

Abstract
Bupropion (2a) analogues were synthesized and tested for their ability to inhibit monoamine uptake and to antagonize the effects of human α3β4*, α4β2, α4β4, and α1* nAChRs. The analogues were evaluated for their ability to block nicotine-induced effects in four tests in mice. Nine analogues showed increased monoamine uptake inhibition. Similar to 2a all but one analogue show inhibition of nAChR function selective for human α3β4*-nAChR. Nine analogues have higher affinity at α3β4*-nAChRs than 2a. Four analogues also had higher affinity for α4β2 nAChR. Analogues 2r, 2m, and 2n with AD50 values of 0.014, 0.015, and 0.028 mg/kg were 87, 81, and 43 times more potent than 2a in blocking nicotine-induced antinociception in the tail-flick test. Analogue 2x with IC50 values of 31 and 180 nM for DA and NE, respectively, and IC50 = 0.62 and 9.8 μm for antagonism of α3β4 and α4β2 nAChRs had the best overall in vitro profile relative to 2a.
Keywords: Nicotine, bupropion, structure activity relationship, dopamine uptake, norepinephrine uptake, nAChR antagonism, antinociception, locomotor activity, hypothermia, multiple target
Introduction
Tobacco-related diseases remain as the predominant cause of premature mortality. An estimated one billion individuals worldwide are smokers.1 Despite recent declines, it is estimated that 21% of the adult population in the United States are active smokers.2 Since less than 10% of smokers are capable of quitting unaided, improvements in the clinical management of smoking are needed.3
Nicotine (1) is the main active ingredient in tobacco smoke that causes and maintains tobacco addiction. Nicotine produces a myriad of profound behavioral and physiological effects and is able to initiate and support drug-seeking behavior in humans and in laboratory animals.4 The pharmacological and behavioral effects result from the activation of different nicotinic acetylcholine receptor (nAChR)a subtypes, which are members of an ionotropic neurotransmitter receptor superfamily.5 nAChRs containing α4 and β2 or α7 subunits (α4β2- and α7-nAChR, respectively) are the two major subtypes found in the brain, although appreciable amounts of α3β4*- and α6β2*-nAChRs also are present in brain regions implicated in reward and drug dependence such as the substantia nigra, the ventral tegmental area (VTA), and the medial habenula system.6-10
Although nAChRs are the initial sites of action of nicotine in the brain, downstream events evolving dopaminergic reward pathways may be critical in reinforcing smoking behavior. Nicotine, similar to other abused substances, is thought to be reinforcing because of the stimulation of mesolimbic dopamine reinforcement pathways.11 Cigarette smoking acutely increases dopamine (DA) concentration in the ventral striatum/nucleus accumbens, key brain regions in the reward pathway.12
The first line medications on the market today to treat nicotine addiction are various nicotine replacement (NRT) formulations (nicotine gum, transdermal nicotine patches, vapor inhaler, nicotab, nasal spray, lozenges), presumed to mimic effects of tobacco-derived nicotine, the antidepressant bupropion (2a), and varenicline (3).13 Both varenicline and bupropion SR (a sustained-release formulation) reduce symptoms of withdrawal, cigarette craving and smoking reinforcement and produce smoking cessation with efficacy equal to or better than nicotine replacement.14 However, none of these therapies has proven to be ideal, as smoking relapse still occurs at alarmingly high rates even after successful short-term therapies.
The use of 2a for treating nicotine addiction resulted from serendipitous observations that patients taking 2a as an antidepressant were more successful in smoking cessation attempts. It was known that 2a inhibited DA and norepinephrine (NE) uptake activity, but the discovery that it also preferentially antagonized α3β4*-nAChR15,16 suggested that more than one of these targets might be involved in its smoking cessation efficacy. It is possible that bupropion SR is achieving its effects by increasing dopamine levels through DA uptake inhibition and shielding against nicotine induction of nAChR-mediated dopamine elevation. Although it is not clear what part if any is played by the ability of 2a to inhibit NE uptake in its smoking cessation activity, it is likely to contribute to nicotine withdrawal amelioration. Surprisingly, very little effort has been devoted toward the development of 2a analogues with improved smoking cessation properties.17-19
Compound 2a inhibits DA reuptake, increasing the synaptic levels of DA. Compound 2a also inhibits nicotine-induced DA and NE overflow from superfused striatial and hippocampal slices, respectively.20 Thus, 2a might be functioning as an indirect DA agonist by reward modulation of this downstream action of nicotine. The ability of 2a to alleviate withdrawal symptoms is consistent with an indirect dopamine agonist mechanism.21,22 Compound 2a noncompetitively inhibits carbamylcholine-induced 86Rb+ efflux from human neuroblastoma cells expressing α3β4*-nAChR15 and function of α3β2-, α4β2-, and α7-nAChR heterologously expressed in Xenopus oocytes.16 Other studies suggest that α3β4*-nAChR plays a major role in nicotine-evoked NE release from hippocampus.23,24
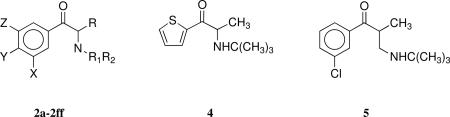
We recently reported the synthesis of a number of 2a analogues that were evaluated for their abilities to inhibit neurotransmitter uptake by the dopamine, norepinephrine, or serotonin (5HT) transporters, DAT, NET, or SERT, respectively, under the National Institute on Drug Abuse (NIDA) Cocaine Treatment Discovery Program (CTDP).25 Those studies sought to identify entities with better potency and selectivity toward DAT than 2a and whose activity as an indirect dopamine agonist might reveal a novel pharmacotherapy for treating cocaine addiction. In the current study, we designed and developed 2a analogues as smoking cessation aids, seeking entities that possess increased inhibitory activity for DA and NE uptake inhibition and/or nAChR antagonism while retaining the drug-like properties of the lead compound. In this study we report the synthesis and biological evaluation of 2a analogues 2b–2ff, 4, and 5. Some of the analogues have higher inhibitory potencies than 2a at DAT and NET as well as at α3β4*-nAChR. In addition, some of the compounds antagonize the antinociceptive, hyperlocomotor, and hypothermic effects of acutely administered nicotine in mice with potencies greater than that of 2a. 2-(N-tert-Butylamino)-3’,4-dichloropentanophenone (2x), which was 41- and 7.5-fold more potent in inhibition of DA and NE uptake and 3-fold more potent as an α3β4*-nAChR antagonist than 2a, is one of the more interesting compounds. Compound 2x is also 9-times more potent than 2a as an antagonist of nicotine-induced antinociception in the mouse tail-flick test.
Chemistry
The 2a analogues 2b–d, 2g–p, 2r, 2w–z, and 2ff were synthesized as previously reported.25 The new 2a analogues 2e, 2f, 2q, 2s–2v, 2aa–2ee, 4, and 5 were prepared using procedures exactly analogous to those used to synthesize the reported analogues. Thus, bromination of the ketones 6a–e with bromine in acetic acid afforded the bromoketones 7a–e. Treatment of 7a–e with tert-butylamine, cyclopentylamine, and piperidine yielded the desired 2a analogues 2e, 2f, 2q, 2s–v, 2aa–2ee, 4, and 5.
In Vitro Assays
The 2a analogues 2b–2ff, 4, and 5 were evaluated for their ability to block reuptake of [3H]dopamine ([3H]DA), [3H]serotonin ([3H]5HT), and [3H]norepinephrine ([3H]NE) using (h)DAT, (h)SERT, and h(NET) stably expressed in HEK293 cells using conditions similar to those previously reported.26,27 The results are given in Table 1. [3H]DA, [3H]5HT, and [3H]NE uptake values for 2a and analogues 2b–d, 2g–p, 2r, and 2n–z were obtained as a part of the NIDA CTDP and previously reported.25 For the most part, the relative potency in both evaluations was the same. However, in general, the efficacy in this study tended to be higher (lower IC50 values) for all analogues than the efficacies obtained in the CTDP program.25 There were some exceptions. For example, in the case of 2o, we obtained IC50 values of 209, 607, and 16,000 nM for the inhibition of [3H]DA, [3H]SERT, and [3H]NE uptake compared to 31 and 969 nM and inactive in the CTDP program.
Table 1.
