

Abstract
Smooth muscle cell (SMC) migration involves interactions of integrin receptors with extracellular matrix (ECM) and is an important process of neointimal formation in atherosclerosis and restenosis after vascular interventions. Previous studies have shown that periostin (PN), a novel ECM protein, is upregulated in rat carotid artery after balloon injury, and growth factor-stimulated expression of PN promotes SMC migration in vitro. Here, we address the mechanism by which PN–integrin interaction mediates SMC migration in vitro. Aortic SMCs isolated from PN null mice exhibited a significantly reduced ability to migrate and proliferate in vitro. Endogenous PN protein was absent and very low in the culture medium from the primary cultures of PN−/− and wildtype SMCs, respectively. In both types of SMCs, adenovirus-mediated overexpression of HA-tagged PN to a similar extent, which induced a robust cell migration concomitantly with an increase in β3-integrin expression and phosphorylation of FAK (Tyr397). Furthermore, in cultured human SMCs, specific integrin blocking antibodies showed that interactions of PN-ανβ3 and PN-ανβ5, but not PN-β1 integrins, are required for SMC migration. Inhibition of FAK signaling by overexpression of an endogenous FAK inhibitor termed FRNK (FAK-related nonkinase) significantly attenuated FAK (Tyr397) phosphorylation and the SMC migration induced by PN. These results reveal a mechanism whereby PN mediates vascular SMC migration through an interaction with alphaV-integrins (mainly ανβ3) and subsequent activation of FAK pathway.
Keywords: Periostin, Integrin receptors, FAK, FRNK, SMC, Migration
1. Introduction
Vascular smooth muscle cell (SMC) migration is a key event in vascular lesion formation, which is a common pathologic feature of atherosclerosis and restenosis after vascular interventions. Growth factors and extracellular matrix (ECM) proteins can stimulate SMC migration [1]. Among the latter, vitronectin and osteopontin exhibit an increased expression after vascular injury, which promote SMC migration and neointima formation through interactions with cell surface integrin receptors (ανβ3 and ανβ5) [2–4]. Blockade of integrin αVβ3 and its ligands (vitronectin and osteopontin) by specific blocking antibodies or peptide antagonists has been shown to reduce SMC migration and neointima formation after vascular injury [2–4].
Periostin (PN) is a secreted 90 kDa ECM protein, originally identified as an osteoblast-specific factor related to the midline fasciclin-1 (mfas-1) gene in Drosophila [5]. It has four repeated fasciclin domains, an alternatively spliced carboxyl tail, an amino terminus cysteine rich region, and a putative signal sequence, suggesting it is a secreted protein [6]. PN is expressed in several normal adult tissues with the highest levels of expression being detected in connective tissues such as bone, skin, and heart valves, but the peripheral vasculature exhibits low or absent expression of PN [7,8]. We and others have previously demonstrated that PN expression is strongly upregulated in rat carotid artery after balloon injury [9,10]. In cultured vascular SMCs, PN expression was stimulated by multiple vascular injury-related factors, including PDGF-BB, FGF-1, FGF-2, TGF-β1, and angiotensin II [9–11]. Although PN has been shown to serve as a ligand for select integrins, including ανβ3, ανβ5, and α6β4, where it can affect the ability of cells (e.g. fibroblasts or cancer cells) to migrate and/or undergo a mesenchymal transformation in select tissues [12–14], the interaction between PN and integrin receptors in vascular SMCs has not yet been elucidated. In the present study, we extended these observations by demonstrating an enhanced expression of PN in a mouse carotid artery injury model, and then examined the role of enhanced PN in SMC migration using both adenovirus gene transfer and recombinant human PN protein, and finally defined the role of alphaV-integrin receptors and focal adhesion kinase (FAK) pathway in PN-mediated SMC migration in vitro.
2. Materials and methods
2.1. Cells and cell culture
Aortic SMCs were obtained from thoracic aortas of C57Bl6 mice (The Jackson Laboratory, Bar Harbor, Me) and PN-deficient mice (on a C57BL/6 genetic background; provided by Dr. Simon Conway, IUPUI, IN) at 8–10 weeks of age. After removing the adventitia and endothelium, the media of aortas were minced and digested in 5 ml of the digestion solution (2 mg/ml collagenase I, 0.125 mg/ml elastase, 0.25 mg/ml soybean trypsin inhibitor, 2.0 mg/ml crystallized bovine albumin) for 45 min at 37 °C. The cellular digests were filtered through sterile 100-μM nylon mesh, centrifuged at 1000 rpm for 10 min, and washed twice in DMEM medium containing 10% fetal bovine serum (FBS). Washed cells were grown in DMEM containing 0.5 ng/ml EGF, 5 μg/ml insulin, 2 ng/ml bFGF, 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin and incubated at 37°C with 5% CO2/95% air. Primary human aortic smooth muscle cells (hASMCs) were obtained from Clonetics (Lonza) and cultured on 0.2% gelatin-coated culture dishes in the complete SmGM-2 medium (Clonetics #CC-3182) containing 15% FBS. All SMC lineage was confirmed by the presence of immunoreactivity for SM α-actin (clone 1A4, Sigma) in >95% of the cells. Experiments were performed with mouse and human SMCs from passages 3–6.
