

Abstract
Archaea use a variety of small basic proteins to package their DNA. One of the most widespread and highly conserved is the Alba (Sso10b) protein. Alba interacts with both DNA and RNA in vitro, and we show in the present study that it binds more tightly to dsDNA (double-stranded DNA) than to either ssDNA (single-stranded DNA) or RNA. The Alba protein is dimeric in solution, and forms distinct ordered complexes with DNA that have been visualized by electron microscopy studies; these studies suggest that, on binding dsDNA, the protein forms extended helical protein fibres. An end-to-end association of consecutive Alba dimers is suggested by the presence of a dimer–dimer interface in crystal structures of Alba from several species, and by the strong conservation of the interface residues, centred on Arg59 and Phe60. In the present study we map perturbation of the polypeptide backbone of Alba upon binding to DNA and RNA by NMR, and demonstrate the central role of Phe60 in forming the dimer–dimer interface. Site-directed spin labelling and pulsed ESR are used to confirm that an end-to-end, dimer–dimer interaction forms in the presence of dsDNA.
Keywords: Alba, archaea, ESR, NMR, site-directed spin labelling
Abbreviations: cw-ESR, continuous wave ESR; DEER, double electron–electron resonance; dsDNA, double-stranded DNA; EMSA, electrophoretic mobility-shift assay; FRET, fluorescence resonance energy transfer; HSQC, heteronuclear single-quantum coherence; SDSL, site-directed spin labelling; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA
INTRODUCTION
In contrast with the eukaryotic histone, there is no universal archaeal chromatin protein. Instead, archaea tend to utilize two or more unrelated proteins to package their DNA. The archaeal histone is conserved in the euryarchaeal branch of the archaeal lineage and, like its eukaryotic counterpart, it forms a nucleosome that wraps dsDNA (double-stranded DNA) (reviewed in [1]). Histone proteins are also present in the marine archaeon Cenarchaeum symbiosum [2], but are absent from most crenarchaea and also from euryarchaeal thermoacidophiles such as Thermoplasma acidophilum [3]. The second widely distributed chromatin protein, Alba (also known as Sac10b or Sso10b), is present in one or two copies in the genomes of all archaea sequenced to date with the exception of the Methanosarcinas and Halophiles [4].
Alba is a dimeric, highly basic protein with a subunit size of approx. 10 kDa. The Alba1 protein is highly abundant in the Sulfolobus species from which it was originally purified [5], representing approx. 4% of the total soluble protein [6]. The crystal structure of Alba1 from Sulfolobus solfataricus revealed a compact α/β structure similar to DNaseI, translation initiation factor IF3 and other bacterial proteins [7]. The structural similarity to RNA-binding proteins, coupled with the observation that Alba can bind to RNA in vivo and in vitro [8], has prompted the suggestion that Alba may also function as an RNA-binding protein in vivo [9]. A role in DNA binding in vivo has been confirmed by chromatin immunoprecipitation experiments, which show a wide distribution of S. solfataricus Alba1 at different gene loci [10]. In S. solfataricus a second Alba paralogue, Alba2, is expressed at 5–10% of the Alba1 protein level. Alba2 forms heterodimers with Alba1, resulting in a reduction in DNA-binding affinity, and it has been suggested that this may provide a mechanism for the control of chromatin packaging in this organism [11].
The Alba1 protein in S. solfataricus is reversibly acetylated on a single lysine residue, and this results in a reduction in the DNA-binding affinity [12]. In vitro, acetylation of Alba1 reduces the repressive effects of the protein on transcription [12] and on strand separation by the replicative helicase MCM (minichromosome maintenance) [13]. However, the relevance of these findings in other archaeal species is not yet clear [4]. DNA binding by Alba is a highly co-operative process, with a final stoichiometry of one Alba dimer for every 6 bp of dsDNA bound, suggesting a high binding density [5,6]. Electron microscopy studies of DNA binding by Alba from Sulfolobus acidocaldarius revealed the formation of fibre-like structures that were thought to indicate extended interwound helical protein fibres [5]. These findings were later confirmed using the recombinant protein from S. solfataricus [11], where it was further shown that the presence of the Alba2 protein caused subtle changes in the chromatin structures visualized. The crystal structures of Alba1 from S. solfataricus and Alba1 proteins from other species have highlighted a strongly conserved dimer–dimer interface centred on the Phe60 residue, which stacks with its counterpart in the adjacent Alba1 dimer in the crystal lattice. It has been proposed that this interaction has biological relevance [14] and may relate to the fibre formation observed by electron microscopy [11]. It has also been suggested that one role of the Alba2 subunit is to weaken or disrupt the dimer–dimer interface in protein fibres, as this interface is not conserved in Alba2 [11].
