

Abstract
The US9 gene of herpes simplex virus 1 encodes a virion tegument protein with a predicted Mr of 10,000. Earlier studies have shown that the gene is not essential for viral replication in cells in culture. We report that (i) US9 forms in denaturing polyacrylamide gels multiple overlapping bands ranging in Mr from 12,000 to 25,000; (ii) the protein recovered from infected cells or purified virions reacts with anti-ubiquitin antibodies; (iii) autoradiographic images of US9 protein immunoprecipitated from cells infected with [35S]methionine-labeled virus indicate that the protein is stable for at least 4 h after entry into cells (the protein was also stable for at least 4 h after a 1-h labeling interval 12 h after infection); (iv) antibody to subunit 12 of proteasomes pulls down US9 protein from herpes simplex virus-infected cell lysates; and (v) the US9 gene is highly conserved among the members of the alpha subfamily of herpes viruses, and the US9 gene product lacks lysines. We conclude that US9 is a lysine-less, ubiquitinated protein that interacts with the ubiquitin-dependent pathway for degradation of proteins and that this function may be initiated at the time of entry of the virus into the cell.
Keywords: conserved protein, tegument
In this report, we show that the herpes simplex virus 1 (HSV-1) protein product of the US9 gene is ubiquitinated and in this form packaged in the virion. We also report that the protein is stable for several hours both in the cell in which it is made and in the cell in which it is translocated during infection; further, we report that immune complexes consisting of proteasomal proteins and an antibody against one of the proteasome subunits pull down US9 protein. The relevant background to this report is as follows.
The US9 ORF is predicted to encode a protein with an Mr of approximately 10,000 (1). The protein is a constituent of the virion tegument (2). In the course of HSV-1 reproductive cycle, tegument proteins are released into the cell subsequent to the fusion of the viral envelope with the plasma membrane. Many of the tegument proteins facilitate the initiation of gene expression, and in many instances, they are dispensable for viral replication at least in cells in culture (reviewed in ref. 3). The function of US9 protein is not known, but its gene, like many others, is dispensable for viral replication in cells in culture (4). A property of US9 protein that led us to investigate ubiquitination is that it forms very wide bands on electrophoresis in denaturing polyacrylamide gels.
Ubiquitin is a small, highly conserved polypeptide covalently attached to target proteins by the sequential activity of three classes of enzymes (reviewed in ref. 5). The resulting hybrid is recognized and degraded in the proteinase complex by the catalytic core, the proteasome (6–8). Although this mechanism has been well characterized, not all of the ubiquitin conjugates follow this pathway, and there is general agreement that ubiquitination, especially if only one molecule of ubiquitin is attached to the target protein, represents a form of posttranslational modification whose function is not fully understood (9, 10). The ubiquitin-mediated protein degradation has been shown to be involved in many cellular mechanisms, from cell cycle regulation to break down of proteins for major histocompatibility complex (MHC) class 1 antigen presentation to T cells (reviewed in refs. 11 and 12).
Ubiquitin and the ubiquitin pathway are common targets of viral products (13). With respect to HSV, of particular interest is the observation that viral-infected cell protein No. 0 (ICP0) has recently been shown to bind and stabilize cyclin D3 (14) and also to bind a ubiquitin-specific protease (15). The ICP0 binding sites of cyclin D3 and of the ubiquitin-specific protease differ. Both cyclin D3 and the ubiquitin-specific protease are translocated to a nuclear structure known as ND10. These observations suggested cyclin D3, a protein involved in the transition through the G1 phase of the cell cycle known to have a relatively short half-life, is stabilized by deubiquitination by the recruitment to the same macromolecular complex of the ubiquitin-specific protease. Furthermore, HSV-1 also encodes a protein (ICP47) that interacts with TAP1/TAP2 (16–18) and thereby blocks the presentation of antigenic peptides on the surface of infected cells. It could be predicted that HSV may require the stabilization of other macromolecules and, conversely, the degradation of others by other mechanisms than those described to date.
MATERIALS AND METHODS
Cell Lines and Viruses.
HEp-2 cells were obtained from the American Type Culture Collection and grown in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal calf serum. HSV-1(F) is the prototype HSV-1 strain used in this laboratory (19). Recombinant R7023 lacking the genes US8 to US12 (α47) was described elsewhere (4).
Labeling of Infected Cells with [35S]Methionine.
