

Abstract
Our laboratory is interested to develop oncolytic adenoviral vectors that can be administered systemically for the treatment of breast cancer. To restrict viral replication in breast tumor cells, we have constructed mhTERTAd.sTβRFc, a 01/07 based adenoviral vector expressing the soluble form of TGFβ receptor II fused with human Fc IgG1 (sTGFβRIIFc) gene, in which viral replication is under the control of modified human telomerase reverse transcriptase (mhTERT) promoter. In addition, mhTERTAd.sTβRFc-mediated sTGFβRIIFc production would target growth factor-β (TGFβ) pathway known to contribute to the tumor progression breast cancer metastasis. We chose to use mhTERT promoter because it was found to be relatively more active (approximately 20-times) in breast cancer cells compared to normal human cells. We showed that infection of MDA-MB-231 and MCF-7 breast cancer cells for 48 hrs with mhTERTAd.sTβRFc produced high levels of sTGFβRIIFc (greater than 1 μg/ml) in the medium. Breast cancer cells produced nearly 6,000-fold increase in the viral titers during 48 hrs infection period. However, mhTERTAd.sTβRFc replication was attenuated in normal cells. Infection of breast cancer cells with a replication deficient virus Ad(E1-).sTβRFc also produced high levels of sTGFβRIIFc, but under these conditions no detectable viral replication was observed. Adenoviral-mediated production of sTGFβRIIFc was shown to bind with TGFβ-1, and abolished the effects of TGFβ-1 on downstream SMAD-3 phosphorylation. The administration of mhTERTAd.sTβRFc intravenously into MDA-MB-231 human xenograft bearing mice resulted in significant inhibition of tumor growth, and production of sTGFβRIIFc in the blood. On the other hand, intravenous injection of Ad(E1-).sTβRFc did not exhibit significant inhibition of the tumor growth, but resulted in the sTGFβRIIFc in the blood, suggesting that viral replication along with sTGFβRIIFc protein production play a critical role in inducing inhibition of tumor growth. These results warrant future investigation of mhTERTAd.sTβRFc as an anti-tumor agent in vivo.
Introduction
Nearly 200,000 women are diagnosed with breast cancer resulting in approximately 30,000 deaths each year in the United States.1 Thus, there is a tremendous need to develop novel approaches to treat breast cancer. In recent years, tumor specific replicating adenoviruses have been proposed as potential gene therapy vehicles to target cancer.2-5 Our long-term goals are to develop gene therapy approaches to treat breast cancer, and have chosen to develop oncolytic adenoviral (dl01/07 based) vectors. The adenoviral mutant 01/07 has two deletions in the E1A (Rb and p300 binding) and replicates in cancer cells regardless of their genetic background.6 In addition, we would target growth factor-β (TGFβ) pathway because high levels of TGFβ protein and activated SMAD signaling are known to contribute to tumor progression, and are associated with metastasis in breast cancer patients.7-14 Recently, we have developed an oncolytic adenoviral vector (dl01/07 based) expressing the soluble form of TGFβ receptor II fused with human Fc IgG1 (sTGFβRIIFc) (Ad.sTβRFc).6 To further restrict dl01/07 viral replication in breast tumor cells, we report here the construction of mhTERTAd.sTβRFc, a modified 01/07 based adenoviral vector expressing sTGFβRIIFc gene in which viral replication is under the control of a modified human telomerase reverse transcriptase (mhTERT) promoter. To examine the potential application of mhTERTAd.sTβRFc as an anti-tumor agent, we have conducted in vitro and in vivo experiments using mhTERTAd.sTβRFc in human breast cancer models. Our data presented here shows that it is feasible to create mhTERTAd.sTβRFc; achieve mhTERTAd.sTβRFc replication in breast cancer cells, and simultaneously produce sTGFβRIIFc that can inhibit TGFβ signaling in human breast cancer cells, and that intravenous administration of mhTERTAd.sTβRFc can inhibit tumor growth. These results warrant future investigation of mhTERTAd.sTβRFc as an anti-tumor agent in vivo.
Materials and Methods
Cell culture
HEK-293, MCF-7, IMR-90 cells were obtained from ATCC, MDA-MB-231 cells were obtained as described earlier.13 Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Mediatech, Inc., Manassas, VA) containing 10% fetal bovine serum (FBS) (Mediatech, Inc., Manassas, VA) and 1% penicillin/streptomycin (Invitrogen).
Construction of Recombinant viral vectors
To create mhTERTAd.sTβRFc, A 0.7 kb fragment of mhTERT15 was cloned in p01/07 shuttle plasmid to produce p01/07mhTERT shuttle vector in which E1A 01/07 gene was controlled by mhTERT promoter. The shuttle vector (PmeI digested) was recombined with PacI cut p01/07/sTβRFc in E. coli BJ5183 to produce pmhTERT01/07/sTβRFc viral backbone plasmid. The resulting plasmid was digested with PacI and transfected into HEK-293 cells to generate mhTERTAd.sTβRFc adenovirus. Ad(E1-).sTβRFc, an E1 minus, replication-deficient adenovirus expressing sTGFβRIIFc was created by homologous recombination procedures using established methods (Clontech, Palo Alto, CA). A replication-deficient adenorvirus devoid of any transgene Ad(E1-).Null has been described previously. 16 The adenoviruses were grown in HEK-293 cells and purified by double CsCl2 density gradient.