Analog Inhibition of Monoamine Uptake and Nicotinic Acetylcholine Receptor (nAChR) Function
 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Monoamine Uptake Inhibitiona | nAChR Inhibitionb | ||||||||||||
| IC50 (nM) |
IC50 (μM) |
||||||||||||
| Compd | R | R1 | R2 | X | Y | z | [3H]DA | [3H]NE | [3H]SERT | α3β4*- | α4β2- | α4β4- | α1*- |
| 2a | CH3 | H | C(CH3)3 | Cl | H | H | 658 ± 178 | 1850 ± 300 | IA | 1.8 (1.15) | 12 (1.15) | 15 (1.07) | 7.9 (1.12) |
| 2b | CH3 | H | C(CH3)3 | F | H | H | 2320 ± 860 | 6500 ± 270 | IA | 4.4 (1.07) | 21 (1.12) | 52 (1.07) | 32 (1.07) |
| 2c | CH3 | H | C(CH3)3 | Br | H | H | 511 ± 33 | 5600 ± 1300 | IA | 1.3 (1.07) | 15 (1.12) | 13 (1.15) | 10 (1.07) |
| 2d | CH3 | H | C(CH3)3 | CH3 | H | H | 1470 ± 170 | 6200 ± 3500 | IA | 1.5 (1.07) | 19 (1.07) | 17 (1.15) | 11 (1.07) |
| 2e | CH3 | H | C(CH3)3 | CH3O | H | H | 3620 ± 460 | IA | IA | 2.0 (1.05) | 37 (1.07) | 23 (1.10) | 16 (1.15) |
| 2f | CH3 | H | C(CH3)3 | NO2 | H | H | IA | IA | IA | 11 (1.07) | 36 (1.10) | 74 (1.07) | 41 (1.12) |
| 2g | CH3 | H | C(CH3)3 | H | Cl | H | 1090 ± 150 | 2070 ± 660 | 9800 ± 4700 | 2.4 (1.10) | 33 (1.12) | 18 (1.15) | 14 (1.07) |
| 2h | CH3 | H | C(CH3)3 | H | Br | H | 689 ± 229 | 2540 ± 740 | 4508 ± 1722 | 1.4 (1.07) | 23 (1.10) | 13 (1.15) | 7.6 (1.07) |
| 2i | CH3 | H | C(CH3)3 | H | CH3 | H | 1950 ± 390 | 2350 ± 560 | IA | 2.4 (1.10) | 17 (1.07) | 16 (1.29) | 12 (1.15) |
| 2j | CH3 | H | C(CH3)3 | F | F | H | 7978 ± 4437 | 6480 ± 2100 | IA | 2.6 (1.10) | 45 (1.12) | 40 (1.35) | 24 (1.07) |
| 2k | CH3 | H | C(CH3)3 | Cl | Cl | H | 463 ± 104 | 1670 ± 250 | 8800 ± 1030 | 6.8 (1.07) | 29 (1.10) | 12 (1.15) | 9.8 (1.10) |
| 21 | CH3 | H | C(CH3)3 | Cl | CH3 | H | 410 ± 75 | 2040 ± 280 | IA | 0.65 (1.10) | 9.2 (1.07) | 4.8 (1.15) | 5.7 (1.07) |
| 2m | CH3 | H | C(CH3)3 | CH3 | Br | H | 2810 ± 590 | 7250 ± 2370 | IA | 2.9 (1.07) | 32 (1.10) | 18 (1.20) | 12 (1.07) |
| 2n | CH3 | H | C(CH3)3 | Cl | H | Cl | IA | 14000 ± 4700 | IA | 3.1 (1.12) | 19 (1.10) | 22 (1.35) | 17 (1.10) |
| 2o | C2H5 | H | C(CH3)3 | Cl | H | H | 209 ± 28 | 607 ± 190 | 16000 ± 6700 | 0.58 (1.23) | 8.6 (1.05) | 6.2 (1.26) | 3.9 (1.07) |
| 2p | C3H7 | H | C(CH3)3 | Cl | H | H | 56 ± 18 | 370 ± 80 | IA | 0.70 (1.17) | 7.7 (1.07) | 4.9 (1.20) | 2.0 (1.07) |
| 2q | CH3 | H | CH(CH2CH2CH2CH2) | F | H | H | 693 ± 270 | 2850 ± 520 | IA | 11 (1.07) | 81 (1.15) | IA | 26 (1.07) |
| 2r | CH3 | H | CH(CH2CH2CH2CH2) | Cl | H | H | IA | IA | IA | 26 (1.10) | IA | IA | IA |
| 2s | CH3 | H | CH(CH2CH2CH2CH2) | Br | H | H | 845 ± 120 | 2530 ± 930 | IA | 3.9 (1.07) | 42 (1.15) | 27 (1.07) | 6.5 (1.07) |
| 2t | CH3 | H | CH(CH2CH2CH2CH2) | CH3 | H | H | 842 ± 190 | 8700 ± 3200 | IA | 3.7 (1.10) | 25 (1.05) | 23 (1.10) | 5.8 (1.10) |
| 2u | CH3 | H | CH(CH2CH2CH2CH2) | CH3O | H | H | 2270 ± 330 | 8200 ± 2370 | 6900 ± | 4.7 (1.07) | 31 (1.10) | 46 (1.07) | 9.2 (1.07) |
| 2v | CH3 | H | CH(CH2CH2CH2CH2) | H | H | IA | IA | IA | 15 (1.05) | IA | 60 (1.07) | 19 (1.10) | |
| 2w | C2H5 | H | C(CH3)3 | Cl | Cl | H | 118 ± 40 | 389 ± 83 | 1090 ± 480 | 0.51 (1.20) | 10 (1.07) | 3.3 (1.17) | 2.7 (1.07) |
| 2x | C3H7 | H | C(CH3)3 | Cl | Cl | H | 31 ± 9.4 | 180 ± 69 | 2300 ± 370 | 0.62 (1.29) | 9.8 (1.07) | 4.2 (1.20) | 1.5 (1.12) |
| 2y | CH3 | CH3 | C(CH3)3 | Cl | H | H | 6000 ± 316 | 1570 ± 80 | IA | 1.2 (1.20) | IA | 16 (1.23) | 16 (1.10) |
| 2z | CH3 | CH3 | CH3 | Cl | H | H | 1750 ± 370 | 1520 ± 250 | IA | 37 (1.12) | IA | 33 (1.23) | IA |
| 2aa | CH3 | CH2CH2CH2CH2CH2 | F | H | H | 156 ± 46 | 135 ± 36 | IA | 35 (1.07) | 71 (1.66) | IA | IA | |
| 2bb | CH3 | CH2CH2CH2CH2CH2 | Br | H | H | 750 ± 173 | 1180 ± 270 | IA | 11 (1.07) | 74 (1.15) | 64 (1.05) | 19 (1.07) | |
| 2cc | CH3 | CH2CH2CH2CH2CH2 | CH3 | H | H | 414 ± 89 | 577 ± 61 | IA | 11 (1.10) | 56 (1.10) | IA | 53 (1.10) | |
| 2dd | CH3 | CH2CH2CH2CH2CH2 | CH3O | H | H | 452 ± 82 | 718 ± 130 | IA | 16 (1.10) | 57 (1.10) | IA | 87 (1.10) | |
| 2ee | CH3 | CH2CH2CH2CH2CH2 | NO2 | H | H | 10000 ± 1900 | 4100 ± 540 | IA | 53 (1.10) | IA | IA | IA | |
| 2ff | CH3 | CH2CH2CH2CH2CH2 | Cl | H | H | 852 ± 220 | 2520 ± 900 | IA | 7.7 (1.12) | 30 (1.12) | 83 (1.23) | 28 (1.07) | |
| 4 | IA | IA | IA | 10 (1.12) | 32 (1.10) | IA | 69 (1.07) | ||||||
| 5 | IA | IA | IA | 3.3 (1.17) | 35 (1.05) | 36 (1.23) | 16 (1.05) | ||||||
Values for mean ± standard error of three independent experiments, each conducted with triplicate determination.
Mean micromolar IC50 values (to two significant digits) for 2a and the indicated analogs from three independent experiments for inhibition of functional responses to an EC80–EC90 concentration of carbamylcholine mediated by nAChR subtypes composed of the indicated subunits (where * indicates that additional subunits are or may be additional assembly partners with the subunits specified; see Methods and Materials). Numbers in parentheses indicate S.E.M. as a multiplication/division factor of the mean micromolar IC50 values shown [i.e., the value 1.8 (1.15) reflects a mean IC50 value of 1.8 μM with an S.E.M. range of 1.8 × 1.15 μM to 1.8/1.15 μM or 1.6-2.1 μM]. IA: IC50 >100 μM.
Compound 2a analogues 2b–2ff, 4, and 5 were also evaluated for their ability to antagonize functional responses of α3β4*-, α4β2-, α4β4-, and α1*-nAChR using previously reported methods27 modified as described in Methods and Materials. Results are given in Table 1 and in Figures 1–2.27
Figure 1.

Specific 86Rb+ efflux (ordinate; percentage of control) was determined for functional, human muscle-type α1β1γδ-nAChR (△), ganglionic α3β4*-nAChR (▼), α4β2-nAChR (○), or α4β4-nAChR (▲) naturally or heterologously expressed in human cell lines in the presence of a receptor subtype-specific, EC80-EC90 concentration of the full agonist, carbamylcholine, either alone or in the presence of the indicated concentrations (abscissa, log molar) of 2a or its analogs having phenyl substitutions (compounds 2c, 2l, and 2h) as indicated. Mean micromolar IC50 values and SEM as a multiplication/division factor of the mean micromolar IC50 value are provided in Table 1.
Figure 2.

Specific 86Rb+ efflux (ordinate; percentage of control) was determined for functional, human muscle-type α1β1γδ-nAChR (△), ganglionic α3β4*-nAChR (▼), α4β2-nAChR (○), or α4β4-nAChR (▲) naturally or heterologously expressed in human cell lines in the presence of a receptor subtype-specific, EC80-EC90 concentration of the full agonist, carbamylcholine, either alone or in the presence of the indicated concentrations (abscissa, log molar) of 2a analogs having alkyl or phenyl plus alkyl substitutions (compounds 2y, 2o, 2w, and 2x) as indicated. Mean micromolar IC50 values and SEM as a multiplication/division factor of the mean micromolar IC50 value are provided in Table 1.