HEp-2 monolayer cultures containing 2 × 106 HEp-2 cells infected as described in Results were overlaid 10 h after infection with medium lacking methionine but supplemented with 400 μCi of [35S]methionine (Amersham; 10 mCi/ml; 1Ci = 37 GBq). Cells were harvested 22 h after infection, disrupted by sonication, and used as virus inoculum for the analysis of input virus proteins. For the pulse and chase experiments, 2 × 106 HEp-2 cells infected as described in Results were overlaid at 12 h after infection with medium lacking methionine but supplemented with 100 μCi of [35S]methionine. After 1 h of incubation at 37°C, the labeling medium was removed, cells were washed once with complete medium, overlaid with complete medium, and harvested at the times indicated in Results.
Antibodies.
Antibodies used in these studies were a rabbit polyclonal antibody to US9, generated in this laboratory; a rabbit polyclonal antibody to the capsid protein VP19C encoded by UL38 (21); a rabbit polyclonal antibody to ubiquitin purchased from Sigma (no. U5379); an affinity-purified rabbit polyclonal antibody to ubiquitin (a kind gift of M. Hochstrasser, University of Chicago); a mouse monoclonal antibody to subunit 12 of proteasome, MCP34 (20) (kindly donated by Klavs B. Hendil, University of Copenhagen, Denmark). To generate antibody against the US9 protein, the US9 gene was amplified from the HSV-1(F) genome by PCR (Fig. 1), and a fragment containing codons 1–59 was cloned into pGEX-4T1 (Pharmacia) in the EcoRI-SalI sites, in frame with glutathione S-transferase (GST). The resulting pRB5172 plasmid was transformed in Escherichia coli, strain RR1, and the expression of the chimeric protein GST-US9 was induced by addition of 5 mM isopropyl β-d-thiogalactopyranoside for 3 h at 37°C. Cells were washed in 20 mM Tris⋅HCl (pH 7.4)/1 mM EDTA/100 mM KCl, resuspended in lysis buffer [10 mM Tris⋅HCl (pH 7.4)/100 mM KCl/1 mM EDTA/5 mM dithiothreitol/0.5% Nonidet P-40/0.1 mM 7-amino-1-chloro-3-tosylamido-2-heptanone (TLCK)/0.1 mM 1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK)/1 mM phenylmethylsulfonyl fluoride (PMSF)], and sonicated. After 15 min of centrifugation at 12,000 × g, supernatants were adsorbed to glutathione-agarose beads (Sigma), and bound GST-US9 fusion protein was eluted with 20 mM reduced glutathione (Sigma). The purified protein was dialyzed against PBS (0.14 M NaCl/3 mM KCl/10 mM Na2HPO4/1.5 mM KH2PO4) and finally used to immunize rabbits at Josman Laboratories (Napa, CA).
Figure 1.
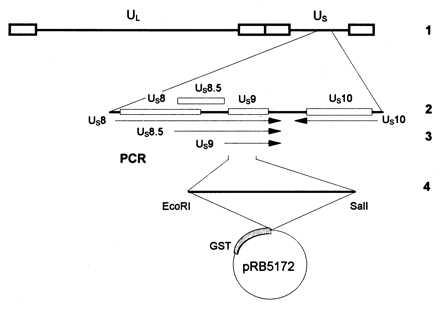
Diagrammatic representation of the sequence arrangement of the HSV-1 genome and of the location of the US9 gene. Line 1, representation of the HSV-1 genome. The single lines represent the unique long (UL) and unique short (US) DNA sequences, whereas the terminal repeats are shown as open rectangles. Line 2, expansion of the domain containing the US9 gene. Line 3, polarity and domain of US9 mRNA. The US11 and US12 mRNAs (not shown) are coterminal with the US10 mRNA. Line 4, representation of the US9 codons 1–59 amplified by PCR and fused to GST for production of antibody.
Electrophoretic Separation and Immunoblotting of Proteins.