In the present study we used NMR to map the amide backbone chemical-shift changes of S. solfataricus Alba1 upon binding to dsDNA, ssDNA (single-stranded DNA) and RNA. This highlights differences between the three nucleic acid types, both in terms of the residues that are perturbed and the nature (size and shape) of the complexes formed. We showed, using gel EMSAs (electrophoretic mobility-shift assays), that dsDNA is bound more tightly than either ssDNA or RNA. SDSL (site-directed spin labelling) was used as a sensitive probe of dimer–dimer formation on DNA binding, and the relevance of the Alba1 crystallographic dimer–dimer interface was confirmed using an F60A site-directed mutant form of the Alba1 protein.
EXPERIMENTAL
Protein expression, purification and mutagenesis
Recombinant Alba1 proteins were expressed in Escherichia coli BL21 Rosetta strain and purified as described previously [11,15]. Protein concentrations were determined by measuring the absorbance at 280 nm. The F60A and R59C mutant versions of Alba1 were constructed using the QuikChange® mutagenesis protocol (Stratagene), following the manufacturer's instructions. The oligonucleotides used for mutagenesis, F60Af, F60Ar, R59Cf and R59Cr, are available from the corresponding author on request. Mutated genes were sequenced fully to confirm that no spurious mutations had been introduced. The mutant proteins were purified as for the wild-type protein.
Oligonucleotides for binding studies
DNA and RNA oligonucleotides were purchased from MWG Biotech and Operon Biotechnologies. The sequence of the 39 mer ssDNA was 5′-CCTCGGTGCTAAGTTGATGCTGGTACTCGGAGTATCCCG-3′ and that of the 16 mer ssDNA and ssRNAs (single-stranded RNAs) were 5′-CCCGGCGT(U)GCGGCCCG-3′, where uracil (U) replaces thymine in the RNA oligomer. The 16 and 39 mer ssDNA sequences correspond to one complimentary strand of the 16 and 39 bp DNA duplexes respectively. 28 mer polyadenine (28A) and 30 mer polyuridine (30U) ssRNA oligonucleotides were also used.
DNA electrophoretic gel-retardation assays
Binding assays were performed, and the data analysed as described previously [11]. Apparent dissociation constants for the interaction between wild-type Alba1 and a 39 bp DNA duplex, 39 mer ssDNA, 30U, 28A and 16 mer ssRNAs, and for the interaction between F60A Alba1 and 16 and 39 bp DNA duplexes were determined. Data were fitted to a two-state binding model where the fraction bound was equal to:
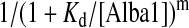 |
and the exponent, m, corrects for deviations in slope resulting from co-operativity. Data-fitting and graphical representations were generated using the program KaliedaGraph (Synergy Software).
NMR spectroscopy
The backbone assignment of Alba1 was obtained as described previously [11]. A 420 μM solution of 15N-labelled Alba1 in 20 mM Bis-Tris, 0.3 M NaCl, 1 mM NaN3 (pH 6.5) containing 10% 2H2O was titrated with a solution of 1.2 mM 16 bp DNA duplex to give a final molecular ratio (duplex:Alba dimer) of 0.2. Similar titrations were performed with the 16 mer ssDNA, 16 mer ssRNA and 28A ssRNA oligomers to final molecular ratios of 0.21, 0.33 and 0.14 respectively. Sub-stoichiometric quantities were used in order to maximize any potential contributions from additional protein–protein interactions formed upon binding of Alba1 to each oligonucleotide. After each addition the samples were allowed to equilibrate at 55 °C for ~15 min after which a 1H-15N HSQC (heteronuclear single-quantum coherence) spectrum was recorded. Spectra were acquired on a 500 MHz Brüker DRX spectrometer. All spectra were referenced in the direct dimension to the carrier frequency at 4.75 p.p.m. and to the relative gyromagnetic ratios of 1H and 15N in the indirect dimension. NMR data were processed using FELIX 2000 (Accelrys).
Plots of the average relative signal intensity as a function of the molar fraction of oligonucleotide were generated using KaliedaGraph (Synergy Software). The initial intensity of each amide resonance was normalized to 1. In order to reflect only the effect on correlation time, data relating to residues with an associated chemical-shift change (where √[(Δδn/10)2+Δδnh2] >0.015) were excluded.