2.2. Adenovirus construction and cell infection
A full-length mouse PN cDNA (generated by Dr. Simon Conway, IUPUI) was cloned into the pDNR-CMV shuttle vector (Clontech) as previously described [15]. To facilitate detection of the adenovirus-produced protein, a hemagluttinin (HA) tag was inserted at the carboxyl-terminus through standard PCR techniques. The mouse PN cDNA insert and the proximal CMV promoter were moved into the Adeno-X Acceptor Vector through a Cre/Lox-mediated recombination. The adenoviral HA-tagged FRNK construct was kindly provided by Dr. Qiang Ding (University of Alabama at Birmingham, AL) [16]. Both PN construct (Ad-PN) and FRNK construct (Ad-FRNK) was packaged in HEK293 cells (ATCC) and amplified to obtain high-titer stocks. The viral particles were purified using the Adeno-X Viral Purification Kit (Clontech). Adenoviral control vectors encoding β-galactosidase (Ad-LacZ) and green fluorescent protein (Ad-GFP) were constructed in the same manner. In some experiments, mouse or human SMCs (∼70% confluence) were infected with Ad-PN, Ad-FRNK, or control vectors as previously described [15,16]. Following a 5-h infection, the cells were maintained in fresh normal growth media. The following day, infection efficiency was evaluated by X-Gal staining for LacZ expression and by direct observation of GFP expression under a fluorescence microscope.
2.3. Western blotting
The cells were lysed in a modified radioimmunoprecipitation assay (RIPA) buffer (50 mM HEPES, 0.15 M NaCl, 2 mM EDTA, 0.1% NP-40, 0.05% sodium deoxycholate, pH 7.2) containing protease inhibitor cocktail (Roche). Cell lysates were maintained for 1 h on ice, twice passed through an 18-gauge needle, and centrifuged at 2000 rpm for 10 min at 4°C. Protein concentration in the supernatant of cell lysates was determined using the BCA kit (Pierce Biochemicals). Equal amount of protein from each lysate was separated using 10% SDS-PAGE and the proteins transferred to nitrocellulose. FAK was detected by a rabbit polyclonal antibody (Cell Signaling) and FRNK was detected by a rabbit polyclonal antibody generated against the part of C-terminal sequence of FAK (Upstate Biotechnology). FAK autophosphorylation was detected using polyclonal rabbit anti-pY397 FAK (BioSource International). The HA-tagged FRNK or PN protein was detected using monoclonal anti-HA-tag antibody (HA-7 clone, Sigma). For measurement of HA-tagged PN protein or endogenous PN released into the medium, the cell culture conditioned medium was collected and concentrated approximately 2-fold using an ultra concentrator (50 kDa cut-off, Amicon). Immunopreciptation (IP) and Western blot analysis of endogenous PN was performed using the method as previously described [9] using a rabbit anti-PN polyclonal antibody (BioVendor, NC). Immunopositive bands of HRP-conjugated secondary antibodies were detected by ECL (GE Healthcare).
2.4. Flow cytometry
SMCs were suspended in PBS/0.2% BSA at 106 cells/ml, and 200 μl of cell suspension was incubated with 2 μg fluorescein-conjugated mAb or isotype-matched control antibody on ice for 45 min in the dark. For the analysis of integrins on hASMCs, the following monoclonal antibodies (mAb) antibodies (Chemicon) were used: mAb against integrin αvβ3 (clone LM609), integrin αvβ5 (clone P1F6), integrin β1 (clone P4C10) and control IgG1 (clone DD7). The β3-integrin on mouse SMCs was analyzed by anti-mouse CD61 mAb (clone 2C9.G2, BD Biosciences) and control Armenian hamster IgG1 (Santa Cruz). Cells were washed three times with PBS containing 0.1% NaN3 and 1.0% FBS and then analyzed using a FACSCalibur (Becton–Dickinson).
2.5. Wound migration assay
A scratch-wound migration assay was performed to assess the effect of endogenous overexpression of PN on SMC migration in response to injury. Mouse SMCs were plated on 6-well tissue culture plates and infected with Ad-PN or Ad-lacZ as described above. After a 5-h infection, the cells were allowed to grow to subconfluence (90%) in fresh normal growth media, and then made quiescent by serum-free incubation for 48 h. After that, a scratch-wound (≈4 mm wide) was made across the diameter of the well using the end of a 200 μl pipette tip and scraped cells were removed by washing three times with PBS. The cells were maintained in DMEM/0.25% FBS for the observation of migration. For each well, images were taken at 0 and 20 h after the wound, and the number of cells (per μm2 area) migrated into the wound space were manually counted in 5 fields per well.
2.6. Chemotaxis and haptotaxis
A modified Boyden chamber (8-μm pores, Costar Corp) was used for both assays under 37°C and 5% CO2. With chemotaxis assay, a suspension of the wildtype or PN−/− SMCs (5 × 104 cells in 0.2 ml migration buffer: DMEM/0.2% BSA) was plated in the upper chamber. The bottom chambers were filled with 0.5 ml migration buffer containing vehicle or indicated concentrations of PDGF-BB. Migration was allowed to proceed for 6 h. With haptotaxis assay, the undersides of the transwell membranes were pre-coated (4°C, overnight) with recombinant human PN protein (rh-PN, kindly provided by Dr. Meriu Dai) [9]. A suspension of hASMCs (1 × 105 cells in 0.2 ml migration buffer) was plated in the upper chamber. The migration buffer (0.5 ml/well) was added to each bottom chamber. Migration towards immobilized rh-PN, in the absence or presence of blocking monoclonal antibodies against individual integrin receptors, was allowed to proceed for 6 h. In both migration assays, cells on the upper membrane surface were removed by a cotton tip applicator, chambers were washed with PBS, and migratory cells on the lower membrane surface were fixed with methanol and stained with hematoxylin. Migration was quantified by cell counts of four random high-power (20×) fields per chamber. Each assay was performed in quadruplicate.
2.7. Cell proliferation assay
Cells at 70% confluence were serum starved in a 0.1% FBS for 72 h and then plated at a concentration of 5000 cells per well on a 96-well plate. Cells were incubated with fresh DMEM containing 20 ng/ml PDGF-BB, 10% fetal bovine serum (FBS), or 0.1% FBS (basal control). The number of viable cells was determined after 48 h by using the Promega Cell Titer 96 MTT assay method. Each assay was performed in quadruplicate.