Infected or mock-infected cells were solubilized in denaturing buffer, and the lysates were subjected to electrophoresis in denaturing polyacrylamide gels, transferred to nitrocellulose membranes, blocked in PBS containing 5% skim milk for 1 h at room temperature or overnight at 4°C, rinsed three times in PBS, and then reacted with the primary antibody. Before exposure to the anti-ubiquitin antibody, the proteins were first completely denatured by boiling the membranes for 1 h in distilled water at 100°C. The membranes were then washed three times, incubated with a goat anti-rabbit or a goat anti-mouse antibody conjugated to alkaline phosphatase (Bio-Rad) or with a goat anti-rabbit antibody conjugated to peroxidase (Sigma), rinsed again, and allowed to react with the alkaline phosphatase colorimetric reaction (Bio-Rad) or with the enhanced chemiluminescent detection system (Amersham), according to the protocols provided by the manufacturers.
Immunoprecipitations.
HEp-2 cells grown in 25-cm2 flask cultures were scraped, washed once in PBS, harvested by short centrifugation in a Microfuge, lysed either in PBS* (PBS/1% Nonidet P-40/1% deoxycholate/0.1 mM TLCK/0.1 mM TPCK/1 mM PMSF for immunoprecipitation with the anti-US9 or the anti-ubiquitin antibody) or in a lysis buffer containing 50 mM Tris (pH 7.2), 1 mM dithiothreitol, 5 mM MgCl2, 2 mM ATP, 12% glycerol, 0.5% Nonidet P-40, 0.1 mM TLCK, 0.1 mM TPCK, 1 mM PMSF (for immunoprecipitations of proteasomes), and briefly sonicated. Cell debris were pelleted by centrifugation for 5 min in a microcentrifuge at 12,000 × g, and supernatant fluids were reacted with the antibodies indicated in Results for 3 h at 4°C with constant agitation. The immune complexes were harvested by reacting with a mixture of protein A and protein G beads (Sigma), 5 mg of each protein/sample, for 1 h. Samples were extensively rinsed in the proper buffer, and proteins bound to the resin were eluted in denaturing buffer and boiled for 4 min, electrophoretically separated in a denaturing (sodium dodecyl sulfate) polyacrylamide gel, transferred to a nitrocellulose sheet, and subjected to autoradiography on X-Omat AR films (Kodak).
RESULTS
HSV-1 US9 Protein Is Ubiquitinated.
HSV-1 US9 protein has a predicted Mr of 10,000 and an apparent Mr ranging from 12,000 to 25,000 in denaturing polyacrylamide gels. Fig. 2A shows the reactivity of electrophoretically separated lysates of HEp-2 cells infected with HSV-1(F) or R7023, an HSV-1 mutant carrying a large deletion that includes the US9 gene, with a polyclonal antibody made against US9 protein. Fig. 2B shows a nitrocellulose membrane containing the same samples as those shown in Fig. 2A but denatured extensively by boiling as described in Materials and Methods and allowed to react with the anti-ubiquitin antibody (Sigma). The results show that the anti-ubiquitin antibody reacted with protein bands that overlapped the slower migrating US9 protein bands and, furthermore, that the reactivity of these antibodies were dependent on the presence of US9 protein inasmuch as the anti-ubiquitin antibody did not react with the corresponding bands in lysates of cells infected with a virus lacking the US9 gene (Fig. 2B, lane 4). In these experiments, the capsid protein VP19C (21) encoded by the UL38 gene and present in the lysates of the infected cells served as a loading control. The same results were obtained with the second, affinity-purified antibody to ubiquitin obtained from M. Hochstrasser (data not shown).
Figure 2.
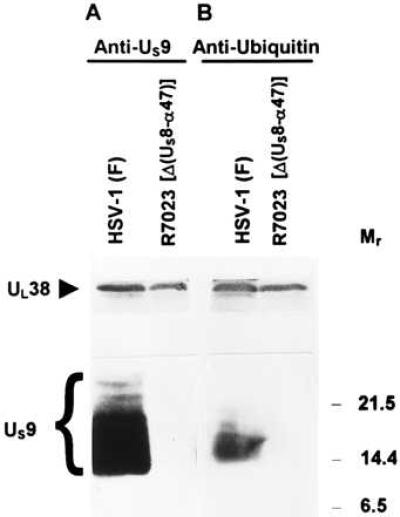
Immunoblots of infected cell proteins, electrophoretically separated on a denaturing gel, and allowed to react with rabbit polyclonal antibodies to US9 protein (A) or to ubiquitin (B). HEp-2 cells infected with HSV-1(F) or with R7023 were harvested at 22 h, solubilized, subjected to electrophoresis on a denaturing gel, transferred to nitrocellulose, and allowed to react with the indicated antibodies as described in Materials and Methods. The UL38 protein served as a loading control. Molecular weights (in thousands) are shown on the right.