SDSL
Site-directed mutagenesis was used to convert residue Arg59 into a cysteine residue. As there were no cysteine residues in the native S. solfataricus Alba1 sequence, the R59C mutant could be directly modified with the thiol-specific methanethiosulfonate spin label MTSSL (Toronto Research Chemicals). The protein was purified as for the wild-type protein, and dialysed against 10 mM Tes buffer (pH 7.4) and 100 mM NaCl overnight at 4 °C. The spin-labelling reaction was carried out using 20 μM R59C protein, 100 μM MTSSL in 10 mM Tes buffer (pH 7.4) and 100 mM NaCl in a volume of 2.5 ml [16]. After incubation at 4 °C for 1 h, the unincorporated label was removed from the protein sample using a Sephadex G-25 minicolumn (GE Healthcare) according to the manufacturer's instructions. The purified protein was concentrated to achieve a final concentration of 100–150 μM in a 300 μl total volume, and the incorporation of the spin label was confirmed by MS. For experiments in the presence of DNA, a 16 bp duplex assembled from the oligonucleotide 5′-CCCGGCGTGCGGCCCG-3′ and its complement, was added to the protein sample to a final concentration of 100 μM.
ESR spectroscopy
Samples were prepared for ESR by buffer-exchanging them into 2H2O/deuterated ethylene-glycol medium [Tes (pH 7.4) and 100 mM NaCl]. The spin-labelled protein was exchanged into 2H2O (Aldrich) buffer containing 20% deuterated ethylene glycol (Cambridge Isotope Laboratories) by sequential concentration and dilution with a Centricon spin concetrator. The protein solution was then transferred into a clear fused quartz ESR tube, with a 3 mm internal diameter under an argon atmosphere. The samples in ESR tubes were quick-frozen by immersion into a 2-methylbutane/hexane freezing mixture (~−70 °C) and then stored under liquid nitrogen until use. cw-ESR (continuous wave ESR) spectra were measured at X-band using a Brüker ESP 300 ESR spectrometer as described previously [17]. Spectra acquisition conditions are given in the legend to Figure 5. The pulsed DEER (double electron–electron resonance) experiments were carried out using a Brüker ELEXSYS E580 spectrometer operating at X-band with a dielectric ring resonator (ER 4118X-MD5-EN) and a Brüker 400U second microwave source unit. All measurements reported in the present study were made at 50 K with an over-coupled resonator giving a Q factor of approx. 100. The measurements used the four pulse, dead-time free, sequence with the pump pulse frequency positioned at the centre of the nitroxide spectrum; the frequency of the observer pulses was increased by 70 MHz [16]. The observer sequence used a 32 ns π-pulse; the pump π-pulse was typically 30 ns. The resulting dipolar coupling evolution data was analysed by Tikhonov regularization to a distance distribution using DeerAnalysis2006 developed and made freely available by Gunnar Jeschke (http://www.epr.ethz.ch/software/index) [18].
Figure 5. SDSL confirms the DNA-dependent assembly of consecutive Alba dimers.

(A) Overlaid cartoon and surface representations of three Alba1 dimers in the end-to-end arrangement found in various crystal structures are shown in grey. The side chains of Arg59 and Phe60 for each molecule, coloured blue and orange respectively, are shown as sticks. The distance (Å) between two Arg59 guanidinium N atoms belonging to the same dimer unit is highlighted in black. This image was generated using MacPyMOL (DeLano Scientific; http://www.pymol.org). (B) The interspin distance profile is obtained from transformation of the background corrected DEER spectrum, using Tikhonov regularization of the four pulse DEER spectrum of spin-labelled Alba R59C (no DNA). The minor peak is not significant. The normalized background-corrected DEER spectrum is also shown (insert), with the best fit by Tikhonov regularization shown in red. The experimental conditions and spectral analysis procedure are outlined in the Experimental section. A four pulse DEER sequence was used at X-band frequencies (approx. 9.5 GHz) and a sample temperature of 50 K. (C) First derivative cw-ESR spectra of spin-labelled Alba R59C are shown in the presence and absence of 16 bp DNA and 21 mer ssDNA. The black trace shows the cw-ESR spectrum in the absence of DNA, the green trace the spectrum in the presence of 16 bp DNA and the red trace in the presence of 21 mer ssDNA. The inserts show an amplification (×10) of salient features of the spectrum. ESR conditions: temperature, 140 K; modulation frequency, 100 kHz; modulation amplitude, 0.4 mT, microwave frequency, 9.5 GHz; and microwave power, 0.2 mW.