Western blot analysis of sTGFβRIIFc produced by mhTERTAd.sTβRFc or Ad(E1-).sTβRFc -infected cells
Breast tumor cells (0.2×106 cells per well in 6-well plates) were plated in DMEM containing 10% FBS and incubated at 37°C overnight. The next morning, cells were infected with 100 plaque forming units (pfu)/cell of mhTERTAd.sTβRFc, Ad(E1-).sTβRFc or Ad(E1-).Null for 24 hours. Cells were washed and incubated with DMEM without FBS for 24 hours. Media and cells were subjected to Western blot analysis as previously described16,17. Blots were probed with antibody reactive against TGFβRII (H-567; Santa Cruz Biotechnology, Santa Cruz, CA) or actin protein (I-19; Santa Cruz Biotechnology, Santa Cruz, CA).
ELISA assays of sTGFβRIIFc produced by mhTERTAd.sTβRFc and Ad(E1-).sTβRFc - infected cells
Supernatants obtained from the mhTERTAd.sTβRFc, Ad(E1-).sTβRFc or Ad(E1-).Null infected cells were analyzed for sTGFβRIIFc expression by ELISA using published method.18 In brief, 96-well plates (Nunc, Roskilde, Denmark) were coated with the anti-human IgG-Fc specific capture antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), incubated with various dilutions of the samples, followed by detection with biotinylated anti-human TGFβ RII antibody (R&D systems, Minneapolis, MN). The detection was carried out with streptavidin conjugate peroxidase using TMB/HRP substrate (BioFX Laboratories, Owing Mills, MD). After stopping the reaction with 1N HCl, the absorbance was measured at 450 nm using a SPECTRA max Plus ELISA plate reader (Molecular Devices, Sunnyvale, CA). Standard curves of sTGFβRIIFc were used to calculate the sTGFβRIIFc concentrations in the test samples.
Affinity purification of sTGFβRIIFc and binding of sTGFβRIIFc and TGFβ1
MDA-MB-231 cells were infected with Ad.sTβRFc (100 pfu/cell) for 24 hrs. Medium was changed into serum free medium for additional 24 hrs. The medium was collected and applied to Protein A Sepharose column. The sTGFβRIIFc was eluted with 100mM glycine buffer (pH 4). The eluted protein solution was neutralized by Tris buffer, pH 9.0, dialyzed against 20mM Tris (pH 7.5) and stored at -70°.
For binding of TGFβ-1 with sTGFβRIIFc, TGFβ-1 (Sigma, St. Louis, MO) protein was mixed with sTGFβRIIFc, and incubated for 1 hr at 4°C. The complexes were mixed with Protein A Sepharose beads (Vector Laboratories, Burlingame, CA), and incubated at 4°C for 1 hr. Samples were centrifuged at 13,000g, 4°C, 1 min, and the beads washed 4 times with wash buffer (50mM NaCl, 10 mM Tris-Cl, 5 mM EDTA, 1% Triton X-100 pH 7.4). Samples were resuspended in elution buffer (100mM glycine buffer pH 4) and centrifuged at 4°C, 5 min. Proteins were neutralized with TrisCl (pH 9.0), subjected to Western blot analysis and probed with anti-TGFβ-1 (R & D systems, Minneapolis, MN).
SMAD phosphorylation studies
Cells were plated in 6-well plate (4×105 cells/well), and starved for 24 hours in serum free media. The cells were then washed with serum free media before treatment with the indicated concentrations of recombinant TGFβ-1 (Sigma, St. Louis, MO) for one hour. Cells were lysed in 250 μL of lysis buffer containing 0.1mM phenylmethylsulfonyl fluoride, sonicated and stored at -80°C until further use. Equal amounts of cell lysates were separated by SDS-PAGE on 10% acrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked in Blotto A for one hour at room temperature and treated with phospho-SMAD3 or SMAD2/3 primary antibodies (Cell Signaling, Danvers, MA) followed by horseradish peroxidase-conjugated bovine anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The blots were visualized by enhanced chemiluminescence substrate (Amersham Biosciences, Piscataway, NJ).