In Vivo Assays
Compound 2a and analogues 2b–2ff, 4, and 5 were also evaluated for their ability to antagonize behavioral responses to acute nicotine administration as previously described.27 Results are given in Table 2.
Table 2.
Pharmacological evaluation of 2a analogs as noncompetitive nicotinic antagonistsa
| AD50 (mg/kg) |
||||
|---|---|---|---|---|
| compd | Tail-flick | Hot-plate | Locomotion | Hypothermia |
| 2a | 1.2 (1–1.8) | 15 (6–19) | 4.9 (0.9–46) | 9.2 (4–23) |
| 2b | IA | IA | IA | IA |
| 2c | 0.12 (0.03–0.5) | 14.9 (6.5–34) | 27.1 (2.9–46) | 11.5 (8.2–16.1) |
| 2d | 0.05 (0.03–0.2) | 6.8 (4.2–11.1) | 4.3 (0.7–28) | 7.8 (7.4–8.2) |
| 2e | 1.6 (0.4–5.4) | IA | IA | IA |
| 2f | IA | IA | IA | IA |
| 2g | 0.43 (0.08–2.3) | IA | 31 (12.8–74.2) | 32 (10.1–100.1) |
| 2h | 0.15 (0.04–0.6) | 23.5 (12.3–44.6) | IA | 29 (11.8–69.1) |
| 2i | 0.09 (0.03–0.9) | 7.75 (2.8–21) | 11.8 (5.8–23) | 8.8 (2–38.2) |
| 2j | 0.05 (0.02–0.11) | IA | IA | IA |
| 2k | 0.28 (0.08–0.9) | IA | IA | IA |
| 21 | 0.2 (0.13–1.3) | 7.2 (0.13–13) | 11 (1.5–83) | 10 (5.1–18.2) |
| 2m | 0.015 (0.005–0.04) | IA | IA | IA |
| 2n | 0.028 (0.01–0.07) | 4.3 (0.2–13.5) | IA | IA |
| 2o | 0.5 (0.1–2.1) | IA | 11.7 | IA |
| 2p | 10 (3.7–26) | IA | IA | IA |
| 2q | IA | IA | IA | IA |
| 2r | 0.014 (0.001–0.08) | IA | IA | IA |
| 2s | IA | IA | IA | IA |
| 2t | IA | IA | IA | IA |
| 2u | IA | IA | IA | IA |
| 2v | n/a | n/a | n/a | n/a |
| 2w | 0.05 (0.03–0.8) | IA | IA | IA |
| 2x | 0.13 (0.06–0.3) | 15 (0.9–25) | IA | 17.5 (13–24) |
| 2y | 0.46 (0.44–0.48) | 8.7 (2–37) | IA | IA |
| 2z | 5.2 (1.5–17.2) | IA | IA | IA |
| 2aa | 3.84 (3.3–4.4) | IA | IA | 7.5 (2.4–24) |
| 2bb | IA | IA | IA | IA |
| 2cc | IA | IA | IA | IA |
| 2dd | 4.5 (1.4–14.3) | IA | IA | IA |
| 2ee | IA | IA | IA | IA |
| 2ff | 0.05 (0.005–0.5) | IA | IA | IA |
| 4 | 0.077 (0.015–0.4) | IA | IA | IA |
| 5 | 10.5 (9.5–11.5) | IA | IA | 16 (9.3–26) |
Results were expressed as AD50 (mg/kg) ± confidence limits (CL) or % effect at the highest dose tested. Dose–response curves were determined using a minimum of four different doses of test compound, and at least eight mice were used per dose group.
Results
Compound 2a and its analogues were evaluated for their ability to inhibit DA, NE, and 5-HT uptake inhibition using human DAT, NET, and SERT heterologously expressed by HEK293 cells. Compound 2a has IC50 values of 658 nM and 1850 nM for inhibition of DA and NE uptake, respectively, and is inactive at SERT (Table 1). Very few 2a analogues had appreciably better IC50 values for DA uptake inhibition than 2a. Compounds 2o and 2p, where the α-methyl group in 2a was replaced by ethyl and propyl groups, respectively, have IC50 values for inhibition of DA uptake of 209 and 56 nM, respectively, indicating that replacement of the α-methyl group with larger alkyl chains produced ligands with better efficacy, a finding that also is borne out by the improvement in inhibitory potency at DAT for 2w (IC50 = 118 nM) and 2x (IC50 = 31 nM) relative to 2k (IC50 = 463 nM) in the 3,4-dichlorophenyl analogue group. In each case, the 3,4-dichlorophenyl analogue has higher inhibitory potency for DA uptake inhibition than the otherwise equivalent monochlorophenyl analogue. Each 3,4-dichlorophenyl compound (2k, 2w, 2x) also has slightly, but not always statistically significant, inhibitory potency for NE uptake (1670, 389, and 180 nM IC50 values, respectively) greater than the equivalent monochlorophenyl compounds (2a, 2o, 2p; 1850, 607, 370 nM IC50 values, respectively). The propyl 2a analogue (2p) and 3,4-dichlorophenyl propyl analogue 2x have 7- and 6-fold selectivity for DA over NE uptake inhibition.
Compound 2aa has 4- and 14-fold elevated inhibitory potency relative to 2a for DA uptake inhibition (IC50 = 156 nM) and NE uptake inhibition (IC50 = 135 nM). Compound 2y is the only compound tested that has significantly higher selectivity (~4-fold) for NE uptake inhibition relative to DA inhibition, but it is only marginally more potent as an NE uptake inhibitor than 2a (IC50s = 1570 and 1850 nM, respectively). None of these analogues has IC50 values for inhibition of SERT less than 1 μM, although the dichlorophenyl analogues are more potent at this target than their monochlorophenyl equivalents.
Thus, 2w, 2x, and 2p are the most potent for DA uptake inhibition, with 2aa and 2o also being more potent for DA uptake inhibition than 2a. Compounds 2w and 2x also are more selective for DA uptake inhibition relative to inhibitions of 5-HT and NE uptake. Compounds 2aa, 2x, and 2p are the most potent analogues for NE uptake inhibition, and analogues 2w, 2o, and 2dd are also more potent than 2a for NE uptake inhibition. With the exception of 2y, which is about 4-fold selective for NE over DA uptake inhibition uptake inhibition (although being about equipotent with 2a at NET), none of the analogues is selective for NE over DA uptake inhibition.
Isotopic 86Rb+ efflux assays were used to determine effects of 2a and analogues on function of diverse human nAChR subtypes naturally or heterologously expressed by human cell lines. These assays repeatedly have been shown to be specific only for nAChR function in the cells used. In the case of SH-SY5Y cells, function of only α3β4*-nAChR is measured because α7-nAChRs that also are expressed by these cells activate and inactivate too quickly to detectably contribute to ion flux. Neither 2a nor any of the analogues tested possess intrinsic activity as agonists at α1*-, α3β4*-, α4β2-, or α4β4-nAChR. 86Rb+ efflux in the presence of these ligands alone at concentrations from ~5 nM to 100 μM (data not shown here) was indistinguishable from responses in cells exposed only to efflux buffer.
86Rb+ efflux assays also were used to assess whether 2a or its analogues had activity as antagonists at human nAChR. With few exceptions (noted in Table 1), 2a and each of its analogues exhibited concentration-dependent inhibition of ion flux responses elicited by EC80–EC90 concentrations of carbamylcholine for α1*-, α3β4*-, α4β2-, and α4β4-nAChR. Representative concentration response curves for 2a itself, for compounds having substituted phenyl groups (2c, 2h, 2l; Figure 1), or for compounds having amine or alkyl charges (2y, 2o, 2x, 2w; Figure 2) illustrate some of the features of these ligands (see also Table 1).
Compound 2a has IC50 values of 1.8, 12, 15, and 7.9 μM for α3β4*-, α4β2-, α4β4-, and α1*-nAChRs, respectively and, thus, is 4–8-fold selective for the α3β4* relative to the other nAChRs. All of the analogues except 2z (~equipotent at α3β4*- and α4β4-nAChR) block function of α3β4*-nAChR at concentrations lower than those needed to inhibit function of the other nAChR subtypes. That is, all of the analogues are selective in their antagonism for α3β4*-nAChR. Compounds 2l, 2o, 2p, and 2x have slightly, but not statistically significant, higher potencies than 2a the α4β2 nAChR (IC50 = 7.7–9.8 μm). The 4-fold selectivity of 2a for the α3β4*-nAChR over other subtypes is increased to >7-fold for the 3-bromo, 3-methyl, and 3-methoxyphenyl analogues 2c, 2d, and 2e, respectively, and to >10-fold for the 3,4-difluorophenyl and N-methyl analogues 2j and 2y, respectively.