Ubiquitin is a small, highly conserved polypeptide of 76 amino acids. There is no obvious amino acid homology between US9 protein and ubiquitin, and the observation that two different antibodies to ubiquitin react in the same fashion rules out the possibility that the double immunostaining of US9 protein is due to a shared epitope. Consistent with the hypothesis that US9 protein is ubiquitinated is the observation that the reaction of the anti-ubiquitin antibody with the bands shown in Fig. 2, lane 3, required extensive denaturation of the sample. Extensive denaturation is required to expose the ubiquitin epitopes, which are otherwise unavailable for the interaction with the antibody (22). All together, these results indicate that US9 protein is ubiquitinated.
US9 Protein Is Incorporated As a Ubiquitin Conjugate into the HSV-1 Virions.
US9 protein was reported to be a virion component localized in the tegument (2). To determine whether US9 protein packaged into virions is ubiquitinated, we purified HSV-1 virions from HEp-2 cells infected for 24 h with HSV-1(F). The procedure for virion purification was as that described by Spear and Roizman, (23). The virion preparations were split in two aliquots, electrophoretically separated on sodium dodecyl sulfate-polyacrylamide gel, transferred to a nitrocellulose, and allowed to react with either the antibody against the US9 protein or the anti-ubiquitin antibody (Sigma). The results, shown in Fig. 3, indicate that US9 protein packaged into virions is ubiquitinated. The apparent discrepancy between the position and pattern of the US9 forms recognized by the ubiquitin and the US9 antibodies is probably because of distortion of the filter, which has been extensively boiled before exposure to the anti-ubiquitin antibody.
Figure 3.
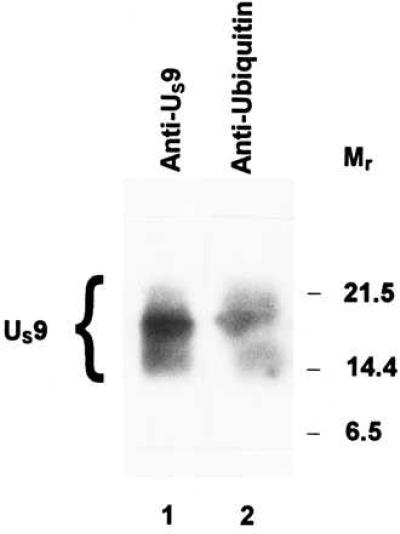
Immunoblots of virion proteins electrophoretically separated on a denaturing gel and allowed to react with rabbit polyclonal antibodies to US9 protein (lane 1) or to ubiquitin (lane 2). The virions were purified on dextran gradients from HEp-2 cells infected with HSV-1(F) (23), solubilized, subjected to electrophoresis, allowed to react with the indicated antibodies, and detected with the enhanced chemiluminescent detection system. Molecular weights in thousands are shown on the right.
US9 Protein Is Released As a Ubiquitin Conjugate into the Cytoplasm of Newly Infected Cells.
The results presented above show that US9 protein is ubiquitinated late in infection and is also incorporated as a ubiquitin conjugate into HSV-1 virions. US9 protein is expected to be released into the cytoplasm of newly infected cells together with other components of the viral tegument. To determine whether US9 protein is present as a ubiquitin conjugate after release in the cytoplasm of the newly infected cell, HEp-2 cells were exposed for 90 min to 100 pfu of [35S]methionine-labeled HSV-1(F) or R7023 per cell. After removal of the inoculum and an additional 30-min incubation, cells were lysed and allowed to react with the antibody to US9 protein (Fig. 4 A and C) or to ubiquitin (Fig. 4 B and D). The immune complexes were collected, rinsed, separated on a denaturing gel, transferred to a nitrocellulose sheet, and subjected to autoradiography. Both antibodies precipitated US9 protein from lysates of cells infected with HSV-1(F) (Fig. 4, lanes 1 and 3, 5 and 7), but in both experiments, the anti-ubiquitin antibody was less effective in precipitating US9 protein than the anti-US9 antibody. One explanation for this observation is a lower affinity of the anti-ubiquitin antibody for the ubiquitinated US9 protein, but we cannot exclude the possibility that some of the US9 protein was deubiquitinated.
Figure 4.