2.8. Statistical analysis
Data are represented as the mean ± SD. Significance of differences was analyzed using one-way analysis of variance or Student's t-test as appropriate. All of the experiments were repeated at least three times. A value of P < 0.05 was considered statistically significant.
3. Results
3.1. Impaired migratory and proliferative activity and FAK autophosphorylation of PN-deficient SMCs
PDGF-BB has been recognized as one of the major migratory factors for arterial SMCs in vitro and during neointima formation in vivo [17,18]. FAK is known to be required for vascular injury- and PDGF-mediated SMC migration [19–21]. Here, we determined whether lack of endogenous PN affects SMC migration towards PDGF-BB. The PN−/− and wildtype SMCs were isolated from aortas of PN−/− mice and C57Bl6 mice, respectively. Using a modified Boyden chamber chemotaxis assay, we showed that the PDGF-BB-induced migration was reduced significantly in PN−/− SMCs (P < 0.05), compared with wildtype SMCs (Fig. 1A). Using the Promega Cell Titer 96 MTT assay, we showed a significant reduction in proliferation of PN−/− SMCs cultured in 10% FBS (P < 0.01) or with PDGF stimulation (P < 0.01), compared with respective wild-type controls (Fig. 1B). By Western blotting, we showed that FAK phosphorylation (p-FAK) at Tyr397 was detected at low levels in both wildtype and PN−/− SMCs under basal conditions, however, the induction of the p-FAK by stimulation with PDGF-BB (20 ng/ml, 15 min) was significantly reduced in PN−/− SMCs versus wildtype SMCs (Fig. 1C).
Fig. 1.

Impaired migratory and proliferative activity and FAK autophosphorylation of PN-deficient SMCs. (A) Comparison of cell migration between wildtype (WT) and PN−/− SMCs in response to PDGF-BB (for 6 h) by modified Boyden chamber method. (B) Comparison of cell proliferation between WT and PN−/− SMCs under the indicated conditions for 72 h. Values shown in (A) and (B) are mean ± SD of three independent experiments. For each experiment, the SMCs were isolated from 2-3 mouse aortas. *P < 0.05 and **P < 0.01 vs. respective WT controls. C, Western blotting demonstrated that level of phosphorylated FAK (Tyr397) was reduced in PN−/− SMCs compared with WT SMCs. Quiescent SMCs (∼90% confluence) were treated with vehicle (PBS) or PDGF-BB (20 ng/ml, for 15 min). Values are mean ± SD of three independent experiments. *P < 0.05 vs. WT.
3.2. Effect of adenovirus-mediated overexpression of PN on migration, β3-integrin expression and FAK autophosphorylation of SMCs
The level of endogenously produced PN in the cell culture conditioned medium obtained from the quiescent unstimulated wildtype SMCs was very low, but markedly increased after stimulation with PDGF-BB for 24 h (Fig. 2A), as evaluated by IP/Western blotting using a rabbit anti-PN polyclonal antibody. As expected, the endogenous PN was absent in the medium obtained from the PN−/− SMCs under basal and PDGF-stimulated conditions (Fig. 2A). To determine the role of PN overexpression in promoting SMC migration in response to injury, we used adenovirus-mediated gene transfer to overexpress a HA-tagged PN in mouse SMCs. The transfection efficiency and expression pattern of the adenovirus-produced PN were similar between wildtype SMCs and PN−/− SMCs (Fig. 2B), as evaluated by Western blotting using anti-HA monoclonal antibody. Data showed that the HA-tagged PN protein was detected abundantly both in the cell lysates and the cell culture medium conditioned by either Ad-PN-infected wildtype SMCs or Ad-PN-infected PN−/− SMCs (Fig. 2B). As expected, the HA-tagged PN protein was not detected in the non-infected and Ad-LacZ-infected cells.
Fig. 2.
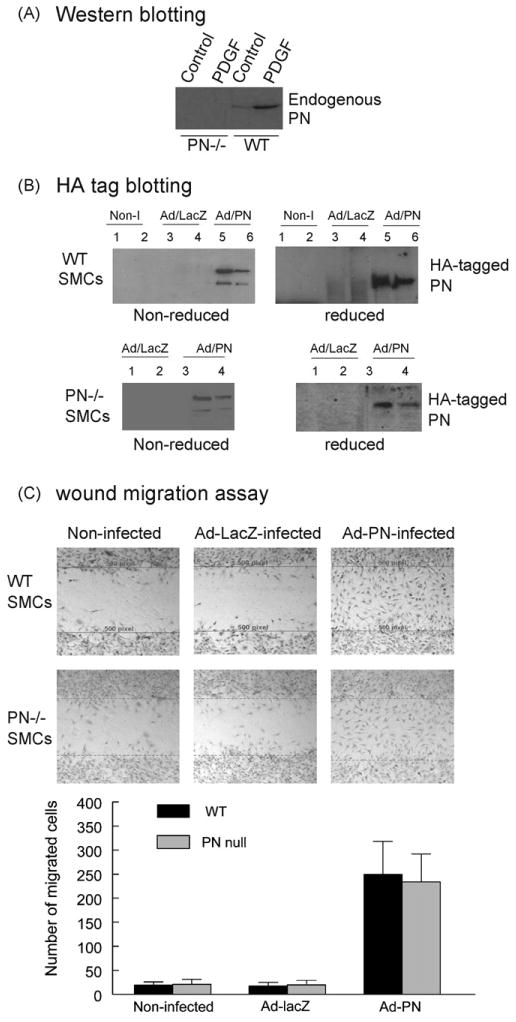
Effect of adenovirus-mediated overexpression of PN on SMC migration. (A) Western blotting demonstrated the expression of endogenous PN detected by using an anti-PN antibody in the conditioned medium obtained from quiescent cultures of WT and PN−/− SMCs, in the absence or presence of PDGF-BB (10 ng/ml) as indicated. (B) Western blotting demonstrated the expression of adenovirus-produced HA-tagged PN detected by using an anti-HA antibody either in the conditioned medium (lanes 1, 3 and 5) or the cellular lysates (lanes 2, 4 and 6) obtained from cultures of WT (top) and PN−/− (bottom) SMCs, non-infected (Non-I), infected with Ad-LacZ or Ad-PN as indicated. Under reducing conditions (with β-mercaptoethanol: β-ME), the PN migrates as a monomer (∼90 kDa). Under non-reducing conditions (without β-ME), the PN migrates as both a monomer and a dimer. (C) Migration by a scratch-wound assay showed that both WT (top) and PN−/− (bottom) SMCs infected by Ad-PN migrated into the wound area at 20 h after wounding, almost completely filling the wound space, whereas few non-infected or Ad-LacZ-infected cells migrated into the wound area. The lines delimit the initially wounded regions. The number of cells migrated into the wound space was measured from digital images. Results are representative of three separate experiments.