In addition to the improvements in selectivity for α3β4*-nAChR over other subtypes, the 3-chloro, 4-methyl substituted phenyl analogue 2l (IC50 = 0.65 μM) has ~3-fold higher inhibitory potency at α3β4*-nAChR relative to 2a. Both mono- and dichloro-substituted phenyl compounds 2o, 2p, 2w, and 2x (IC50 = 0.51–0.70 μM), having extended alkyl side chains, have ~3-fold higher potency at α3β4*-nAChR relative to 2a with the 3,4-dichlorophenyl ethyl and propyl compounds (2w and 2x), respectively, being >10-fold more potent than the unextended 3,4-dichlorophenyl analogue (2k; IC50 = 6.8 μM). These compounds also are remarkable because they also had higher inhibitory potency at and selectivity for DA uptake inhibition and higher potency for NE uptake inhibition than 2a, so they all represent analogues with higher affinity than 2a for three of its molecular targets.
Compound 2a blocks nicotine-induced antinociception in the tail-flick and hot-plate tests with AD50 values of 1.2 and 15 mg/kg, respectively (Table 2). It also blocks nicotine-induced locomotor activity and hypothermia with AD50 values of 4.9 and 9.2 mg/kg, respectively. Seventeen of the 33 analogues had AD50 values of 0.014 to 0.5 mg/kg in the tail-flick test, showing higher inhibitory potency than 2a. Analogues 2r, 2m, and 2n with AD50 values of 0.014, 0.015, and 0.028 mg/kg were 86-, 80-, and 43-times more potent than 2a in blocking nicotine-induced antinociception in the tail-flick test. Surprisingly, none of these analogues has higher potency than 2a for DA, NE, and SERT uptake inhibition or any nAChR subtype. Analogues 2d, 2j, 2w, and 2ff with AD values of 0.05 mg/kg were the next most potent in the tail-flick assay, being 24-fold more potent than 2a. Analogues 2c, 2h, 2i, and 2x (AD50 = 0.09–0.15 mg/kg) have ~10-fold better inhibitory potency than 2a against nicotine-mediated analgesia in the tail-flick assay. Thus, 2x is an analogue that has increased potency relative to 2a as an inhibitor of DA and NE uptake and α3β4*-nAChR, analogues 2h and 2c, have potencies comparable to those of 2a across those targets. The 3,4-dichlorophenyl analogues 2k, 2w, and 2x are ~4-, ~10-, and ~77-fold more potent than their 3-chlorophenyl substituted equivalents, 2a, 2o, and 2p, respectively. However, the tail-flick assay results are not particularly illuminating about the molecular targets contributing to analogue effects, perhaps because effects at higher levels might override the presumed spinal level of nicotine-mediated antinociception.
In the hot-plate assay, 2d and 2l (potent and selective at α3β4*-nAChR) and 2y (selective for NE uptake inhibition and α3β4*-nAChR) have about 2-fold more potency than 2a, and 2n (inactive at transporters and less potent than 2a at α3β4*-nAChR) has about 3-fold more potency. These findings suggest that several mechanisms might impact supraspinal mechanisms of nicotine-mediated antinociception.
Of all the analogues, only 2d has potency like 2a in blocking nicotine's effects on locomotion, and only 2d and 2l rival the ability of 2a to inhibit nicotine's effects on body temperature. Similar to 2a, substituted phenyl analogues 2c, 2d, and 2l (roughly sharing the in vitro fingerprint of 2a) had potency in all four behavioral tests as nicotine antagonists. Compound 2x, a potent DA and NE uptake inhibitor and α3β4*-nAChR antagonist, had potency in three of four of the in vivo tests.
Compounds 2p, 2w, and 2x, having the highest potencies as DA uptake inhibitors, differ nearly 200-fold in potency in the tail-flick assay, and only 2x shows activity in the hot-plate or locomotor tests. These analogues also are among the most potent at NE uptake inhibition.
Discussion
Several 2a analogues were synthesized and tested for their ability to inhibit monoamine uptake and to antagonize function of four different nAChR subtypes. The analogues were also evaluated for their ability to block nicotine-induced antinociception, locomotor activity, and hypothermia.
We succeeded in creating and characterizing analogues with significantly lower IC50 values relative to 2a for inhibition of both DA or NE uptake inhibition (2o, 2p, 2w, 2x, and 2aa).
The current efforts also succeeded in generating nine agents with higher (2l, 2o, 2p, 2w, 2x) or slightly higher (2c, 2d, 2h, 2y) antagonist potency relative to 2a at α3β4*-nAChR. Of these, 2c, 2d, 2l, and especially 2y also have improved selectivity for α3β4*-nAChR over other nAChR subtypes relative to 2a. In addition, analogues 2e and 2j show improved selectivity for α3β4*-nAChR although without having lower IC50 values than 2a.
Compounds were also developed that had altered target selectivity between nAChR and transporters. For example, 2e had improved selectivity for α3β4*-nAChR over DA and NE uptake inhibition, 2y improved selectivity for α3β4*-nAChR over DA uptake inhibition, and 2p, 2w, and 2x improved selectivity for DA and NE uptake inhibition over α3β4*-nAChR antagonism relative to 2a.
Thus, several new compounds have been developed that have higher potency and/or selectivity at specific neurotransmitter transporters or α3β4*-nAChR than 2a, thereby providing leads for further target-directed drug development. These compounds also afford, in principle, opportunities to dissect roles of specific molecular targets in nicotine-mediated behavioral effects. Structure-activity relationships are complex and difficult to generalize because phenyl substitutions and amine/alkyl changes seem to interact in terms of altering potencies at and selectivities for specific targets in vitro. However, compounds with alkyl chain extensions in mono-chlorophenyl (like 2a) or di-chlorophenyl configurations (2o, 2p, 2w, and 2x) had higher affinity than 2a for all three targets, DAT, NET, and α3β4*-nAChR, implicated in 2a action. The selectivity increase for 2a analogs in blocking α3β4* nAChRs subtypes is very relevant since these subtypes are highly expressed in the medial habenula and its primary target, the interpeduncular nucleus. Recent data suggest an important role for the habenulo-interpeduncular system and the nicotinic receptor subunits expressed therein in nicotine withdrawal.28
Seventeen of the analogues had higher potency than 2a as antagonists of nicotine-induced antinociception in the tail-flick test, having AD50 values that ranged from ~2- to ~86-fold lower than that for 2a. However, five analogues (2d, 2i, 2l, 2n, and 2y) were only slightly (~2-fold) better than 2a as antagonists of nicotine-induced antinociception in the hot plate assay, just one analogue (2d) rivaled or bettered 2a as an antagonist of nicotine's effects on locomotion, and only 2d, 2i, 2l, and 2aa rivaled or bettered the antagonistic potency of 2a toward nicotine-induced hypothermia. Analogues 2m, 2n, and 2r have the lowest AD50 values in the tail-flick assay, yet only 2n in the hot-plate assay shows any other form of nicotine behavioral antagonism. Every ligand except 2p that had higher potency than 2a at α3β4*-nAChR also showed an improvement over 2a in antagonistic potency in the tail-flick assay as did three of the ligands (2o, 2w, and 2x; but not 2aa and 2p) that had higher potency than 2a as antagonists for DA and NE uptake inhibition. Thus, increased potency at α3β4*-nAChR correlates with but is not necessary for improvement in ligand antagonist potency in the tail-flick assay. Moreover, when improvement in activity for DA and NE uptake inhibition is dissociated from improvement in activity at α3β4*-nAChR (i.e., for 2aa), there is no improvement in behavioral antagonism in the tail-flick assay. Perhaps compounds like 2g, 2i, 2k, 2j, 2ff, 3, 4, or even 2m, 2n, and 2r that are not as active as 2a in vitro are metabolized in vivo to forms that are active behaviorally and might also have higher affinity for DA and NE uptake inhibition and/or α3β4*-nAChR antagonism, but drug exposure times in vivo are short.
In summary, several 2a analogues were synthesized and tested for their ability to inhibit monoamine uptake and to antagonize the effects of α3β4*, α4β2, α4β4, and α1β1 nAChRs. The analogues were also evaluated for their ability to block nicotine-induced antinociception, locomotor activity, and hypothermia. Analogues 2o, 2p, and 2x had significantly better IC50 values for DA uptake inhibition relative to 2a. Analogue 2x also had a significantly better IC50 value for NE uptake inhibition relative to 2a. Analogues 2l, 2o, 2p, 2s, 2w, and 2x had IC50 values of 2.6- to 3.6-times better than 2a for the antagonism of the α3β4* nAChR. Seventeen of the 2a analogues had better AD50 values for blocking nicotine-induced antinociception in the tail-flick test with analogues 2m, 2n, and 2r having the lowest AD50 values. Analogue 2x with IC50 values of 31 and 180 nM for DA and NE uptake inhibition compared to 658 and 1850 nM for 2a and an IC50 = 0.62 and 9.8 μM for antagonism of the α3β4* and α4β2 nAChRs, respectively, compared to 1.8 and 12 μM for 2a had the best overall in vitro profile. This compound also had an AD50 = 0.13 mg/kg in the tail-flick test compared to an AD50 = 1.2 mg/kg for 2a.