RESULTS
Comparison of nucleic-acid-binding affinities of Alba1
To compare the binding of Alba1 to dsDNA, ssDNA and ssRNA, we determined the binding affinities by EMSA using 39 bp dsDNA, 39 mer ssDNA and 30U, 28A and 16 mer ssRNA oligonucleotides (Figure 1). The RNA oligonucleotides were designed to test for sequence-specific binding and to avoid the possibility of base pairing. The apparent dissociation constants were calculated from experiments carried out in triplicate. The data show that Alba1 binds approx. 5-fold less tightly to ssDNA (Kd, 270 nM) and approx. 20-fold less tightly to ssRNA (Kd, 960 nM) than it does to dsDNA (Kd, 50 nM) (Table 1). In contrast, Guo et al. [8] reported that Kd values for all three species were ‘similar’ (~100–300 nM), although a rigorous quantification of the dissociation constants was not included in their paper. This difference can be accounted for by the fact that our experiments were conducted at relatively high salt (0.3 M compared with 0.01 M), which weakens the interaction with RNA [8], but does not affect the dsDNA-binding affinity (results not shown). Higher ionic strengths are likely to be closer to the physiological condition. Notably, we also found the RNA-binding affinity of Alba1 to be independent of the length of oligonucleotide, in contrast with dsDNA where binding to longer duplexes is tighter [11]. This suggests that the molecular nature of the interaction of Alba1 with ssRNA is also different from that with dsDNA. dsRNA, which adopts the A-form helical conformation, was not studied.
Figure 1. DNA and RNA binding by Alba1.

Binding curves were obtained by gel-retardation analysis. Data relate to binding of Alba to 39 bp dsDNA (data taken from [11]), 39 mer ssDNA and 30U ssRNA, 28A ssRNA and 16 mer ssRNA are denoted by ○, □, ▲, ■ and ● respectively. Values are means±S.E.M. for each data point and curves obtained from fitting the data to the binding equation described in the text are represented as solid black lines.
Table 1. Comparison of the binding affinity of Alba1 for DNA and RNA.
Values are means±S.E.M. for triplicate measurements. Values obtained for the F60A mutant are shown in parentheses.
| Oligonucleotide | Apparent Kd (nM) | m* |
|---|---|---|
| 16 bp (dsDNA) | 200±4† (710±20) | 1.3±0.1 (0.9±0.1) |
| 39 bp (dsDNA) | 56±2† (520±13) | 1.9±0.1 (1.4±0.1) |
| 39 mer (ssDNA) | 270±13 | 2.0±0.2 |
| 30U (ssRNA) | 900±38 | 1.6±0.1 |
| 28A (ssRNA) | 960±30 | 2.1±0.1 |
| 16 mer (ssRNA) | 920±69 | 2.2±0.4 |
*This parameter was used to improve the fit and reflects the steepness of the curve.
† These values are taken from [11].
Residues involved in binding dsDNA, ssDNA and ssRNA
Initial 1H one-dimensional experiments, where Alba1 was titrated with a 28 bp duplex, resulted in the formation of a very large complex suggestive of a DNA-templated assembly process involving multiple protein–protein interactions (results not shown). A much smaller complex was formed upon binding to a shorter duplex (16 bp) under the same conditions (0.3 M NaCl, pH 6.5, 55 °C). Residues perturbed upon binding this duplex and the 16 mer ssDNA and ssRNA oligomers were then determined by measuring the changes in chemical shift of fast exchange peaks observed in the 1H-15N HSQC spectrum of Alba1 upon titration with the relevant oligonucleotide. It was found that amide resonances, corresponding to Gly4 and Arg86, were not present in the 1H-15N HSQC spectrum at pH 6.5, presumably due to intermediate exchange with solvent. In addition, no peaks were observed for Lys16, Arg44 or Gln80 regardless of pH. In Figure 2 the summed, weighted changes in 1H- and 15N-chemical shifts are plotted against residue number. These changes cannot unequivocally be attributed to a direct interaction between protein and oligonucleotide, but their localized nature implies that large changes in protein conformation are not involved. When the sum chemical-shift changes (≥0.015) are plotted on to a surface representation of Alba1, the general binding regions can be clearly identified. It is clear that the basic surface predicted to interact with DNA, following the initial structural studies [7], does constitute the major portion of the binding interface. It is apparent that the residues involved in binding dsDNA and ssDNA are the same, whereas only a subset of these are involved in binding to the equivalent ssRNA. Additionally, there is a distinct region (residues 10–15) that appears to be associated more specifically with RNA binding, as well as a DNA-specific region (residues 19–21). Data obtained from a titration of Alba1 with 28 A ssRNA were very similar to those obtained with the 16 mer ssRNA, suggesting that the changes observed were not strongly sequence dependent.
Figure 2. NMR chemical-shift analysis of the Alba1 interaction with nucleic acids.

The weighted sum of 15N-r and 1H- proton chemical shift changes (√[(Δδn/10)2+Δδnh2]) upon titration of Alba with (A) 16 bp dsDNA, (B) 16 mer ssDNA, (C) 16 mer ssRNA and (D) 28A ssRNA are plotted against residue number. Vertical broken lines show residues associated with a conserved crystallographic dimer–dimer interface. Those residues with a sum chemical-shift change >0.015 are plotted. These are highlighted in red on the corresponding surface representations of Alba1 in order to indicate the regions that are perturbed upon binding. Lys16 and Arg44, which are in intermediate exchange, are shown in dash grey. Lys16 has been shown previously to be involved in binding DNA [7], and Arg44 is likely to be involved given its position and charge. The structural representations were generated using MacPyMOL (DeLano Scientific; http://www.pymol.org).