Functional consequences of overexpressing the HA-tagged PN in SMC migration were examined using a wound migration assay. We demonstrated that the adenovirus-mediated overexpression of PN resulted in a robust migration, to a similar extent, in the Ad-PN-infected wildtype SMCs and PN−/− SMCs, whereas few SMCs migrated in the non-infected or Ad-LacZ-infected cells (Fig. 2C). These results were not likely due to cell proliferation because the cells had been made quiescent for 48 h prior to the migration assay. These findings showed a significant role of PN overexpression in regulation of SMC migration and also indicate that the PN−/− phenotypes can be rescued by PN overexpression.
Integrin-β3-FAK pathway is a key pathway in mediating cell migration induced by ECM proteins (e.g. vitronectin and osteopontin) and growth factors (e.g. PDGF-BB) [21]. To provide insights into mechanism of the PN-induced SMC migration, we examined the effect of the adenovirus-mediated overexpression of PN on this pathway. Flow cytometry showed that the levels of integrin-β3 expression were enhanced in both wildtype (approximately 3-fold) and PN−/− (approximately 2.1-fold) SMCs subjected to Ad-PN infection (Fig. 3A), compared with the non-infected and Ad-LacZ-infected cells. In addition, we observed that the expression of integrin-β5 was not altered by the Ad-PN infection of SMCs, compared with non-infected cells (data not shown). Moreover, Western blotting showed that the adenovirus-enhanced PN increased the phosphorylation level of FAK (Tyr397) in the Ad-PN infected wild-type SMCs (Fig. 3B), compared with the Ad-LacZ-infected and non-infected cells.
Fig. 3.
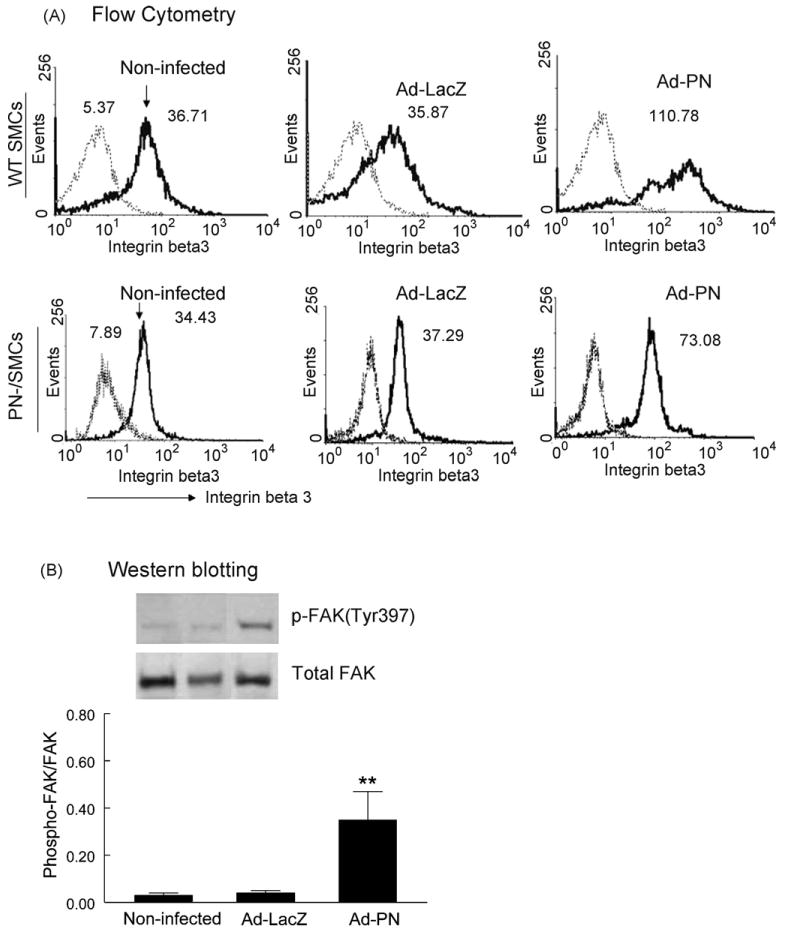
Effect of adenovirus-mediated overexpression of PN on β3-integrin expression and FAK autophosphorylation of SMCs. (A) Flow cytometry demonstrated increased expression of β3-integrin on the cell surface of WT (top) and PN−/− (bottom) SMCs infected by Ad-PN. Integrin-β3 was detected with FITC-conjugated hamster anti-mouse CD61 mAb (thick line), and a rat IgG1 isotype was used as a negative control (thin line). Numbers indicate the mean fluorescence intensity in the FL1 (FITC) channel, showing a nearly 2.1-fold (PN−/− cells) and 3-fold (WT cells) increase in β3-integrin expression on the surface of Ad-PN-infected SMCs (lane 3), compared with either non-infected (lane 1) or Ad-lacZ-infected (lane 2) cells. (B) Western blotting demonstrated that phosphorylation level of FAK (Tyr397) was increased in Ad-PN-infected WT SMCs (lane 3), compared with non-infected (lane 1) or Ad-lacZ-infected (lane 2) SMCs. Values are mean ± SD of three independent experiments. **P < 0.01 vs. bars 1 and 2.