Overall, the findings support the idea that multiple molecular targets can play roles in mediating nicotine's behavioral effects and that these new 2a analogues have potential not only as pharmacological tools to study targets and mechanisms involved but also as new pharmacotherapies with potentially higher efficacy as aids to smoking cessation.
Experimental
Chemistry
Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded on a 300 MHz (Bruker AVANCE 300) spectrometer. Chemical shift data for the proton resonances were reported in parts per million (δ) relative to internal (CH3)4Si (δ 0.0). Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research. Elemental analyses were performed by Atlantic Microlab, Norcross, GA. Purity of compounds (>95%) was established by elemental analysis. Analytical thin-layer chromatography (TLC) was carried out on plates precoated with silica gel GHLF (250 μM thickness). TLC visualization was accomplished with a UV lamp or in an iodine chamber. All moisture-sensitive reactions were performed under a positive pressure of nitrogen maintained by a direct line from a nitrogen source. Anhydrous solvents were purchased from Aldrich Chemical Co.
2-(N-tert-Butylamino)-3’-methoxypropiophenone (2e) Fumarate
To a stirred solution of 2-bromo-3’-methoxypropiophenone 7d (400 mg, 1.5 mmol) in 3 mL of CH3CN was added 0.315 mL (3.0 mmol) of tert-butyamine. The mixture was stirred for 10 h at room temperature. The reaction solution was diluted with EtOAc, washed with aqueous NaHCO3, water and brine then dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography on silica gel using MeOH-CH2Cl2 (1:50 to 1:10 w/ 1% NH4OH) as the eluent to afford 138 mg (39%) of 2e. The 2e was immediately dissolved in Et2O, 1.0 eq of fumaric acid (dissolved in minimal amount of MeOH) was added dropwise, and the mixture was stirred overnight. The solid was collected by filtration, washed with Et2O, and vacuum dried to yield 150 mg (28%) of 2e fumarate as a white solid: mp 152–153 °C. 1H NMR (CD3OD) δ 7.75 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.54 (t, J = 8.1 Hz, 1H), 7.32 (dd, J = 8.0, 2.5 Hz, 1H), 6.68 (s, 2H), 5.20 (q, J = 7.1 Hz, 1H), 4.88 (s, 3H), 1.57 (d, J = 7.1 Hz, 3H), 1.35 (s, 9H). 13C NMR (CD3OD) δ 196.8, 170.8, 161.5, 135.7, 134.5, 131.2, 122.0, 114.2, 59.0, 55.7, 54.3, 26.2, 18.5. LCMS (ESI) m/z 236.5 (M+1)+. Anal. (C18H25NO6) C, H, N.
2-(N-tert-Butylamino)-3’-nitropropiophenone (2f) Fumarate
To a stirred solution of 2-bromo-3’-nitropropiophenone 7e (300 mg, 1.16 mmol) in 3 mL of CH3CN was added tert-butyamine (0.170 g, 3.0 mmol). The mixture was stirred for 6 h at 40 °C. The solution was filtered to remove the white precipitate. The reaction solution was diluted with EtOAc, washed with aqueous NaHCO3, water, and brine then dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography on silica gel using MeOH-CH2Cl2 (1:50 to 1:10 w/ 1% NH4OH) as the eluent to afford 211 mg (73%) of 2f. The 2f was immediately dissolved in Et2O, 1.0 eq of fumaric acid (dissolved in minimal amount of MeOH) was added dropwise, and the mixture was stirred overnight. The solid was collected by filtration, washed with Et2O, and vacuum dried to give 270 mg (64% from 2f) of 2f•fumarate as a white solid: mp 166–167 °C. 1H NMR (CD3OD) δ 8.94 (s, 1H), 8.59 (dd, 2H), 7.90 (t, 1H), 6.68 (s, 2H), 5.30 (q, 1H), 1.61 (d, 3H), 1.37 (s, 9H). 13C NMR (CD3OD) δ 198.5, 172.8, 152.1, 137.9, 137.5, 133.9, 131.9, 126.5, 60.9, 56.8, 29.5, 28.7, 20.6. LCMS (ESI) m/z 252.3 (M+1)+. Anal. (C17H22N2O7) C, H, N.
2-(N-Cyclopentylamino)-3’-fluoropropiophenone (2q) Hydrochloride
To a CH3CN solution (3 mL) of 2-bromo-3’-fluoropropiophenone 7a (290 mg, 1.26 mmol) was added cyclopentylamine (125 μL, 1.26 mmol), and the mixture was allowed to react for 3 h at room temperature. The precipitate was filtered off. The filtrate was concentrated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 2q as a light yellow oil. The 2q was dissolved in 50 mL of Et2O, a solution of hydrochloric acid in Et2O was added dropwise, and the mixture was stirred overnight. The precipitate was filtered, washed with Et2O, and dried under vacuum to give 50 mg (15%) of 2q•HCl as a white solid: mp 185–186 °C. 1H NMR (CD3OD) δ 7.95 (d, 1H), 7.85 (d, 1H), 7.64 (m, 1H), 7.51 (m, 1H), 5.16 (m, 1H), 3.64 (m, 1H), 2.15 (m, 2H), 1.61–1.85 (m, 6H), 1.60 (d, 3H). 13C NMR (CD3OD) δ 196.4, 166.5, 163.2, 136.8, 136.7, 133.0, 126.6, 123.5, 123.2, 117.0, 116.7, 59.2, 58.9, 31.2, 25.2, 17.0. LCMS (ESI) m/z 236.4 (M+1)+. Anal. (C14H19ClFNO) C, H, N.
2-(N-Cyclopentylamino)-3’-bromopropiophenone (2s) Fumarate
To the CH3CN solution (5 mL) of 2-bromo-3’-bromopropiophenone 7b (480 mg, 1.65 mmol) was added cyclopentylamine (330 μL, 3.31 mmol), and the mixture was allowed to react for 6 h at 40 °C. After cooling to room temperature, the precipitate was removed by filtration. The filtrate was concentrated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 182 mg (37%) of 2s as light yellow oil. The 2s was dissolved in 50 mL of Et2O, a MeOH solution (1 mL) of fumaric acid (71 mg) was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 240 mg (35%) of 2s•fumarate as a white solid: mp 152–153 °C. 1H NMR (CD3OD) δ 8.25 (s, 1H), 8.07 (d, 1H), 7.89 (d, 1H), 7.54 (t, 1H), 6.68 (s, 2H), 5.13 (q, 1H), 3.30 (m, 1H), 2.13 (m, 2H), 1.83 (m, 2H), 1.68 (m, 4H), 1.56 (d, 3H). 13C NMR (CD3OD) δ 196.8, 171.6, 139.2, 136.7, 136.6, 133.1, 132.6, 129.2, 124.8, 59.2, 59.2, 32.5, 31.3, 25.2, 17.2, 14.8. LCMS (ESI) m/z 296.2 (M+1)+. Anal. (C18H22BrNO5) C, H, N.
2-(N-Cyclopentylamino)-3’-methylpropiophenone (2t) Fumarate
To a CH3CN solution (4 mL) of 2-bromo-3’-methylpropiophenone 7c (400 mg, 1.76 mmol) was added cyclopentylamine (175 μL, 1.76 mmol), and the mixture was allowed to react for 6 h at 40 °C. After cooled to room temperature, the precipitate was filtered off. The filtrate was concentrated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 226 mg (56%) of 2t as light yellow oil. The free base was dissolved in 50 mL of Et2O, MeOH solution (1 mL) of fumaric acid (1 eq) was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 260 mg (43%) of 2t•fumarate as a white solid: mp 171–172 °C. 1H NMR (CD3OD) δ 7.90 (m, 2H), 7.56 (d, J = 7.5 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 6.68 (s, 2H), 5.14 (q, J = 7.1 Hz, 1H), 3.59 (m, 1H), 2.45 (s, 3H), 2.13 (m, 2H), 1.90-1.65 (m, 6H), 1.58 (d, J = 7.1 Hz, 3H). 13C NMR (CD3OD) δ 201.0, 175.1, 144.4, 140.5, 139.9, 138.1, 134.0, 130.9, 62.6, 61.9, 34.5, 28.6, 25.0, 20.7, 10.8. LCMS (ESI) m/z 232.8 (M+1)+. Anal. (C19H25NO5) C, H, N.
2-(N-Cyclopentylamino)-3’-methoxypropiophenone (2u) Fumarate
To a CH3CN solution (6 mL) of 2-bromo-3’-methoxypropiophenone 7d (600 mg, 2.47 mmol) was added cyclopentylamine (245 μL, 2.47 mmol), and the mixture was allowed to react for 6 h at 40 °C. After cooling to room temperature, the precipitate was filtered off. The filtrate was concentrated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 2u as a light yellow oil. The 2u was dissolved in 50 mL of Et2O, and a MeOH solution (1 mL) of fumaric acid (1 eq) was added dropwise and stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 290 mg (33%) of 2u•fumarate as an off white solid: mp 144–145 °C. 1H NMR (DMSO-d6) δ 7.66 (d, J = 7.7 Hz, 1H), 7.50 (m, 2H), 7.27 (dd, J = 8.2, 2.4 Hz, 1H), 6.53 (s, 2H), 4.72 (m, 1H), 3.84 (s, 3H), 3.18 (m, 1H), 1.77 (m, 2H), 1.64 (m, 2H), 1.48 (m, 4H), 1.29 (d, J = 6.9 Hz, 3H). 13C NMR (CD3OD) δ 196.5, 170.7, 161.3, 135.6, 135.1, 131.0, 121.8, 121.5, 113.9, 58.3, 57.7, 55.5, 30.3, 24.3, 16.4. LCMS (ESI) m/z 248.3 (M+1)+. Anal. (C19H25NO6) C, H, N.