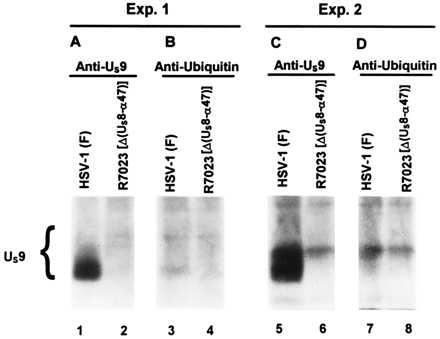
Autoradiographic image of electrophoretically separated [35S]methionine-labeled proteins, immunoprecipitated with a rabbit polyclonal antibody to US9 protein (A and C), or to ubiquitin (B and D) from infected cell lysates. HEp-2 cells were exposed for 90 min to [35S]methionine-labeled HSV-1(F) or R7023 generated as described in Material and Methods and then incubated for an additional 30 min in medium containing excess unlabeled methionine. The cells were lysed, and immune complexes containing US9 protein were subjected to electrophoresis in denaturing gels and autoradiography on X-Omat AR film.
Stability of US9 Protein.
As indicated in the Introduction, ubiquitination is the signal for ATP-dependent protein degradation. Even though this is not its only function, ubiquitination signals the degradation of the protein by proteasomes (reviewed in ref. 24). Inasmuch as US9 protein is ubiquitinated, it was of interest to determine whether it was stable. US9 protein is packaged in the virion tegument and is detected in infected cells at approximately 12 h after infection (data not shown). We have therefore tested the stability of the US9 protein immediately after infection and after labeling the protein at 12 h after infection.
In the first series of experiments, replicate cultures of HEp-2 cells were exposed at 10°C for 90 min to 100 pfu of [35S]methionine-labeled HSV-1(F) or R7023 per cell. Radiolabeled virus was prepared as described in Materials and Methods. The cells were then rinsed, overlaid with complete medium, harvested at 0, 2, 3, or 4 h after incubation at 37°C, then solubilized and allowed to react with the antibody to US9 protein. The immune complexes were collected, solubilized, and subjected to electrophoresis in denaturing gels. The procedures were as described in the legend to Fig. 4. The results (Fig. 5) indicate that the US9 protein released into the cytoplasm of newly infected cells is stable for at least 4 h after infection. (Fig. 5, lanes 2–5).
Figure 5.
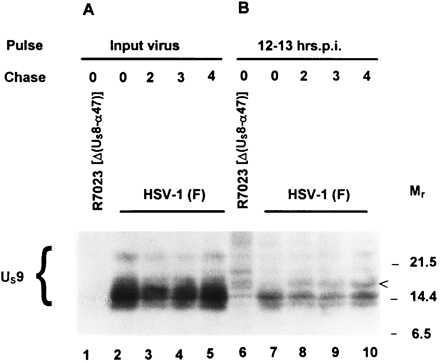
Autoradiographic image of [35S]methionine-labeled proteins immunoprecipitated with a rabbit polyclonal antibody to US9 protein from infected cell lysates and subjected to electrophoresis on a denaturing gel. (A) HEp-2 cells were incubated at 37°C for 0, 2, 3, and 4 h after exposure to radiolabeled HSV-1(F) or R7023 at 10°C for 90 min and then lysed and allowed to react with the antibody. The immune complexes were subjected to electrophoresis on a denaturing gel, transferred to a nitrocellulose sheet, and subjected to autoradiography. (B) At 12 h after infection with HSV-1(F) or R7023, HEp-2 cells were labeled for 1 h with [35S]methionine as described in Materials and Methods and then incubated in medium containing excess unlabeled methionine and harvested immediately or at hourly intervals for 4 h. The cells were subjected to the same procedures as described for A. Molecular weights (in thousands) are shown on the right.
In the second series of experiments, replicate cultures of HEp-2 cells were infected with HSV-1(F) or R7023. At 12 h after infection, the cells were labeled for 1 h with [35S]methionine and then rinsed and incubated in medium containing unlabeled methionine. At 0, 2, 3, or 4 h after the labeling interval, the cells were harvested, solubilized, and allowed to react with the rabbit antiserum made against the US9 protein. The results (Fig. 5, lanes 7–10) show that US9 protein harvested after the 1-h labeling period was post-translationally processed to higher apparent molecular weight forms (arrowhead), but the amount of the labeled US9 protein did not change throughout the 4-h chase interval.