Viral titer assay
Cells were plated in six-well plates at about 70% confluence and the viral titers were examined using the published method6,19 with some modifications. Cells were infected with mhTERTAd.sTβRFc, Ad(E1-).sTβRFc or Ad(E1-).Null for 3 hrs at a multiplicity of infection (MOI) of 100. Cells were washed with DMEM and incubated in 1 ml DMEM for additional one hr at 37°C. At the end of the incubation, cells were washed and either collected in 0.5 ml growth media and frozen at -70°C, or were maintained in 2 ml of growth media for additional 48 hrs. Media and cells were collected, and cells were subjected to three cycles of freezing and thawing to release the viruses. The total viruses from media and cells were serially diluted and added to monolayers of HEK-293 cells respectively. After 3 hrs of incubation at 37°C, the infected HEK-293 cells were overlaid with 3 ml of 1.25% SeaPlaque agarose (Cambrex, East Rutherford, NJ) in growth media. Plaques were counted following 7-8 days incubation using published methods.20 To calculate the viral burst size, viral titers at 48 hrs were divided by the viral titers present in 3 hrs samples of various cell lines.
β-gal assays
Cells were plated in 6-well plate (105 cell per well), and next day were infected with adenoviral vectors expressing β-galactosidase (β-gal) for 24 hrs and examined for β-gal as described earlier.16,21 At the end of the incubation, cells were washed three times with PBS (pH 7.5) and lysed in 200 μl of 0.1 M Tris (pH 7.5) containing 0.1% Triton x-100. An aliquot (30 μl) was assayed for β-gal activity. Samples were treated with 100 μl 20 mM Tris (pH 7.5) containing 1 mM MgCl2, 450 mM β mercaptoethanol, and 150 mM O-nitrophenyl-3-galactopyranoside. Incubations were performed at 37°C for 20 min, and the reaction was stopped by the addition of 100 μl/well of 1 M Na2CO3. The absorbance was determined at 420 nm. An A420 of 1.0 was defined as 1.0 unit of enzyme activity.
Evaluating anti-tumor responses in vivo in breast tumor model
MDA-MB-231 cells were injected subcutaneously in 4 weeks old nude mice (Nu/Nu) (5 × 106 cells/mouse). After about a week, when the tumor sizes reached about 50 mm3 (day 0), mhTERTAd.sTβRFc or Ad(E1-).sTβRFc or Ad(E1-).Null vectors were injected into the tail vein (2×108 pfus in 0.1 ml buffer per injection, two injections total on days 0 and 3). Tumor sizes were measured once a week by a digital caliper. Tumor volumes were calculated using the formula (a × b2) × 0.523, whereby a is the larger dimension, and b is the smaller dimension. At the end of the experiment blood was collected by intracardiac puncture. All animal protocols were approved by IACUC of the NorthShore Research Institute.
Statistical analysis
For in vitro studies, experimental groups were compared by t-tests, two tailed analysis, using GraphPad Prism software version 5 (GraphPad Software, San Diego, CA). For in vivo studies, comparisons among different treatment groups were done by the two-way ANOVA method and the Bonferroni post hoc test using the commercial software GraphPad Prism 5 (GraphPad Software, San Diego, CA). P < 0.05 was considered to be significant difference. All the data points were presented as mean ± SE (standard error).
Results
mhTERT promoter is much more active in breast tumor cells compared to the normal cells
Before creating mhTERT promoter-driven oncolytic viruses, we wanted to examine whether mhTERT promoter would have enhanced transcriptional activity in breast tumor cells as compared to the normal cells. Breast tumor (MDA-MB-231, MCF-7) and normal human cells (IMR-90) were exposed to E1 deleted Ad(E1-).mhTERTLacZ in which mhTERT promoter drives the expression of a marker β-gal gene15, or E1 deleted Ad(E1-).CMVLacZ in which CMV promoter drives β-gal expression15. β-gal activity was quantified, and the results are shown in Fig 1. All three cell lines produced high levels of β-gal activity when infected with Ad(E1-).CMVLacZ. β-gal activity was relatively lower in each cell line infected with Ad(E1-).mhTERTLacZ. However, the β-gal activity was markedly reduced in IMR-90 cells (Fig. 1A). To evaluate the relative activities of the CMV and mhTERT promoters, the ratios of β-gal activities obtained from Ad(E1-)CMVLacZ vs. Ad(E1-)mhTERTLacZ viruses were calculated in each cell line and are shown in Fig.1 B. While, CMV promoter was 6.1 and 7.7 fold more active than mhTERT promoter in MDA-MB-231 and MCF-7 breast tumor cells respectively, it was 115- fold more active than mhTERT promoter in IMR-90 normal cells (Fig.1 B). These results indicate that mhTERT promoter incorporated in adenovirus backbone is about 20-times more active in breast cancer cells compared to normal human cells (p values < 0.05), favoring the use of mhTERT promoter in the oncolytic adenoviruses.
Fig. 1.

A. CMV and mhTERT promoter driven β-gal activities in breast tumor and normal cells. Various cells were plated in 6-wells (2×104 cells/well) and infected with Ad(E1-).CMVLacZ or Ad(E1-).mhTERTLacZ viruses (100 pfus/cell). Forty eight hrs later, cells were lysed and β-gal activities measured by staining with o-nitrophenyl-β-D-galactopyranoside (ONPG) using published methods16,21. Shown are the averages of triplicate samples (±SE). B. Ratios of β-gal obtained from CMV promoter-directed and mhTERT promoter-directed β-gal activities. Ratios shown were obtained from Panel A. These ratios were significantly lower in IMR-90 cells compared to MDA-MB-231 or MCF-7 cells (p values < 0.05).