The NMR acquisition parameters, processing and experimental conditions for the titrations of Alba1 with DNA and RNA were identical. The degree to which peaks are attenuated upon addition of the various oligonucleotides therefore provides further evidence that the interaction of Alba1 with DNA is distinct from that with RNA (Figure 3). Provided that contributions from chemical and conformational exchange resulting from direct binding interactions are removed, the degree of line-broadening reflects the tumbling rate (i.e the size and shape) of the complex. The greater degree of line-broadening indicates that a larger complex is formed upon binding of Alba1 to the 16 mer ss- and ds-DNAs than to 16 mer ssRNA. These data clearly show that the increase in correlation time upon binding to DNA is larger than that upon binding to RNA. This indicates that the DNA complex is larger and/or more elongated than the RNA complex and is consistent with the formation of additional protein–protein interactions.
Figure 3. Global NMR line-broadening effects during titration of Alba1 with nucleic acid.

Alba1 was titrated with a 16 bp duplex (●), a 16 mer ssDNA (■) and the corresponding 16 mer ssRNA (▲). For each point in the titration, the average relative signal intensity for peaks in the 15N-1H HSQC spectrum of Alba1 was plotted (values are means±S.E.M.). Residues with an associated chemical-shift change were omitted from the calculation to remove contributions from chemical and conformational exchange resulting from direct binding interactions. Plots of the average relative signal intensity as a function of the molar fraction of oligonucleotide were generated using KaliedaGraph (version 3.6, Synergy Software). S.E.M.s were calculated for each experimental condition, where only cross-peaks satisfying the criterion √[(Δδn/10)2+Δδnh2]>0.015 were included in the analysis (corresponding to a total of 61, 58 or 67 cross-peaks in dsDNA, ssDNA and ssRNA experiments respectively).
Importance of the dimer–dimer interface for DNA binding
Upon binding of Alba1 to DNA, chemical-shift changes were observed for residues associated with a conserved crystallographic dimer–dimer interface (Figure 2). These observations are consistent with the formation of additional protein–protein interactions in the Alba1–DNA complex that may indicate a mechanism for the assembly of nucleoprotein filaments. Crystal structures of the Alba1 protein from several species highlight the conserved dimer–dimer interface [14]. In contrast, the S. solfataricus Alba2 protein is quite divergent in this region, and this interface is not present in the crystal structure [11]. Phe60 is central to the dimer–dimer crystallographic interface of Alba1, where it forms a π–π stacking interaction with an adjacent dimer. Mutation of this position (F54R) in Archaeoglobus fulgidus Alba1 resulted in a qualitative decrease in the apparent binding affinity for plasmid DNA [14]. The F60A mutant version of Alba1 was made to test the significance of this residue with respect to DNA binding. The apparent binding affinities of the mutant with 16 and 39 bp duplexes were determined by EMSA and compared with the wild-type protein (Figure 4 and Table 1). The F60A mutant bound the 16 mer and 39 mer duplexes more weakly than the wild-type protein (3.5-fold and 9-fold respectively), resulting in an equal affinity for both duplexes. This was consistent with the hypothesis that the dimer–dimer interface is involved in the assembly of Alba1–DNA nucleoprotein complexes and suggests that additional protein–protein interactions formed upon binding of the wild-type protein to longer duplexes are not present in the F60A complexes. These data therefore support the assertion that a protein–protein interaction surface, similar to the crystallographic dimer–dimer interface, is involved in the assembly of Alba1 nucleoprotein filaments.
Figure 4. The F60A Alba1 mutant binds DNA more weakly than the wild-type protein.

Alba–DNA-binding curves obtained by gel-retardation analysis. Open and closed symbols are used to denote binding of wild-type (circles) and F60A (squares) Alba1 to either a 39 bp or 16 bp duplex respectively. The sequence of the 39 mer ssDNA was 5′-CCTCGGTGCTAAGTTGATGCTGGTACTCGGAGTATCCCG-3′, which was annealed with a complementary strand to produce the 39 bp duplex. Gel-shift analysis was carried out in triplicate and analysed as described previously [11]. Values are means±S.E.M. The apparent dissociation constants measured for the wild-type and F60A mutant proteins with the 39 bp duplex were 56±2 μM and 520±13 μM respectively. The equivalent values for the 16 bp duplex were 100±4 μM and 710±20 μM. A significant increase in apparent binding affinity (Kd) for the 39 bp duplex when compared with the 16 bp duplex is observed for the wild-type, but not for the F60A mutant (Table 1). Data for the wild-type protein are taken from [11], but were measured at the same time as those for the F60A mutant.