3.3. Role of alphaV-integrin receptors in PN-induced migration of human SMCs
Although arterial SMCs are known to express multiple integrin receptors, the expression of particular integrin heterodimers can vary depending on the origin of the cells and experimental conditions [22]. Thus, we detected the expression of integrin receptors on human aortic SMCs (hASMCs) by flow cytometry. Compared to cells incubated with an isotype-matched control antibody, flow cytometry demonstrated the presence of ανβ3, ανβ5, and β1-integrins on the cell surface (Fig. 4A). Using a modified Boyden chamber haptotaxis assay, we demonstrated that recombinant human PN (rh-PN) immobilized on the underside of the membrane filters stimulated hASMC migration in a dose-dependent manner (Fig. 4B). Further, blocking experiments were performed to investigate whether ανβ3, ανβ5, and β1-integrins were required for the PN-stimulated migration. The presence of anti-ανβ3, or -ανβ5 integrin mAbs induced, respectively, a 49 ± 8% (P < 0.05) and 28 ± 5% (P < 0.05) decrease of SMC migration (Fig. 4C). We also observed significant additional inhibition of the migration in the presence of both mAb anti-ανβ3 and -ανβ5 (75 ± 10%) (Fig. 4C). In contrast, blocking with the anti-β1 integrin mAb failed to inhibit SMC migration, suggesting that there is no significant role for β1-integrin in this process.
Fig. 4.

Role of alphav-integrin receptors in PN-induced human ASMC migration. (A) Flow cytometry demonstrated the presence of ανβ3, ανβ5, and β1-integrin in human aortic SMCs (passage 5ht). Cells were labeled with isotype (shaded area) or integrin antibodies (thin line) as described in Materials and Methods. (B) Recombinant human PN (rh-PN) induces haptotactic migration of human ASMCs in a dose-dependent manner. (C) Blocking experiment with specific integrin antibodies showed that interactions of PN-ανβ3 and PN-ανβ5, but not PN-β1, are required for rhPN-induced SMC migration. Values are mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 vs. bar 1.
3.4. Role of FRNK in PN-induced FAK activation and migration of human SMCs
FAK activity is required for integrin- and growth factor-induced SMC migration. Expression of FRNK, an endogenous inhibitor of FAK, has been shown to inhibit FAK activity and growth factor- and integrin-induced SMC migration [20]. We thus determined the involvement of FAK/FRNK in the PN-induced FAK (Tyr397) phosphorylation and migration of human hASMCs. Using adenovirus-mediated gene transfer, overexpression of FRNK was achieved in cultured hASMCs. Western blotting showed that the HA-tagged FRNK protein was abundantly detected in whole cell lysates obtained from the Ad-FRNK-infected SMCs (Fig. 5A). As expected, the HA-FRNK was not detectable in the Ad-GFP-infected cells.
Fig. 5.
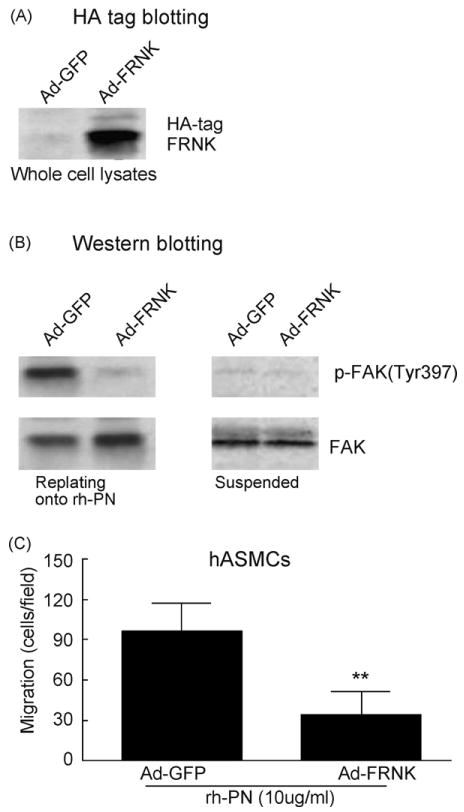
FRNK inhibition of PN-induced FAK (Tyr397) phosphorylation and migration of human ASMCs. (A) Western blotting demonstrated adenovirus-mediated over-expression of FRNK in human aortic SMCs that were infected with Ad-GFP (lane1) or Ad-FRNK (lane 2). (B) Adenovirus-mediated overexpression of FRNK reduced PN-induced FAK (Tyr397) phosphorylation. Ad-FRNK- or Ad-GFP-infected hASMCs were replated onto dishes that were pre-coated with rh-PN (20 μg/ml) or suspended in serum-free medium. After 60 min attachment, cells were washed once with ice-cold PBS and lysed, and whole cell lysates were analyzed by Western blot analysis of phosphorylated FAK (Tyr397) as described above. (C) Adenovirus-mediated over-expression of FRNK reduced PN-induced haptotactic migration of hASMCs. Values are mean ± SD of three independent experiments. **P < 0.01 vs. control.