2-(N-Cyclopentylamino)-3’-nitropropiophenone (2v) Hydrochloride
To a CH3CN solution (3 mL) of 2-bromo-3’-nitropropiophenone 7e (300 mg, 1.16 mmol) was added cyclopentylamine (230 μL, 2.32 mmol), and the mixture was allowed to react for 6 h at room temperature. The precipitate was filtered off. The filtrate was concentrated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 2v as a light yellow oil. The 2v was dissolved in 50 mL of Et2O, a solution of hydrochloric acid in Et2O was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 50 mg (15%) of 2v•HCl as a white solid: mp 149–150 °C. 1H NMR (CD3OD) δ 8.88 (s, 1H), 8.60 (s, 1H), 8.51 (s, 1H), 7.89 (s, 1H), 5.28 (s, 1H), 3.68 (s, 1H), 2.19 (s, 2H), 1.64–1.87 (m, 9H). 13C NMR (CD3OD) δ 195.6, 150.4, 135.9, 135.6, 132.2, 130.1, 124.7, 59.0, 58.8, 31.0, 25.0, 16.7. Anal. (C17H19ClN2O3·0.5H2O) C, H, N.
2-(N-Pyrrolidinyl)-3’-fluoropropiophenone (2aa) Hydrochloride
To a CH3CN solution (3 mL) of 2-bromo-3’-fluoropropiophenone 7a (300 mg, 1.30 mmol) was added pyrrolidine (108 μL, 1.30 mmol), and the mixture was allowed to react for 3 h at room temperature. The precipitate was filtered off. The filtrate was concentrated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give the 2aa as a light yellow oil. The 2aa was dissolved in Et2O, a solution of hydrochloric acid in Et2O was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 180 mg (54%) of 2aa•HCl as a white solid: mp 184–185 °C. 1H NMR (CD3OD) δ 7.91 (d, 1H), 7.81 (d, 1H), 7.64 (m, 1H), 7.52 (m, 1H), 5.32 (m, 1H), 3.73 (m, 2H), 3.07 (m, 2H), 2.09–2.16 (m, 4H), 1.63 (d, 3H). 13C NMR (CD3OD) δ 196.3, 166.2, 162.9, 136.5, 132.7, 132.6, 126.2, 123.3, 123.1, 116.7, 116.4, 66.9, 55.8, 53.4, 24.4, 16.8. LCMS (ESI) m/z 222.6 (M+1)+. Anal. (C13H17ClFNO) C, H, N.
2-(N-Pyrrolidinyl)-3’-bromopropiophenone (2bb) Fumarate
To a CH3CN/H2O solution (4 mL/2 mL) of 2-bromo-3’-bromopropiophenone 7b (700 mg, 2.4 mmol) was added pyrrolidine (200 μL, 2.4 mmol), and the mixture was allowed to react for 4 h at room temperature. The reaction solution was diluted with EtOAc, washed with aq. NaHCO3, water and brine then dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 452 mg (67%) of 2bb as a light yellow oil. The 2bb was dissolved in 50 mL of Et2O, a MeOH solution (1 mL) of fumaric acid (1 eq) was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 2bb•fumarate as a white solid: mp 134–135 °C. 1H NMR (CDCl3) δ 8.25 (s, 1H), 8.05 (d, 1H), 7.67 (d, 1H), 7.33 (t, 1H), 3.91 (q, 1H), 2.61 (m, 4H), 1.80 (m, 4H), 1.37 (d, 9H). 13C NMR (CDCl3) δ 199.7, 137.8, 135.8, 131.8, 130.1, 127.3, 122.8, 65.0, 51.1, 23.6, 15.8. LCMS (ESI) m/z 284.7 (M+1)+. Anal. (C17H20BrNO5) C, H, N.
2-(N-Pyrrolidinyl)-3’-methylpropiophenone (2cc) Fumarate
To a CH3CN-H2O solution (4 mL-2 mL) of 2-bromo-3’-methylpropiophenone 7c (400 mg, 1.76 mmol) was added pyrrolidine (150 μL, 1.76 mmol), and the mixture was allowed to react for 4 h at room temperature. The reaction solution was diluted with EtOAc, washed with aq. NaHCO3, water and brine then dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 2cc as a light yellow oil. The 2cc was dissolved in Et2O, a MeOH solution (1 mL) of fumaric acid (1 eq) was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 320 mg (55%) of 2cc•fumarate as a white solid: mp 131–132 °C. 1H NMR (CD3OD) δ 7.86 (m, 2H), 7.57 (d, J = 7.6 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 6.69 (s, 2H), 5.23 (q, J = 7.1 Hz, 1H), 3.55–3.33 (m, 4H), 2.45 (s, 3H), 2.12 (m, 4H), 1.60 (d, J = 7.1 Hz, 3H). 13C NMR (CD3OD) δ 201.2, 175.0, 144.4, 140.6, 139.9, 138.2, 134.0, 130.9, 70.3, 58.0, 28.0, 25.0, 20.6. LCMS (ESI) m/z 218.1 (M+1)+. Anal. (C18H23NO5) C, H, N.
2-(N-Pyrrolidinyl)-3’-methoxypropiophenone (2dd) Fumarate
To a CH3CN-H2O solution (4 mL-2 mL) of 2-bromo-3’-methyoxypropiophenone 7d (600 mg, 2.47 mmol) was added pyrrolidine (200 μL, 2.4 mmol), and the mixture was allowed to react for 4 h at room temperature. The reaction solution was diluted with EtOAc, washed with aq. NaHCO3, water and brine then dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 300 mg (52%) of 2dd as a light yellow oil. The 2dd was dissolved in 50 mL of Et2O, a MeOH solution (1 mL) of fumaric acid (1 eq) was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 340 mg (39%) of 2dd•fumarate as a white solid: mp 121–122 °C. 1H NMR (DMSO-d6) δ 7.67 (d, J = 7.7 Hz, 1H), 7.56 (s, 1H), 7.47 (t, J = 8.1 Hz, 1H), 7.24 (dd, J = 8.2, 2.6 Hz, 1H), 6.58 (s, 2H), 4.46 (m, 1H), 3.82 (s, 3H), 2.76 (s, 4H), 1.74 (s, 4H), 1.29 (d, J = 6.8 Hz, 3H). 13C NMR (DMSO-d6) δ 197.7, 165.0, 157.8, 135.0, 132.7, 128.3, 119.4, 117.8, 111.6, 61.2, 53.7, 48.6, 21.6, 12.8. LCMS (ESI) m/z 234.3 (M+1)+. Anal. (C18H23NO6) C, H, N.
2-(N-Pyrrolidinyl)-3’-nitropropiophenone (2ee) Hydrochloride
To a CH3CN-H2O solution (4 mL-2 mL) of 7e (310 mg, 1.2 mmol) was added pyrrolidine (100 μL, 1.2 mmol), and the mixture was allowed to react for 2 h at room temperature. The reaction solution was diluted with EtOAc, washed with aq. NaHCO3, water and brine then dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography on silica gel using cyclohexane-EtOAc (2:1 to 1:0 w/ 1% NH4OH) as the eluent to give 120 mg (40%) of 2ee as a light yellow oil. The 2ee was dissolved in 50 mL of Et2O, a solution of hydrochloric acid in ether was added dropwise, and the mixture was stirred overnight. The precipitate was collected, washed with Et2O, and dried under vacuum to give 60 mg (18%) of 2ee•HCl as an off-white solid: mp 35–36 °C. 1H NMR (CD3OD) δ 8.86 (s, 1H), 8.59 (s, 1H), 8.50 (s, 1H), 7.91 (s, 1H), 5.50 (s, 1H), 3.80 (s, 2H), 3.40 (m, 2H), 2.14 (m, 4H), 1.70 (s, 3H). 13C NMR (CD3OD) δ 196.2, 150.7, 136.8, 136.1, 132.9, 130.6, 125.3, 67.9, 56.8, 54.6, 25.3, 25.2, 17.7. LCMS (ESI) m/z 249.2 (M+1)+. Anal. (C13H17ClN2O3·H2O) C, H, N.
General procedure for 2-Bromo-3-substituted propiophenones 7a–e
To a solution of the appropriate propiophenone, 6a–e (25 mmol) in acetic acid (45 mL), bromine (25 mmol) was added dropwise. After stirring over night, the acetic acid was removed under vacuum, and the resulting residue was dissolved in EtOAc, washed with saturated NaHCO3 solution, brine, dried (Na2SO4), and concentrated to give the bromoketones 7a–e. The unpurified 7a–e was used to prepare the 2a analogues without further purification.