SDSL as a probe for DNA-templated protein assembly
In the absence of DNA, Alba1 exists as a dimer in solution, and Arg59 in one subunit is separated from the same residue in the second subunit of the dimer by a distance of approx 40 Å (1 Å=0.1 nm) (Figure 5A). The optimum form of ESR spectroscopy to measure interspin distances depends on the separation of the spin labels: below 20 Å, dipolar splittings observed by conventional cw-ESR can be used; above 20 Å, more sophisticated ESR methods are required. We used pulsed DEER spectroscopy to measure the distance between spin labels in the R59C SDSL sample, estimating an average inter-spin distance of 41 Å with a half-height distribution of ±3.5 Å (Figure 5B), in good agreement with the crystal structure (Figure 5A).
When 16 bp duplex DNA was added to the spin-labelled Alba R59C to a final concentration of 100 μM, a dramatic change in the spin–spin interaction between spin-labels on adjacent monomers occurred. A splitting of the cw-ESR spectrum due to a dipolar interaction between close paramagnets was observed. In Figure 5(C) the cw-ESR spectrum of the spin-labelled Alba R59C is shown in the absence (top spectrum) and the presence (middle spectrum) of 16 bp duplex DNA or 21 mer ssDNA (bottom spectrum). Splitting of the spectrum due to dipolar interactions between closely adjacent paramagnets was induced by dsDNA binding (these are indicated in Figure 5 and also shown at ×10 amplification). Not all of the Alba R59C present was expected to exhibit this interaction, as the 16 bp duplex is only long enough to bind three dimers and there was also more DNA present than required for saturation of binding. Therefore the ESR spectrum shows a mixture of the interacting and non-interacting species because some of the spin-label will be at an exposed ‘free-end’ and not involved in a close dipolar interaction with an adjacent dimer. The dipolar splitting observed is in the order of 72 Gauss peak-to-peak (7.2×10−3 T) which corresponds to an interspin distance of approx. 7.5 Å [16]. This confirms the close assembly of two spin labels, as would be expected if consecutive Alba dimers stack end-to-end on DNA binding as predicted from the crystal structure. The addition of ssDNA (21 bases) appeared to have a grossly similar, but less well resolved, effect to the addition of dsDNA. There are splittings due to interactions between close spin-labels, but these are broader and less well resolved, indicating that the structure is more heterogeneous, perhaps due to a greater flexibility in the Alba–ssDNA complexes.
DEER spectra could not be readily obtained in the presence of dsDNA or ssDNA because the close dipolar interactions enhance the transverse relaxation times, which shortens the available experimental timing window to an extent that the technique cannot be readily used. Direct measurement of the transverse relaxation (results not shown) confirmed a considerable enhancement of the relaxation due to the close interactions between spin labels when bound to DNA.
DISCUSSION
The majority of sequenced archaeal genomes contain at least one gene encoding the Alba protein [3], making it perhaps the most conserved nucleic-acid-binding protein in the archaea. Although Alba clearly binds dsDNA in vitro, there has been some debate about its true role in vivo. On the one hand, UV cross-linking data has suggested that an interaction with RNA might be more physiologically relevant [8]. On the other, chromatin immunoprecipitation experiments have demonstrated that Alba1 is associated with dsDNA [10]. Alba1 from S. solfataricus has been shown to stabilize dsDNA against DNA melting by the cognate ssDNA-binding protein, consistent with a role in the stabilization of dsDNA in vivo [19]. Our data demonstrate that Alba1 binds dsDNA more tightly than either ssDNA or RNA in vitro, and suggests that differences exist in the interface with DNA and RNA species that are still not fully understood. The stabilization of RNA, as well as DNA, is likely to be particularly important in thermophiles, where chemical and physical damage to nucleic acid is accelerated by elevated growth temperatures [20]. A previous study of nucleic-acid-binding proteins in the crenarchaeote Thermoproteus tenax, which lacks any clear ssDNA-binding protein encoding gene, revealed that the two main detectable ssDNA-binding proteins in this organism were the Alba protein and a novel protein named CC1 [21]. Both proteins bound ssDNA and dsDNA. Thus it is possible that archaea utilize a number of highly expressed general nucleic-acid-binding proteins, such as Alba, to stabilize all classes of nucleic acid in vivo.