Construction of mhTERT driven 01/07 based oncolytic adenovirus
To examine the use of mhTERT promoter in the context of 01/07 oncolytic viral replication, an oncolytic adenovirus mhTERTAd.sTβRFc was constructed using homologous recombination method (Fig. 2). In this viral construct, mhTERT promoter regulates E1A expression. The key elements in this viral genome are mhTERT driving a mutant E1A region (01/07) and the expression cassette containing sTGFβRIIFc (part of E3 containing adenovirus death protein is left intact) (Fig. 2). Rest of the genomic structure of mhTERTAd.sTβRFc is identical to wild type Ad5.
Fig. 2.

A. Construction of mhTERTAd.sTβRFc. A 0.7 kb fragment of mhTERT was cloned in p01/07 shuttle plasmid to produce p01/07mhTERT shuttle vector in which E1A 01/07 gene was controlled by mhTERT promoter. The shuttle vector (PmeI cut) was recombined with PacI cut p01/07/sTβRFc in E. coli BJ5183 to produce pmhTERT01/07/sTβRFc viral backbone plasmid. The resulting plasmid was cut with PacI and transfected into HEK-293 cells to generate mhTERTAd.sTβRFc adenovirus. The Adenovirus was grown in HEK293 cells and purified by double CsCl2 density gradient. B. Schematic structure of recombinant mhTERTAd.sTβRFc. The key elements in this viral genome are mhTERT promoter driving a mutant E1A region (01/07). The expression cassette containing sTGFβRIIFc was introduced in E3 region. Rest of the genome (E1B, E2B, L1-L5, and E4) is similar to wild type Ad5.
Infection of human breast cancer cells with mhTERTAd.sTβRFc and Ad(E1-).sTβRFc produce sTGFβRIIFc that is secreted into the medium
To investigate if the infection of breast cancer cells with mhTERTAd.sTβRFc and Ad(E1-).sTβRFc can produce sTGFβRIIFc protein, breast cancer cells were infected with mhTERTAd.sTβRFc or Ad(E1-).sTβRFc and the protein expression was examined using Western blot analysis. As shown in Fig. 3A, infection of MDA-MB-231 and MCF-7 cells with mhTERTAd.sTβRFc showed 60-80 Kd protein band of sTGFβRIIFc. Similarly, the extracellular media of mhTERTAd.sTβRFc also showed the presence of sTGFβRIIFc. Infection of cells with Ad(E1-).sTβRFc also produced sTGFβRIIFc as shown by the presence of protein band in the Ad(E1-).sTβRFc-infected cells. However, sTGFβRIIFc protein was not detectable in the cell lysates or the media of the tumor cells infected with a Ad(E1-).Null (Fig. 3A). The amounts of sTGFβRIIFc in the supernatants of mhTERTAd.sTβRFc and Ad(E1-).sTβRFc -infected breast cancer cells were quantified by ELISA method. These amounts were found to be 2.22 μg/ml and 1.13 μg/ml in MDA-MB-231 and MCF-7 cells respectively in mhTERTAd.sTβRFc infected cells (Fig. 3B). Similar amounts of sTGFβRIIFc were found in Ad(E1-).sTβRFc-infected MDA-MB-231 and MCF-7 cells (Fig. 3B). These results indicate that infection of breast tumor cells with mhTERTAd.sTβRFc or Ad(E1-).sTβRFc can produce sTGFβRIIFc that is subsequently released into the media. Thus, incorporation of mhTERT promoter driving E1A expression, did not adversely affect the expression of sTGFβRIIFc.
Fig. 3.
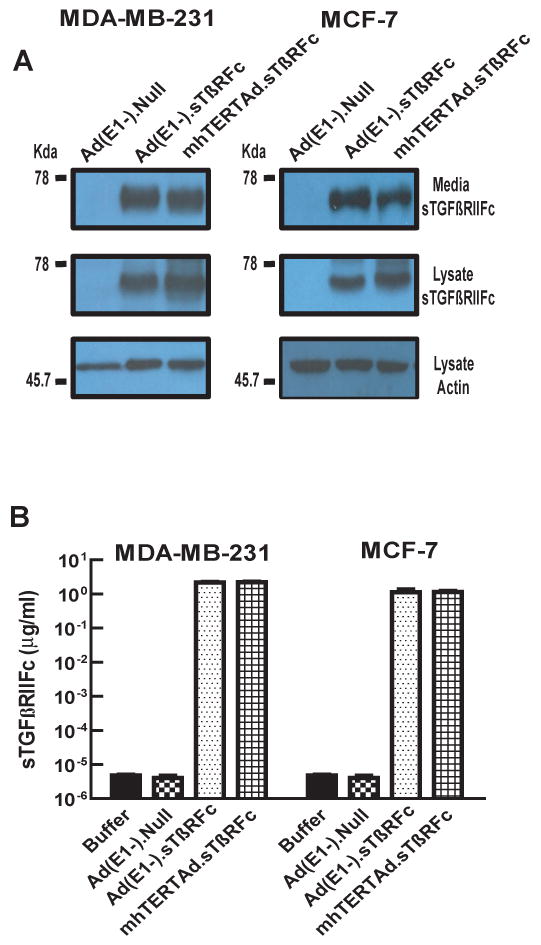
mhTERTAd.sTβRFc and Ad(E1-).sTβRFc -mediated expression of sTGFβRIIFc protein in breast cancer cells. Breast cancer cells were infected with either mhTERTAd.sTβRFc, Ad(E1-).sTβRFc or Ad(E1-).Null (100 pfu/cell) for 24 hrs. At the end of the incubation, media were changed into serum free media, and the incubations continued for an additional 24 hrs. A. Western blot analysis of sTGFβRIIFc protein. Cells and media were subjected to Western blot analysis for sTGFβRIIFc expression and are shown in the Figure. B. ELISA assays of sTGFβRIIFc protein. Aliquots of media samples were analyzed for sTGFβRIIFc by ELISA as described in materials and methods. Shown are the averages of triplicate samples (±SE).