Cell lines and culture: HEK-293 cells stably expressing human DAT, NET or SERT were maintained as previously described26
Use was made of several human cell lines that naturally or heterologously express specific, functional, human nAChR subtypes.29 Cells of the TE671/RD line naturally expresses muscle-type nAChR (α1β1γδ- or α1*-nAChR), and SH-SY5Y neuroblastoma cells naturally expresses autonomic α3β4*-nAChRs (containing α3, β4, probably α5, and sometimes β2 subunits). Different clones of SH-EP1 epithelial cell lines have been engineered to heterologously express either α4β2-nAChR, which are thought to be the most abundant, high affinity nicotine-binding nAChR in mammalian brain, or α4β4-nAChR, another possible brain nAChR subtype (SH-EP1-hα4β2 or α4β4 cells, respectively).30,31 These cells were maintained as low passage number (1–26 from our frozen stocks) cultures to ensure stable expression of native or heterologously-expressed nAChR as previously described.29 Cells were passaged once weekly by splitting just-confluent cultures 1/300 (TE671/RD), 1/5 (SH-SY5Y), or 1/20 (transfected SH-EP1) in serum-supplemented medium to maintain log-phase growth.
Transporter Assays
The abilities of 2a and its analogues to inhibit uptake of [3H]dopamine ([3H]DA), [3H]serotonin ([3H]5-HT), or [3H]norepinephrine ([3H]NE) by the respective human transporters were evaluated using the appropriate HEK-293 cell line as previously reported.26
nAChR functional assays
Cells were harvested at confluence from 100-mm plates by mild trypsinization (Irvine Scientific, Santa Ana, CA) and trituration or (for SH-SY5Y cells) by trituration alone before being suspended in complete medium and evenly seeded at a density of 1.25–2 confluent 100-mm plates per 24-well plate (Falcon; ~100–125 μg of total cell protein per well in a 500 μL volume). After cells had adhered (generally overnight, but no sooner than 4 h later), the medium was removed and replaced with 250 μL per well of complete medium supplemented with ~350,000 cpm of 86Rb+ (PerkinElmer Life and Analytical Sciences, Boston, MA) and counted at 40% efficiency using Cerenkov counting (TriCarb 1900 liquid scintillation analyzer, 59% efficiency; PerkinElmer Life Sciences). After at least 4 h and typically overnight, 86Rb+ efflux was measured using the “flip-plate” technique.29 Briefly, after aspiration of the bulk of 86Rb+ loading medium from each well of the “cell plate,” each well containing cells was rinsed 3× with 2 mL of fresh 86Rb+ efflux buffer (130 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 5 mM glucose, 50 mM HEPES, pH 7.4) to remove extracellular 86Rb+. Following removal of residual rinse buffer by aspiration, the flip-plate technique was used again to simultaneously introduce 1.5 mL of fresh efflux buffer containing drugs of choice at indicated final concentrations from a 24-well “efflux/drug plate” into the wells of the cell plate. After a 5-min incubation, the solution was “flipped” back into the efflux/drug plate, and any remaining buffer in the cell plate was removed by aspiration. Cells remaining in the cell plate were lysed and suspended by addition of 1.5 mL of 0.1 M NaOH, 0.1% sodium dodecyl sulfate to each well. Suspensions in each well were then subjected to Cerenkov counting (Wallac Micobeta Trilux 1450; 25% efficiency) after placement of inserts (Wallac 1450-109) into each well to minimize cross-talk between wells.
For quality control and normalization purposes, the sum of 86Rb+ in cell plates and efflux/drug plates was defined to confirm material balance (i.e., that the sum of 86Rb+ released into the efflux/drug plates and 86Rb+ remaining in the cell plate were the same for each well). This assured that 86Rb+ efflux was the same whether measured in absolute terms or as a percentage of loaded 86Rb+. Similarly, the sum of 86Rb+ in cell plates and efflux/drug plates also determined the efficiency of 86Rb+ loading (the percentage of applied 86Rb+ actually loaded into cells).
Control, total 86Rb+ efflux was assessed in the presence of only a fully efficacious concentration of carbamylcholine (1 mM for SH-EP1-hα4β2, SH-EP1-hα4β4 cells or TE671/RD cells; 3 mM for SH-SY5Y cells). Control, non-specific 86Rb+ efflux was measured either in the presence of the fully efficacious concentration of carbamylcholine plus 100 μM mecamylamine, which gave full block of agonist-induced and spontaneous nAChR-mediated ion flux, or in the presence of efflux buffer alone. Either determination of non-specific efflux was equivalent. Specific efflux was then taken as the difference in control samples between total and non-specific 86Rb+ efflux. Any intrinsic agonist activity of test drugs was ascertained using samples containing test drug only at different concentrations and was normalized, after subtraction of non-specific efflux, to specific efflux in test drug-free, control samples. Antagonism of carbamylcholine-evoked 86Rb+ efflux was assessed in samples containing the full agonist at a concentration where it stimulates 80-90% of maximal function (i.e., its EC80–EC90 value) when exposed alone to a given nAChR subtype (i.e, 460 μM for TE671/RD cells, 2 mM for SH-SY5Y cells; 200 μM for SH-EP1-hα4β2 or -α4β4 cells) and test drugs at the concentrations shown. After subtraction of non-specific efflux, results were normalized to specific ion flux in control samples. For studies of mechanism of antagonism, concentration-response curves were obtained using samples containing the full agonist, carbamylcholine, at the indicated concentrations alone or in the presence of a concentration of the test ligand close to its IC50 value for inhibition of nAChR function. In other studies, cells were pre-exposed to analogues for 1 h (over the last hour of 86Rb+_loading) or 1 day (with 86Rb+_loading occurring during the final 4 h of drug pretreatment) before effects on nAChR function were assessed after analogue was removed (during extracellular 86Rb+_removal) or in the continued presence of drug.
Ion flux assay results were fit using Prism (GraphPad) to the Hill equation, F = Fmax / (1 + (X/Z)n), where F is the test sample specific ion flux as a percentage of control, Fmax is specific ion flux in the absence of test drug (i.e., for control samples), X is the test ligand concentration, Z is the EC50 (n>0 for agonists) or IC50 (n<0 for antagonists), and n is the Hill coefficient. All concentration-ion flux response curves were simple and fit well allowing maximum and minimum ion flux values to be determined by curve fitting, but in cases where antagonists had weak functional potency, minimum ion flux was set at 0% of control. Note that because agonist concentrations used for test ligand antagonism assessments were EC80-EC90 values, not all of the data, even at the lowest concentrations of test antagonist, approaches 100% of specific efflux as separately determined in sister samples exposed to fully efficacious concentrations of agonist.
Behavior
All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committee guidelines.
Animals
Male Institute of Cancer Research (ICR) mice (weighing 20–25 g) obtained from Harlan (Indianapolis, IN) were used throughout the study. Animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility, were placed in groups of six, and had free access to food and water. Studies were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Tail-Flick Test
Antinociception for pain mediated at the spinal level was assessed by the tail-flick method of D'Amour and Smith.32 In brief, mice were lightly restrained while a radiant heat source was shone onto the upper portion of the tail. To minimize tissue damage, a maximum latency of 10 s was imposed. Latency to remove the tail from the heat source was recorded for each animal. A control response (2–4 s) was determined for each mouse before treatment, and a test latency was determined after drug administration (nicotine as an analgesic 5 min after subcutaneous administration at 2.5 mg/kg; nicotine administration 15 min after exposure to saline of 2a analogue to assess the latter drug's ability to block nicotine-mediated antinociception). Antinociceptive response was calculated as the percentage of maximum possible effect (%MPE), where %MPE = [(test control)/(10 control)] × 100.
Hot-Plate Test
Mice were placed into a 10-cm wide glass cylinder on a hot plate (Thermojust Apparatus) maintained at 55 °C for assessment of pain responses mediated at supraspinal levels. To minimize tissue damage, a maximum exposure to the hot plate 40 s was imposed. Measures of control latencies (time until the animal jumped or licked its paws; typically 8–12 s) were done twice for stimuli applied at least 10 min apart for each mouse. Antinociceptive responses after test drug administrations were determined and calculated as the %MPE, where %MPE = [(test latency in s – control latency in s)/(40 s – controllatency in s) × 100]. Groups of 8 to 12 animals were used for each drug condition. Antagonism studies were carried in mice pretreated with either saline or 2a metabolites 15 min before nicotine. The animals were then tested 5 min after administration of a subcutaneous dose of 2.5 mg/kg nicotine.
Locomotor Activity
Mice were placed into individual Omnitech photocell activity cages (28 × 16.5 cm; Omnitech Electronics, Columbus, OH) 5 min after subcutaneous administration of either 0.9% saline or nicotine (1.5 mg/kg). Interruptions of the photocell beams (two banks of eight cells each) were then recorded for the next 10 min. Data were expressed as the number of photocell interruptions. Antagonism studies were carried out by pretreating the mice with either saline or 2a metabolites 15 min before nicotine.