The DNA-interaction region, as determined by NMR, is only partially consistent with previous binding models [7,14] and differs significantly from that determined by Cui et al. [22]. Our experiment is different in that sub-stoichiometric quantities of duplex were used in order to maximize the contribution from protein–protein interactions, which were not evident at the 3:1 excess of duplex used previously [22]. We also found widespread changes in chemical shift upon the addition of salt, which we measured prior to commencing our titrations. We found that although the basic, positively charged surface is important, the extended R-loop region (residues 78–84), previously thought to bind in the minor groove of the double helix does not appear to be involved. The evidence for this is 2-fold; first, the lack of changes in chemical shifts in this region, and secondly, that the loop remains flexible upon binding. This flexibility is exemplified by Arg83, at the tip of the loop region, which gives rise to a distinctive sharp resonance throughout the titrations (results not shown). The NMR data also provided evidence that higher-order complexes of Alba1 are a feature of DNA binding. Oligomerization of Alba1, through a dimer–dimer interface, into linear rod-like structures has been observed in all but one of the five crystal structures published to date [7,14,23]. This has been argued to be of functional importance as the residues involved at the oligomerization interface are highly conserved within the archaea [14]. The NMR data suggest that this protein–protein interface might indeed be important for the assembly of the Alba–DNA nucleoprotein complex. This was confirmed by site-directed mutagenesis of the central residue of the interface, Phe60, which resulted in weaker DNA binding.
Additionally, we have shown using SDSL that Alba1 dimers assemble on DNA with consecutive dimers binding end-to-end, consistent with the crystal structures. This presumably relates to the extended helical protein fibres observed by electron microscopy when Alba1 is complexed with plasmid DNA [5,11]. An attractive hypothesis is that the dimer–dimer interface may provide a mechanism by which DNA could be rapidly sequestered into high-order structures. The next challenge is to formulate a molecular description of the chromatin structure formed by DNA-bound Alba1. The present study represents, to the best of our knowledge, the first example of the use of spin labelling to study nucleoprotein formation, and demonstrates the utility of SDSL to study this type of molecular interaction. One advantage of SDSL over FRET (fluorescence resonance energy transfer) is that only a single type of reporter group is required in SDSL, whereas donor and acceptor dyes are required for FRET studies, which can pose a technical challenge.
AUTHOR CONTRIBUTION
Clare Jelinska carried out the majority of the experimental work, analysed the NMR data and co-wrote the paper. Biljana Petrovic-Stojanovska prepared spin-labelled proteins. John Ingledew carried out the ESR measurements. John Ingledew and Malcolm White planned and supervised the experimental approach, analysed the data and co-wrote the paper with Clare Jelinska.
ACKNOWLEDGEMENTS
We thank Dr Catherine Botting and Dr Sally Shirran for MS services (which are funded by the Wellcome Trust), Professor Jonathan P. Waltho for critical discussion of the manuscript, and Dr M. J. Cliff and and Dr Hassane El-Mkami for assistance with data analysis. We are grateful to Dr Graham Smith (School of Physics, University of St. Andrews, Fife, U.K.) for the use of the Brüker ElexSys 580 Spectrometer, and for advice and support.
FUNDING
This work was supported by the Biotechnology and Biological Sciences Research Council [grant number BB/BIC165-09I004583/1]; and by the Engineering and Physical Sciences Research Council [grant number EP/F039034/1].
References
- 1.Sandman K., Soares D., Reeve J. N. Molecular components of the archaeal nucleosome. Biochimie. 2001;83:277–281. doi: 10.1016/s0300-9084(00)01208-6. [DOI] [PubMed] [Google Scholar]
- 2.Cubonova L., Sandman K., Hallam S. J., Delong E. F., Reeve J. N. Histones in crenarchaea. J. Bacteriol. 2005;187:5482–5485. doi: 10.1128/JB.187.15.5482-5485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White M. F., Bell S. D. Holding it together: chromatin in the archaea. Trends Genet. 2002;18:621–626. doi: 10.1016/s0168-9525(02)02808-1. [DOI] [PubMed] [Google Scholar]