mhTERTAd.sTβRFc-dependent production of sTGFβRIIFc in breast cancer cells can bind with TGFβ-1 and inhibit TGFβ-dependent SMAD-3 phosphorylation
To examine the binding capacity of sTGFβRIIFc with TGFβ, sTGFβRIIFc was incubated with TGFβ-1 for 30 minutes at room temperature and mixed with Protein A sepharose beads. The beads were centrifuged, the sTGFβRIIFc-TGFβ complexes were eluted by denaturing buffer and subjected to Western blot analysis. The blots were probed with anti-TGFβ-1 antibody. The detection of TGFβ-1 in the precipitate from sTGFβRIIFc-TGFβ complexes indicating that secreted sTGFβRIIFc can form complexes with TGFβ-1 (Fig 4A).
Fig. 4.
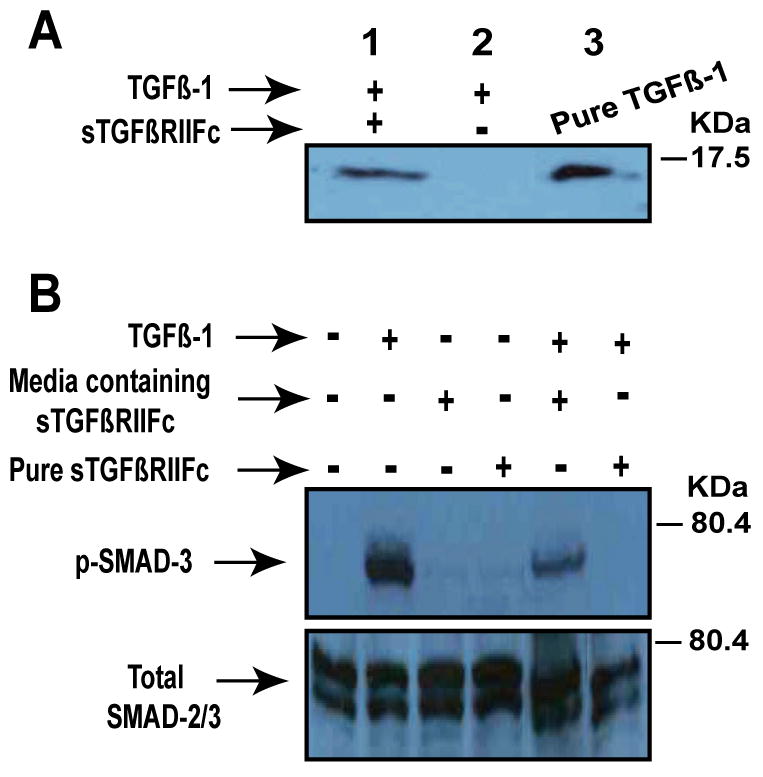
A. The binding of TGβ-1 with sTGFβRIIFc. One μg of sTGFβRIIFc was mixed with 40 ng of TGFβ-1 (Sigma, St. Louis, MO) for 1 hour at 4°C and then incubated with Protein-A Sepharose beads (Vector Laboratories, Burlingame, CA) for 1 hour at 4°C. The bound TGFβ-1 was released with acidic buffer as described in materials and methods. The samples were subjected to Western blot analysis and probed with rabbit anti-TGFβ-1 polyclonal antibody (R & D systems, Minneapolis, MN). Note TGFβ-1 band is present when TGFβ-1 was pre-incubated with sTGFβRIIFc (lane 1), but not without sTGFβRIIFc (Lane 2). Lane 3 received pure TGFβ-1 as a positive control for Western blot. B. Inhibition of TGFβ-dependent SMAD-3 phosphorylation by sTGFβRIIFc. MDA-MB-231 cells were plated in 6-well plates (4×105 cells/well). Next day, media was changed and cells were incubated with TGFβ-1 (1 ng/ml) in the absence or presence of media containing sTGFβRIIFc or purified sTGFβRIIFc (100 ng/ml) for 60 minutes. Cell lysates were prepared and further subjected to Western blot analysis. Blots were probed with phospho SMAD3 antibody, or Smad2/3 antibody (Cell Signaling, Danvers, MA).