Previous studies have shown that FAK (Tyr397) phosphorylation was induced only in adherent human SMCs replating onto vitronectin but not in suspended cells [21]. Similarly, we observed a strong upregulation of FAK (Tyr397) phosphorylation only in the hASMCs replating onto rh-PN but not in suspended cells (Fig. 5B) or replating onto poly-l-lysine (as a negative control, data not shown). In the replating assay, cell culture dishes were coated overnight at 4 °C with 20 μg/ml recombinant human PN (rh-PN) or 10 μg/ml poly-l-lysine (Sigma). Suspensions of hASMCs that had been infected with Ad-FRNK- or Ad-GFP were allowed to attach to the substrates for 60 min. After that, cells were washed once with ice-cold PBS and lysed, and whole cell lysates were analyzed by Western blotting. Furthermore, we showed that enforced expression of FRNK by Ad-FRNK transfer inhibited rh-PN-induced FAK (Tyr397) phosphorylation (Fig. 5B). Finally, we demonstrated that rh-PN-induced migration was reduced significantly in the Ad-FRNK-infected hASMCs (P < 0.01), compared with the Ad-GFP-infected cells (Fig. 5C).
4. Discussion
Previous studies have demonstrated that medial SMCs start to migrate toward the intima in the early phase (usually between 3 and 7 days) after vascular injury, then proliferate there to constitute the neointima [23,24]. PN has been shown to serve as a ligand for select integrins including ανβ3 and ανβ5, both integrin receptors are upregulated at early time points after vascular injury [3,25]. Consistent with the previous observations in rat carotid arteries after balloon injury [9,10], we observed that PN expression is upregulated in mouse carotid arteries after wire injury, especially in the early phase (3d, 7d), and the upregulation is more prominent in the atherogenic apoE−/− mice than in normal C57Bl6 mice (Supplemental Fig. 1). Therefore, we speculate that the enhanced PN in injured vessel wall may stimulate medial SMC migration via interaction with integrin receptors ανβ3 (and ανβ5), eventually contributing to neointima formation after vascular injury (Fig. 6). In this regard, the present study was conducted to explore the molecular mechanism by which the integrin–PN interaction mediates vascular SMC migration using well-defined cell culture and gene transfection system.
Fig. 6.
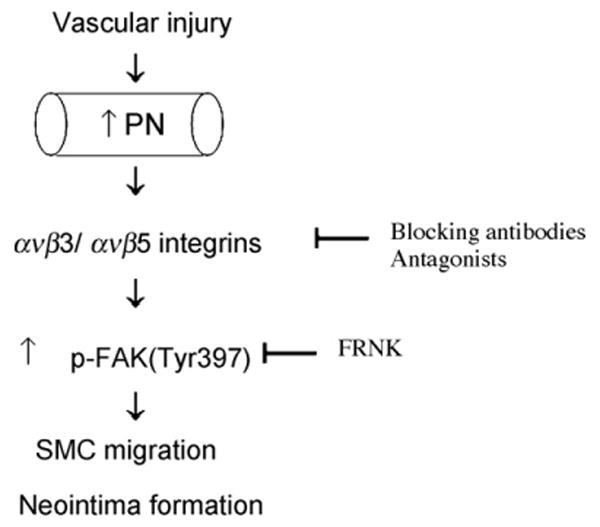
Hypothetical mechanism whereby PN plays a role in the process of SMC migration and neointima induced by vascular injury. PN expression is induced in the injured arterial wall in the early phase after vascular injury. PN stimulates medial SMC migration likely through binding to ανβ3/ανβ5 integrins and subsequent activation of FAK-dependent signaling, eventually contributing to neointima formation.
Our study is the first to provide several lines of evidence demonstrating a significant role for PN in the regulation of SMC migration in vitro. We show that (1) aortic SMCs isolated from PN−/− mice exhibit a significantly reduced ability to migrate and proliferate in vitro, (2) adenovirus-mediated overexpression of PN resulted in a robust migration to a similar extent in PN−/− and wildtype mouse SMCs in the wound migration assay, and (3) recombinant human PN (rhPN) stimulated hASMC migration in a dose-dependent manner in the haptotaxis assay. It is well known that different ECM proteins stimulate SMC migration through distinct integrins. For example, vitronectin and osteopontin stimulate SMC migration predominantly through ανβ3 integrin, whereas fibronectin and collagen stimulate SMC migration through β1 integrins (e.g. α2β1 and α5β1) [26–28]. PN is a unique ligand for integrins in that it does not contain an Arg-Gly-Asp (RGD) sequence motif, instead it binds to integrins through a fasciclin I (FAS1) domain [29]. PN has been shown to be a ligand for several integrins in human tumors, endothelial cells, and human autosomal dominant polycystic kidney cells [9–11,30]. Lindner et al. reported that overexpression of periostin enhanced cell migration in C3H10T1/2 cells (mouse fibroblast cell lines) [10]. However, little is known about which integrin receptor(s) mediates migration of vascular SMCs induced by PN. The present study extends previous studies by demonstrating that interactions of PN-ανβ3, and to a lesser extent PN-ανβ5, but not PN-β1, are required for migration of human SMCs in vitro. It is worth to note that while our data support the view that such integrin subsets are needed for PN-induced migration (i.e. the role of integrins), more work still needs to understand whether PN–integrin interaction is required for PN-dependent migration and the relative effect of the same anti-integrin antibodies on cell migration under more complex conditions in the presence of serum or PDGF, or other integrin ECM ligands. Although it is well established that integrin ανβ3 in the vessel wall is upregulated in response to vascular injury, the mechanism remains poorly understood. In cultured vascular SMCs, αvβ3 expression appears to be stimulated by PDGF-BB, thrombin, and osteopontin [28,31]. In this study, we show that the adenoviral overexpression of PN resulted in an increase in expression of β3-integrin in cultured mouse SMCs. Therefore, we speculate that the enhanced PN in the injured arterial wall might contribute to the upregulation of ανβ3 integrin in the response to vascular injury.