- 4.Sandman K., Reeve J. N. Archaeal chromatin proteins: different structures but common function? Curr. Opin. Microbiol. 2005;8:656–661. doi: 10.1016/j.mib.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Lurz R., Grote M., Dijk J., Reinhardt R., Dobrinski B. Electron microscopic study of DNA complexes with proteins from the Archaebacterium Sulfolobus acidocaldarius. EMBO J. 1986;5:3715–3721. doi: 10.1002/j.1460-2075.1986.tb04705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue H., Guo R., Wen Y., Liu D., Huang L. An abundant DNA binding protein from the hyperthermophilic archaeon Sulfolobus shibatae affects DNA supercoiling in a temperature-dependent fashion. J. Bacteriol. 2000;182:3929–3933. doi: 10.1128/jb.182.14.3929-3933.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wardleworth B. N., Russell R. J., Bell S. D., Taylor G. L., White M. F. Structure of Alba: an archaeal chromatin protein modulated by acetylation. EMBO J. 2002;21:4654–4662. doi: 10.1093/emboj/cdf465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo R., Xue H., Huang L. Ssh10b, a conserved thermophilic archaeal protein, binds RNA in vivo. Mol. Microbiol. 2003;50:1605–1615. doi: 10.1046/j.1365-2958.2003.03793.x. [DOI] [PubMed] [Google Scholar]
- 9.Aravind L., Iyer L. M., Anantharaman V. The two faces of Alba: the evolutionary connection between proteins participating in chromatin structure and RNA metabolism. Genome Biol. 2003;4:R64. doi: 10.1186/gb-2003-4-10-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsh V. L., Peak-Chew S. Y., Bell S. D. Sir2 and the acetyltransferase, Pat, regulate the archael chromatin protein, Alba. J. Biol. Chem. 2005;280:21122–21128. doi: 10.1074/jbc.M501280200. [DOI] [PubMed] [Google Scholar]
- 11.Jelinska C., Conroy M. J., Craven C. J., Hounslow A. M., Bullough P. A., Waltho J. P., Taylor G. L., White M. F. Obligate heterodimerization of the archaeal Alba2 protein with Alba1 provides a mechanism for control of DNA packaging. Structure. 2005;13:963–971. doi: 10.1016/j.str.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 12.Bell S. D., Botting C. H., Wardleworth B. N., Jackson S. P., White M. F. The interaction of Alba, a conserved archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science. 2002;296:148–151. doi: 10.1126/science.1070506. [DOI] [PubMed] [Google Scholar]
- 13.Marsh V. L., McGeoch A. T., Bell S. D. Influence of chromatin and single strand binding proteins on the activity of an archaeal MCM. J. Mol. Biol. 2006;357:1345–1350. doi: 10.1016/j.jmb.2006.01.074. [DOI] [PubMed] [Google Scholar]
- 14.Zhao K., Chai X., Marmorstein R. Structure of a Sir2 substrate, Alba, reveals a mechanism for deacetylation-induced enhancement of DNA binding. J. Biol. Chem. 2003;278:26071–26077. doi: 10.1074/jbc.M303666200. [DOI] [PubMed] [Google Scholar]
- 15.Wardleworth B. N., Russell R., White M. F., Taylor G. L. Preliminary crystallographic studies of the double-stranded DNA binding protein Sso10b from Sulfolobus solfataricus. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001;57:1893–1894. doi: 10.1107/s0907444901015517. [DOI] [PubMed] [Google Scholar]
- 16.Rabenstein M. D., Shin Y. K. Determination of the distance between two spin labels attached to a macromolecule. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hastings S. F., Kaysser T. M., Jiang F., Salerno J. C., Gennis R. B., Ingledew W. J. Identification of a stable semiquinone intermediate in the purified and membrane bound ubiquinol oxidase-cytochrome bd from Escherichia coli. Eur. J. Biochem. 1998;255:317–323. doi: 10.1046/j.1432-1327.1998.2550317.x. [DOI] [PubMed] [Google Scholar]
- 18.Jeschke G., Panek G., Godt A., Bender A., Paulsen H. Data analysis procedures for pulse ELDOR measurements of broad distance distributions. Appl. Magn. Reson. 2004;26:223–244. [Google Scholar]
- 19.Cubeddu L., White M. F. DNA damage detection by an archaeal single-stranded DNA-binding protein. J. Mol. Biol. 2005;353:507–516. doi: 10.1016/j.jmb.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 20.Grogan D. W. Stability and repair of DNA in hyperthermophilic Archaea. Curr. Issues Mol. Biol. 2004;6:137–144. [PubMed] [Google Scholar]
- 21.Luo X., Schwarz-Linek U., Botting C. H., Hensel R., Siebers B., White M. F. CC1, a novel crenarchaeal DNA binding protein. J. Bacteriol. 2007;189:403–409. doi: 10.1128/JB.01246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Q., Tong Y., Xue H., Huang L., Feng Y., Wang J. Two conformations of archaeal Ssh10b. The origin of its temperature-dependent interaction with DNA. J. Biol. Chem. 2003;278:51015–51022. doi: 10.1074/jbc.M308510200. [DOI] [PubMed] [Google Scholar]
- 23.Wang G., Guo R., Bartlam M., Yang H., Xue H., Liu Y., Huang L., Rao Z. Crystal structure of a DNA binding protein from the hyperthermophilic euryarchaeon Methanococcus jannaschii. Protein Sci. 2003;12:2815–2822. doi: 10.1110/ps.03325103. [DOI] [PMC free article] [PubMed] [Google Scholar]