In order to examine if the mhTERTAd.sTβRFc-mediated sTGFβRIIFc expression can lead to the inhibition of TGFβ-dependent signal transduction, the effect of sTGFβRIIFc on the inhibition of TGFβ-dependent SMAD-3 phosphorylation was examined. MDA-MB-231 cells were exposed to TGFβ-1 in the absence or presence of the extracellular media containing the sTGFβRIIFc or the purified sTGFβRIIFc protein. As shown in Fig. 4B, TGFβ-1 induced SMAD-3 phosphorylation in MDA-MB-231 cells, and this phosphorylation was inhibited by the crude media containing sTGFβRIIFc, as well as by the purified sTGFβRIIFc (Fig. 4B). These results suggest that sTGFβRIIFc derived form mhTERTAd.sTβRFc-infected breast tumor cells is functionally active.
mhTERTAd.sTβRFc can replicate in human breast cancer cells, but has reduced replication potential in normal cells
Next, we examined the replication potential of mhTERTAd.sTβRFc in breast cancer cells. Breast cancer cells were exposed to mhTERTAd.sTβRFc and the viral titers measured at 3 hrs (basal level) and at 48 hrs. As shown in Fig. 5A, in MDA-MB-231 cells, at 3 hrs, the mhTERTAd.sTβRFc viral titer was 5.17×102. This titer was increased to 3.2×106 in 48 hrs, indicating a 6,189 fold increase in viral titer. The increase in viral titers from 3 hrs to 48 hrs is described here as the “viral burst size” (Fig. 5A, B). Similar increase (6,285-fold) in burst size was observed in MCF-7 cells (Fig. 5B). On the other hand, infection of MDA-MB-231 or MCF-7 cells with Ad(E1-).sTβRFc or Ad(E1-).Null resulted in less than 2-fold increase in viral burst size (data not shown), suggesting that Ad(E1-).sTβRFc is replication-deficient in the breast cancer cells. Using these viral replication assays, burst size of mhTERTAd.sTβRFc was also compared in normal human cells. The burst size of mhTERTAd.sTβRFc was about 16-fold lower in IMR-90 cells compared to the breast tumor cells (Fig. 5A, B). The burst size of mhTERTAd.sTβRFc was about 16-fold higher in breast tumor cells compared to IMR-90 cells (p <0.001) (Fig. 5 B). These results indicate mhTERTAd.sTβRFc replication is attenuated in normal cells while maintaining the replication potential in the breast tumor cells.
Fig. 5.

A. mhTERTAd.sTβRFc titers in breast tumor and normal human cells. MDA-MB-231, MCF-7 and IMR-90 cells were plated in 6-well dishes (2×105 cells/well). Cells were infected with mhTERTAd.sTβRFc (100 pfu/cell) for either 3 hrs or 48 hrs as described in Materials and Methods. Cell lysates and supernatants were collected and viral titers were measured by plaque forming assays in HEK-293 cells. Results shown are average of three determinations ± SE. B. Comparison of viral burst sizes. Shown are the increases in the viral titers during 48 hrs infection (burst sizes) of the mhTERT.AdsTβRFc in MDA-MB-231, MCF-7 and IMR-90 cells. The mhTERT.AdsTβRFc burst size in IMR-90 cells was significantly lower compared to burst sizes in MDA-MB-231 or MCF-7 cells (p values < 0.001).
Intravenous delivery of mhTERTAd.sTβRFc into MDA-MB-231 tumors bearing mice causes inhibition of tumor growth, and produces high levels of sTGFβRIIFc in the blood
Next, we conducted experiments to evaluate in vivo efficacy of mhTERTAd.sTβRFc in a human breast xenograft model. MDA-MB-231 human xenograft tumors were established in nude mice. mhTERTAd.sTβRFc, Ad(E1-).sTβRFc or Ad(E1-).Null or buffer were injected into the tail vein. Tumor sizes were measured once a week and are shown in Fig 6A. In the control group of animals, tumors grew rapidly. However, in the group of animals that received mhTERTAd.sTβRFc, there was a significant inhibition of tumor growth over time (p value <0.01 compared to buffer control). Intravenous injection of Ad(E1-).sTβRFc showed some inhibition of tumor growth, however, the inhibition of the tumor growth in Ad(E1-).sTβRFc treated group of animals was not statistically significant compared to buffer treated group (p values >0.05). Ad(E1-).Null had no significant inhibition on tumor growth (p value >0.05) compared to buffer group suggesting that the anti-tumor responses of mhTERTAd.sTβRFc require viral replication in addition to sTGβRIIFc production.
Fig. 6.