FAK is a non-receptor cytoplasmic protein tyrosine kinase that localizes to focal adhesion plaques, it becomes phosphorylated and activated in response to integrin-mediated binding of cells to the extracellular matrix proteins [32]. A critical event in integrin-mediated FAK signaling is phosphorylation of Tyr397 [33]. When phosphorylated, Y397 becomes a binding site for the tyrosine kinase Src, which phosphorylates FAK at Y576 and Y577 to further activate FAK kinase activity [32,33]. Several recent studies have demonstrated a role for PN in integrin-mediated FAK signaling. Shimazaki et al. [34] showed that PN is essential for cardiac healing after acute myocardial infarction through FAK-alphaV-integrin signaling. Cardiac healing was impaired in PN−/− mice, caused by a reduced number of alpha smooth muscle actin-positive cells, impaired collagen fibril formation, and decreased phosphorylation of FAK. These phenotypes can be rescued by gene transfer of PN. The inhibition of FAK or alphaV-integrin blocked the PN-promoted cardiac fibroblast migration. Shao et al. [35] showed that PN-stimulated FAK tyrosine phosphorylation in cultured human microvascular endothelial cells via ανβ3 integrin. Altogether these observations reveals that alphaV-integrin and FAK are involved in PN signaling. In the present study, we extend previous findings by demonstrating that activation of FAK pathway is required for PN-mediated SMC migration in vitro. Our results show that (1) the phosphorylation levels of FAK at Tyr397 was reduced in PN−/− SMCs after transient stimulation with PDGF-BB, and (2) the adenovirus-mediated overexpression of PN enhanced the phosphorylation levels of FAK at Tyr397 in Ad-PN infected wildtype SMCs. One limitation of our study was the lack of data about the effect of PN overexpression on FAK phosphorylation in PN−/− SMCs. Furthermore, we investigated the involvement of FAK/FRNK in the PN-induced cell migration in cultured human hASMCs. FAK activity is known to be required for integrin- and growth factor-induced SMC migration. It is well characterized that FRNK, an endogenous inhibitor of FAK, inhibits integrin-mediated cell migration through blocking FAK activation and FAK-mediated signaling [16,21]. Our data show that the rh-PN can induce FAK (Tyr397) phosphorylation and cell migration in cultured hASMCs, and these effects can be inhibited by means of overexpressing FRNK using Ad-FRNK gene transfer. Taken together, these findings reveal a mechanism whereby enhanced PN expression mediates vascular SMC migration through interaction with alphaV-integrins (mainly ανβ3) and subsequent activation of FAK.
In conclusion, the present study demonstrates for the fist time that enhanced PN expression plays a significant role in vascular SMC migration induced either by mechanic injury or PDGF stimulation, through interaction with alphaV-integrins (mainly ανβ3) and subsequent activation of FAK pathway. Whether the enhanced PN in the injured vessel wall substantially contributes to the development of neointima formation in vivo is now under investigation.
Supplementary Material
Acknowledgments
This work was supported by a Cardiovascular Research Center grant (GL) from the University of Virginia Health Science System and by American Heart Association grant 0530166N (GL) and National Institutes of Health grants HL087990 (GL). This work was also supported in part by HL26441 (DNG) and HL078663 (SSS), National Science Foundation FIBRE EF0526854 (RRM and RAN), and SC INBRE 2 P20 RR16461-05A1 (RAN). We acknowledge Dr. Qiang Ding (Medicine, University of Alabama at Birmingham) for providing with Ad-FRNK vector.
Footnotes
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.atherosclerosis.2009.07.046.
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Schwartz S. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest. 1997;99:2814–7. doi: 10.1172/JCI119472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dufourcq P, Couffinhal T, Alzieu P, et al. Vitronectin is up-regulated after vascular injury and vitronectin blockade prevents neointima formation. Cardiovasc Res. 2002;53(4):779–81. doi: 10.1016/s0008-6363(01)00547-8. [DOI] [PubMed] [Google Scholar]
- 3.Corjay MH, Diamond SM, Schlingmann KL, Gibbs SK, Stoltenborg JK, Racanelli AL. Alphavbeta3, alphavbeta5, and osteopontin are coordinately upregulated at early time points in a rabbit model of neointima formation. J Cell Biochem. 1999;75(3):492–504. doi: 10.1002/(sici)1097-4644(19991201)75:3<492::aid-jcb13>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- 4.Srivatsa SS, Fitzpatrick LA, Tsao PW, et al. Selective alpha v beta 3 integrin blockade potently limits neointimal hyperplasia and lumen stenosis following deep coronary arterial stent injury: evidence for the functional importance of integrin alpha v beta 3 and osteopontin expression during neointima formation. Cardiovasc Res. 1997;36(3):408–28. doi: 10.1016/s0008-6363(97)00184-3. [DOI] [PubMed] [Google Scholar]
- 5.Horiuchi K, Amizuka N, Takeshita S, et al. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res. 1999;14:1239–49. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- 6.Takeshita S, Kikuno R, Tezuka K, Amann E. Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J. 1993;294(Pt 1):271–8. doi: 10.1042/bj2940271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Litvin J, Zhu S, Norris R, Markwald R. Periostin family of proteins: therapeutic targets for heart disease. Anat Rec A Discov Mol Cell Evol Biol. 2005;287(2):1205–12. doi: 10.1002/ar.a.20237. [DOI] [PubMed] [Google Scholar]
- 8.Norris RA, Borg TK, Butcher JT, Baudino TA, Banerjee I, Markwald RR. Neonatal and adult cardiovascular pathophysiological remodeling and repair: developmental role of periostin. Ann N Y Acad Sci. 2008;1123:30–40. doi: 10.1196/annals.1420.005. [DOI] [PubMed] [Google Scholar]