Body Temperature
Rectal temperature was measured by a thermistor probe (inserted 24 mm) and digital thermometer (YSI Inc., Yellow Springs, OH). Readings were taken just before and 30 min after subcutaneous injection of either saline or 2.5 mg/kg nicotine. The difference in rectal temperature before and after treatment was calculated for each mouse. The ambient temperature of the laboratory varied from 21 to 24 °C from day to day. Antagonism studies were carried out by pretreating the mice with either saline or 2a metabolites 15 min before nicotine. The animals were then tested 30 min after administration of a subcutaneous dose of 2.5 mg/kg nicotine.
Supplementary Material
Scheme 1a.

Acknowledgments
This work was supported by National Institutes of Health National Cooperative Drug Discovery Group grant U19 DA019377. Other effort was supported by grants (to RJL) from the National Institutes of Health (DA015389) and the Barrow Neurological Foundation.
Footnotes
Abbreviations: DA, dopamine; 5HT, serotonin; NE, norepinephrine; SR, sustained release; HEK, human embryonic kidney; DAT, dopamine transporter; SERT, serotonin transporter; NET, norepinephrine transporter; nAChR, nicotine acetylcholine receptor; VTA, ventral tegmental area; NRT, nicotine replacement therapy; CTDP, Cocaine Treatment Discovery Program; NIDA, National Institute on Drug Abuse; MPE, maximum possible effect.
Supporting Information Available: Elemental analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.World Health Organization . WHO Report on the Global Tobacco Epidemic, 2008: The MPOWER package. Geneva: 2008. p. 15. [Google Scholar]
- 2.Centers for Disease Control and Prevention Cigarette smoking among adults--United States, 2007. MMWR Morb. Mortal. Wkly. Rep. 2007;56:1157–1161. [PubMed] [Google Scholar]
- 3.Benowitz NL. Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin. Pharmacol. Ther. 2008;83:531–541. doi: 10.1038/clpt.2008.3. [DOI] [PubMed] [Google Scholar]
- 4.Lerman C, LeSage MG, Perkins KA, O'Malley SS, Siegel SJ, Benowitz NL, Corrigall WA. Translational research in medication development for nicotine dependence. Nat. Rev. Drug Discov. 2007;6:746–762. doi: 10.1038/nrd2361. [DOI] [PubMed] [Google Scholar]
- 5.Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M. Heterogeneity and complexity of native brain nicotinic receptors. Biochem. Pharmacol. 2007;74:1102–1111. doi: 10.1016/j.bcp.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 6.Rahman S, Lopez-Hernandez GY, Corrigall WA, Papke RL. Neuronal nicotinic receptors as brain targets for pharmacotherapy of drug addiction. CNS Neurol. Disord. Drug Targets. 2008;7:422–441. doi: 10.2174/187152708786927831. [DOI] [PubMed] [Google Scholar]
- 7.Adams DJ, Nutter TJ. Calcium permeability and modulation of nicotinic acetylcholine receptor-channels in rat parasympathetic neurons. J. Physiol. Paris. 1992;86:67–76. doi: 10.1016/s0928-4257(05)80009-9. [DOI] [PubMed] [Google Scholar]
- 8.Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur. J. Neurosci. 1996;8:2428–2439. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 9.Goldner FM, Dineley KT, Patrick JW. Immunohistochemical localization of the nicotinic acetylcholine receptor subunit alpha6 to dopaminergic neurons in the substantia nigra and ventral tegmental area. Neuroreport. 1997;8:2739–2742. doi: 10.1097/00001756-199708180-00019. [DOI] [PubMed] [Google Scholar]
- 10.Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ. Alpha6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology. 2008;33:2158–2166. doi: 10.1038/sj.npp.1301617. [DOI] [PubMed] [Google Scholar]
- 11.Corrigall WA, Franklin KB, Coen KM, Clarke PB. The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology. 1992;107:285–289. doi: 10.1007/BF02245149. [DOI] [PubMed] [Google Scholar]
- 12.Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P, Lee GS, Huang J, Hahn EL, Mandelkern MA. Smoking-induced ventral striatum dopamine release. Am. J. Psychiatry. 2004;161:1211–1218. doi: 10.1176/appi.ajp.161.7.1211. [DOI] [PubMed] [Google Scholar]
- 13.Fant RV, Buchhalter AR, Buchman AC, Henningfield JE. Pharmacotherapy for Tobacco Dependence. In: Henningfield JE, London ED, Pogun S, editors. Nicotine Psychopharmacology. Handbook of Experimental Pharmacology. Vol. 192. Springer-Verlag; Berlin Heidelberg: 2009. p. 487. [DOI] [PubMed] [Google Scholar]
- 14.Gonzales D, Rennard SI, Nides M, Oncken C, Azoulay S, Billing CB, Watsky EJ, Gong J, Williams KE, Reeves KR. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. Jama. 2006;296:47–55. doi: 10.1001/jama.296.1.47. [DOI] [PubMed] [Google Scholar]
- 15.Fryer JD, Lukas RJ. Noncompetitive functional inhibition at diverse, human nicotinic acetylcholine receptor subtypes by bupropion, phencyclidine, and ibogaine. J. Pharmacol. Exp. Ther. 1999;288:88–92. [PubMed] [Google Scholar]
- 16.Slemmer JE, Martin BR, Damaj MI. Bupropion is a nicotinic antagonist. J. Pharmacol. Exp. Ther. 2000;295:321–327. [PubMed] [Google Scholar]
- 17.Harbeson SL. Propiophenone Derivatives. 2009 WIPO Patent WO/2009/105218.
- 18.Hadizadeh F, Ebrahimzadeh MA, Hosseinzadeh H, Motamed-Shariaty V, Salami S, Bekhradnia AR. Antidepressant and Antioxidant Activities of Some 2-Benzoxazolinone Derivatives as Bupropion Analogues. Pharmacologyonline. 2009;1:331–335. [Google Scholar]
- 19.Foley KF, Cozzi NV. Novel aminopropiophenones as potential antidepressants. Drug Dev. Res. 2003;60:252–260. [Google Scholar]
- 20.Miller DK, Sumithran SP, Dwoskin LP. Bupropion inhibits nicotine-evoked [(3)H]overflow from rat striatal slices preloaded with [(3)H]dopamine and from rat hippocampal slices preloaded with [(3)H]norepinephrine. J. Pharmacol. Exp. Ther. 2002;302:1113–1122. doi: 10.1124/jpet.102.033852. [DOI] [PubMed] [Google Scholar]
- 21.Mooney ME, Sofuoglu M. Bupropion for the treatment of nicotine withdrawal and craving. Expert Rev. Neurother. 2006;6:965–981. doi: 10.1586/14737175.6.7.965. [DOI] [PubMed] [Google Scholar]
- 22.Paterson NE, Balfour DJ, Markou A. Chronic bupropion attenuated the anhedonic component of nicotine withdrawal in rats via inhibition of dopamine reuptake in the nucleus accumbens shell. Eur. J. Neurosci. 2007;25:3099–3108. doi: 10.1111/j.1460-9568.2007.05546.x. [DOI] [PubMed] [Google Scholar]
- 23.Reuben M, Boye S, Clarke PB. Nicotinic receptors modulating somatodendritic and terminal dopamine release differ pharmacologically. Eur. J. Pharmacol. 2000;393:39–49. doi: 10.1016/s0014-2999(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 24.Makhay MM, O'Donnell JM. Effects of antidepressants in rats trained to discriminate the beta-2 adrenergic agonist clenbuterol. Pharmacol. Biochem. Behav. 1999;63:319–324. doi: 10.1016/s0091-3057(98)00266-4. [DOI] [PubMed] [Google Scholar]
- 25.Carroll FI, Blough B, Abraham P, Mills AC, Holleman JA, Wolckenhauer SA, Decker AM, Landavazo A, McElroy KT, Navarro HA, Gatch MB, Foster MJ. Synthesis and Biological Evaluation of Bupropion Analogues as Potential Pharmacotherapies for Cocaine Addiction. J. Med. Chem. 2009;52:6768–6781. doi: 10.1021/jm901189z. [DOI] [PubMed] [Google Scholar]
- 26.Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. J. Pharmacol. Exp. Ther. 1999;289:877–885. [PubMed] [Google Scholar]
- 27.Damaj MI, Carroll FI, Eaton JB, Navarro HA, Blough BE, Mirza S, Lukas RJ, Martin BR. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol. Pharmacol. 2004;66:675–682. doi: 10.1124/mol.104.001313. [DOI] [PubMed] [Google Scholar]
- 28.Salas R, Sturm R, Boulter J, De Biasi M. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J. Neurosci. 2009;29:3014–3018. doi: 10.1523/JNEUROSCI.4934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukas RJ, Fryer JD, Eaton JB, Gentry CL. Some methods for studies of nicotinic acetylcholine receptor pharmacology. In: Levin ED, editor. Nicotinic Receptors and the Nervous System. CRC Press; Boca Raton: 2002. pp. 3–27. [Google Scholar]
- 30.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, Buhlman L, Kuo YP, Steinlein O, Lukas RJ. Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol. Pharmacol. 2003;64:1283–1294. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 31.Gentry CL, Lukas RJ. Local anesthetics noncompetitively inhibit function of four distinct nicotinic acetylcholine receptor subtypes. J. Pharmacol. Exp. Ther. 2001;299:1038–1048. [PubMed] [Google Scholar]
- 32.D'Amour FE, Smith DL. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.