A. In vivo evaluation of mhTERTAd.sTβRFc administration on MDA-MB-231 tumor xenograft in nude mice. MDA-MB-231 cells were injected subcutaneously (5×106 cells per mouse). Once tumor sizes reached about 50 mm3, mhTERTAd.sTβRFc (n=13) Ad(E1-).sTβRFc (n= 12), Ad(E1-).Null (n=10) or buffer (n=11) were administered into the tail vein. The virus dose was 2×108 pfus in 0.1 ml buffer per injection; two injections total on days 0 and 3. Tumor volumes were measured using a digital caliper on various days shown. The tumor volumes were calculated using the formula (a × b2) × 0.523. Results show the tumor volumes in animals that received intravenous injections of mhTERTAd.sTβRFc (▲), Ad(E1-).sTβRFc (■) Ad(E1-).Null (▼)or buffer (●). The tumor growth of various groups were compared with buffer control groups using two-way Anova, and further evaluated by Bonferroni post hoc test using Graph Pad Prism 5 software. Compared to buffer group the p value for mhTERTAd.sTβRFc group is <0.01 (*), p value for Ad(E1-).sTβRFc is >0.05, and p value for Ad(E1-).Null is >0.05. B. Blood levels of sTGFβRIIFc in mhTERTAd.sTβRFc or Ad(E1-).sTβRFc treated animals. Blood was collected on day 35 (Fig. 6A) and analyzed for the expression of sTGFβRIIFc by ELISA. Shown are the sTGFβRIIFc levels in animals that received the buffer, Ad(E1-).Null, Ad(E1-).sTβRFc or mhTERTAd.sTβRFc.
To examine if the intravenous delivery of mhTERTAd.sTβRFc in the experiments described above produces high levels of sTGFβRIIFc, blood samples were collected at the end of the experiment (day 35). Aliquots of blood samples were analyzed for the presence of sTGFβRIIFc using ELISA method described in Materials and Methods. In mhTERTAd.sTβRFc treated group, the levels of sTGFβRIIFc present in the blood were 17.64 ± 3.99 mg/ml (Fig 6B). Similarly, E1-Ad.sTβRFc treated group also produced high levels (9.45 ± 2.59 mg/ml) of sTGFβRIIFc (Fig. 6B). As expected animals that received buffer or Ad(E1-).Null had very low levels sTGFβRIIFc (less than 5 pg/ml) in the blood. These results show that the intravenous delivery of mhTERTAd.sTβRFc as well as sTGFβRIIFc into the mice results in the release of sTGFβRIIFc protein in the blood. However, since mhTERTAd.sTβRFc is more effective as an anti-tumor agent, suggests that perhaps viral replication as well as sTGFβRIIFc production play an important role in producing inhibition of tumor growth following intravenous delivery of the viral vectors.
Discussion
There is a tremendous need to create novel approaches to treat breast cancer. In recent years tumor specific replicating adenoviruses have been proposed as potential gene therapy vehicles to target cancer 2-5. Given that breast caner often metastasizes to other organs, there is a need to develop replicating adenoviruses that could be administered systemically for the treatment of breast cancer. 5 Results presented here indicated that modified hTERT promoter is relatively more specific than CMV promoter for targeting breast cancer, strengthening the idea to use oncolytic adenoviruses in which E1A expression would be controlled by mhTERT promoter. mhTERTAd.sTβRFc virus was created by the homologous recombination procedures previously established in our laboratory. In vitro results suggested that in cancer cells supported the mhTERTAd.sTβRFc replication and the viral replication was attenuated in normal cells, indicating a potential advantage to use mhTERTAd.sTβRFc for in vivo research. Another important feature of mhTERTAd.sTβRFc is that it has retained the capacity to express the sTGFβRIIFc protein. These results therefore, suggest that mhTERT-driven E1A expression does not compromise with the ability of mhTERTAd.sTβRFc to replicate or to produce the sTGFβRIIFc in the breast tumor cells.
Another important finding being reported here is that the adenoviral-mediated sTGFβRIIFc produced is functionally active. In vitro assays showed that sTGFβRIIFc can bind with TGFβ-1, and inhibit TGFβ-dependent SMAD-3 phosphorylation. The inhibition of SMAD phosphorylation production are of particular significance as higher levels of SMAD phosphorylation is linked with the progression of breast cancer metastasis.10,14,22,23 We also report that systemic administration of mhTERTAd.sTβRFc via tail vein causes significant inhibition of tumor growth of human xenograft model in nude mice. The mhTERTAd.sTβRFc treated animals also produce high levels of sTGFβRIIFc in the blood, although, in this limited set of animals, it is difficult to correlate mhTERTAd.sTβRFc-mediated sTGFβRIIFc expression and in vivo anti-tumor responses of the local tumors. An important point to note that a replication-defective virus when injected systemically, though also produced high levels of sTGFβRIIFc in the blood, was less effective than mhTERTAd.sTβRFc in inhibiting the tumor growth. These results clearly suggest that while the viral replication is critical for anti-tumor responses at least in this particular tumor model and the route of viral injection. Since TGFβ appears to play a major role in breast cancer associated bone metastasis7-14. Our future studies will be designed to examine the effect of mhTERTAd.sTβRFc-mediated sTGFβRIIFc protein production in causing the inhibition of bone metastasis and osteolytic bone destruction.
