]