- 9.Li G, Oparil S, Sanders JM, et al. Phosphatidylinositol-3-kinase signaling mediates vascular smooth muscle cell expression of periostin in vivo and in vitro. Atherosclerosis. 2006;188(2):292–300. doi: 10.1016/j.atherosclerosis.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindner V, Wang Q, Conley BA, Friesel RE, Vary CP. Vascular injury induces expression of periostin: implications for vascular cell differentiation and migration. Arterioscler Thromb Vasc Biol. 2005;25(1):77–83. doi: 10.1161/01.ATV.0000149141.81230.c6. [DOI] [PubMed] [Google Scholar]
- 11.Li P, Oparil S, Feng W, Chen YF. Hypoxia-responsive growth factors upregulate periostin and osteopontin expression via distinct signaling pathways in rat pulmonary arterial smooth muscle cells. J Appl Physiol. 2004;97(4):1550–8. doi: 10.1152/japplphysiol.01311.2003. [DOI] [PubMed] [Google Scholar]
- 12.Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002;62(18):5358–64. [PubMed] [Google Scholar]
- 13.Butcher JT, Norris RA, Hoffman S, Mjaatvedt CH, Markwald RR. Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev Biol. 2007;302(1):256–66. doi: 10.1016/j.ydbio.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baril P, Gangeswaran R, Mahon PC, et al. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene. 2007;26(14):2082–94. doi: 10.1038/sj.onc.1210009. [DOI] [PubMed] [Google Scholar]
- 15.Norris RA, Damon B, Mironov V, et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. 2007;101(3):695–711. doi: 10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding Q, Gladson CL, Wu H, Hayasaka H, Olman MA. Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J Biol Chem. 2008;283(40):26839–49. doi: 10.1074/jbc.M803645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jalvy S, Renault MA, Leen LL, et al. Autocrine expression of osteopontin contributes to PDGF-mediated arterial smooth muscle cell migration. Cardiovasc Res. 2007;75(4):738–47. doi: 10.1016/j.cardiores.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–32. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 19.Hauck CR, Hsia DA, Schlaepfer DD. Focal adhesion kinase facilitates platelet-derived growth factor-BB-stimulated ERK2 activation required for chemotaxis migration of vascular smooth muscle cells. J Biol Chem. 2000;275(52):41092–9. doi: 10.1074/jbc.M005450200. [DOI] [PubMed] [Google Scholar]
- 20.Sayers RL, Sundberg-Smith LJ, Rojas M, et al. FRNK expression promotes smooth muscle cell maturation during vascular development and after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28(12):2115–22. doi: 10.1161/ATVBAHA.108.175455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varadarajulu J, Laser M, Hupp M, Wu R, Hauck CR. Targeting of alpha(v) integrins interferes with FAK activation and smooth muscle cell migration and invasion. Biochem Biophys Res Commun. 2005;331(2):404–12. doi: 10.1016/j.bbrc.2005.03.175. [DOI] [PubMed] [Google Scholar]
- 22.Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52(3):372–86. doi: 10.1016/s0008-6363(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 23.Clowes AW, Clowes MM, Au YP, Reidy MA, Belin D. Smooth muscle cells express urokinase during mitogenesis and tissue-type plasminogen activator during migration in injured rat carotid artery. Circ Res. 1990;67(1):61–7. doi: 10.1161/01.res.67.1.61. [DOI] [PubMed] [Google Scholar]
- 24.Tulis DA. Histological and morphometric analyses for rat carotid balloon injury model. Methods Mol Med. 2007;139:31–66. doi: 10.1007/978-1-59745-571-8_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kappert K, Blaschke F, Meehan WP, et al. Integrins alphavbeta3 and alphavbeta5 mediate VSMC migration and are elevated during neointima formation in the rat aorta. Basic Res Cardiol. 2001;96(1):42–9. doi: 10.1007/s003950170076. [DOI] [PubMed] [Google Scholar]
- 26.Yue TL, McKenna PJ, Ohlstein EH, et al. Osteopontin-stimulated vascular smooth muscle cell migration is mediated by beta 3 integrin. Exp Cell Res. 1994;214(2):459–64. doi: 10.1006/excr.1994.1282. [DOI] [PubMed] [Google Scholar]
- 27.Stouffer GA, Hu Z, Sajid M, et al. Beta3 integrins are upregulated after vascular injury and modulate thrombospondin- and thrombin-induced proliferation of cultured smooth muscle cells. Circulation. 1998;97(9):907–15. doi: 10.1161/01.cir.97.9.907. [DOI] [PubMed] [Google Scholar]
- 28.Kokubo T, Uchida H, Choi ET. Integrin alpha(v)beta(3) as a target in the prevention of neointimal hyperplasia. J Vasc Surg. 2007;45:A33–38. doi: 10.1016/j.jvs.2007.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kudo Y, Ogawa I, Kitajima S, et al. Periostin promotes invasion and anchorage-independent growth in the metastatic process of head and neck cancer. Cancer Res. 2006;66(14):6928–35. doi: 10.1158/0008-5472.CAN-05-4540. [DOI] [PubMed] [Google Scholar]
- 30.Wallace DP, Quante MT, Reif GA, et al. Periostin induces proliferation of human autosomal dominant polycystic kidney cells through alphaV-integrin receptor. Am J Physiol Renal Physiol. 2008;295(5):F1463–71. doi: 10.1152/ajprenal.90266.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han M, Wen JK, Zheng B, Liu Z, Chen Y. Blockade of integrin beta3-FAK signaling pathway activated by osteopontin inhibits neointimal formation after balloon injury. Cardiovasc Pathol. 2007;16(5):283–90. doi: 10.1016/j.carpath.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99(1):35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 33.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 34.Shimazaki M, Nakamura K, Kii I, et al. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205(2):295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao R, Bao S, Bai X, et al. Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol Cell Biol. 2004;24(9):3992–4003. doi: 10.1128/MCB.24.9.3992-4003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.