Acknowledgments
We thank Dr. Janardan D. Khandekar for his support of this research. We are thankful to Dr. ZG Wang and Murali Ramachandra for their help, and Monica Tsang (R & D systems) for providing sTGFβRIIFc gene. This work was supported by NIH grant R01CA127380 (P. Seth).
References
- 1.Cancer Facts and Figures 2009. American Cancer Society; 2009. http://www.cancer.org/docroot/STT/stt_0.asp. [Google Scholar]
- 2.Seth P, editor. Adenoviruses : Basic Biology to Gene Therapy. R G Landes Company; Austin, TX: 1999. [Google Scholar]
- 3.McCormick F. Cancer gene therapy: fringe or cutting edge? Nat Rev Cancer. 2001;1:130–141. doi: 10.1038/35101008. [DOI] [PubMed] [Google Scholar]
- 4.Seth P. Vector-mediated cancer gene therapy: an overview. Cancer Biol Ther. 2005;4:512–517. doi: 10.4161/cbt.4.5.1705. [DOI] [PubMed] [Google Scholar]
- 5.McCormick F. Future prospects for oncolytic therapy. Oncogene. 2005;24:7817–7819. doi: 10.1038/sj.onc.1209064. [DOI] [PubMed] [Google Scholar]
- 6.Seth P, et al. Development of oncolytic adenovirus armed with a fusion of soluble transforming growth factor-beta receptor II and human immunoglobulin Fc for breast cancer therapy. Hum Gene Ther. 2006;17:1152–1160. doi: 10.1089/hum.2006.17.1152. [DOI] [PubMed] [Google Scholar]
- 7.Teicher BA. Malignant cells, directors of the malignant process: role of transforming growth factor-beta. Cancer Metastasis Rev. 2001;20:133–143. doi: 10.1023/a:1013177011767. [DOI] [PubMed] [Google Scholar]
- 8.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 10.Kang Y, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A. 2005;102:13909–13914. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kingsley LA, Fournier PG, Chirgwin JM, Guise TA. Molecular biology of bone metastasis. Mol Cancer Ther. 2007;6:2609–2617. doi: 10.1158/1535-7163.MCT-07-0234. [DOI] [PubMed] [Google Scholar]
- 12.Kozlow W, Guise TA. Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia. 2005;10:169–180. doi: 10.1007/s10911-005-5399-8. [DOI] [PubMed] [Google Scholar]
- 13.Yin JJ, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akhtari M, et al. Biology of breast cancer bone metastasis. Cancer Biol Ther. 2007;7 doi: 10.4161/cbt.7.1.5163. [DOI] [PubMed] [Google Scholar]
- 15.Kim E, et al. Ad-mTERT-delta19, a conditional replication-competent adenovirus driven by the human telomerase promoter, selectively replicates in and elicits cytopathic effect in a cancer cell-specific manner. Hum Gene Ther. 2003;14:1415–1428. doi: 10.1089/104303403769211637. [DOI] [PubMed] [Google Scholar]
- 16.Katayose D, et al. Cytotoxic effects of adenovirus-mediated wild-type p53 protein expression in normal and tumor mammary epithelial cells. Clin Cancer Res. 1995;1:889–897. [PubMed] [Google Scholar]
- 17.Craig C, et al. A recombinant adenovirus expressing p27Kip1 induces cell cycle arrest and loss of cyclin-Cdk activity in human breast cancer cells. Oncogene. 1997;14:2283–2289. doi: 10.1038/sj.onc.1201064. [DOI] [PubMed] [Google Scholar]
- 18.Yang YA, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607–1615. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang ZG, Zhao W, Ramachandra M, Seth P. An oncolytic adenovirus expressing soluble transforming growth factor-beta type II receptor for targeting breast cancer: in vitro evaluation. Mol Cancer Ther. 2006;5:367–373. doi: 10.1158/1535-7163.MCT-05-0125. [DOI] [PubMed] [Google Scholar]
- 20.Seth P, Higginbotham J. Advantages and disadvantages of multiple different methods of adenoviral vector construction. In: Habib N, editor. Methods in Molecular Medicine. Hunana Press Inc.; Totowa, NJ: 2000. pp. 189–198. [DOI] [PubMed] [Google Scholar]
- 21.Seth P, et al. Adenovirus-mediated gene transfer to human breast tumor cells: an approach for cancer gene therapy and bone marrow purging. Cancer Res. 1996;56:1346–1351. [PubMed] [Google Scholar]
- 22.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 23.Clines GA, Guise TA. Mechanisms and treatment for bone metastases. Clin Adv Hematol Oncol. 2004;2:295–302. [PubMed] [Google Scholar]