We conclude from these series of experiments that the US9 protein is stable for several hours, either late in infection in cells in which it was made or in newly infected cells.
Association of US9 Protein with the Proteasome.
The apparent stability of the ubiquitinated US9 protein raised the possibility that one of its functions may be to interact with components of the ubiquitin pathway, either to enhance or to block protein degradation. A candidate for such an interaction is the proteasome, the catalytic core of the larger 26S proteinase complex, involved in the recognition of the ubiquitin conjugates, and in their degradation (25). To determine whether US9 protein is associated with proteasomes, HEp-2 cells were mock infected (Fig. 6, lane 1) or exposed to 10 pfu of HSV-1(F) (Fig. 6, lanes 2, 3, and 5) or of R7023 (Fig. 6, lane 4) per cell. The cells were harvested at 12 (Fig. 6, lane 2) or 22 h after mock infection or infection (Fig. 6, lanes 1, 3–5), lysed, and allowed to react at 4°C with the MCP34 anti-proteasome antibody, which recognizes subunit 12 of the proteasome (20) (Fig. 6, lanes 1–4), or with the anti US9 antibody (Fig. 6, lane 5). Under the conditions tested (presence of ATP and glycerol in the buffer), the proteasome precipitates as a complex. The proteins specifically precipitated by the antibody were then electrophoretically separated on a polyacrylamide gel, electrically transferred to a nitrocellulose sheet, and allowed to react with the antibody to US9 protein. The results indicate that US9 protein coimmunoprecipitated with the proteasome at 12 and 22 h after infection (Fig. 6, lanes 2 and 3). The antibody did not react with antigens of corresponding size in lysates of mock-infected cells (lane 1) or lysates of R7023-infected cells (lane 4). These results indicate that US9 protein is associated with the proteasomes.
Figure 6.
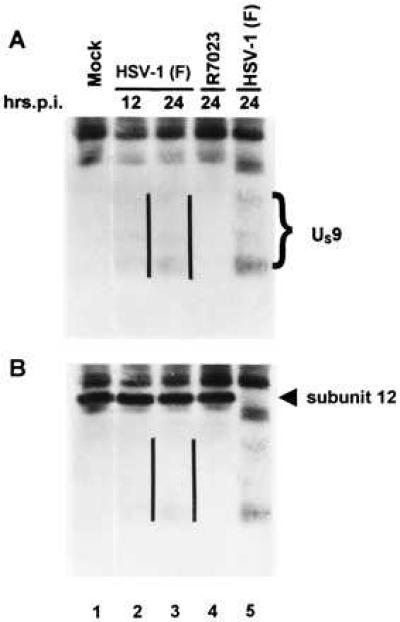
Immunoblot of infected cell proteins immunoprecipitated with a monoclonal antibody to proteasome, electrophoretically separated on a denaturing gel and allowed to react with a rabbit polyclonal antibody to US9 protein (A), photographed, and then allowed to react with a monoclonal antibody to subunit 12 of proteasome (B). The parallel lines are to the right of the US9 protein bands.
DISCUSSION
We report that the US9 protein is ubiquitinated, packaged in virions, introduced into newly infected cells as a ubiquitinated species, stable for at least several hours, and is associated with proteasomes. Relevant to this report are the following:
(i) US9 protein forms an overlapping series of broad bands with an apparent Mr ranging from 12,000 to 25,000. The proteins in the lower range are in the greatest abundance. The anti-ubiquitin antibody usually reacted with the proteins in the lower range, either because the slower migrating forms were not ubiquitinated or because the anti-ubiquitin antibody was less efficient in detecting the US9-ubiquitin conjugate. We prefer the latter explanation inasmuch as in some experiments (Fig. 2 and Fig. 4, experiment 2), the anti-ubiquitin antibody reacted with the slower migrating forms, but these were also in relatively greater abundance than those seen in other experiments. Ubiquitin, characteristically, is conjugated to lysines. As shown in Fig. 7, neither the HSV-1 US9 protein nor the more conserved homologs of the US9 protein encoded by other members of the alphaherpesvirinae subfamily contain lysines. Ubiquitin must be covalently linked to another amino acid, possibly at the amino terminus of the protein. At this time it is unclear whether ubiquitination is solely responsible for the extent of posttranslational modifications evident from the electrophoretic mobility of denatured US9 protein.
Figure 7.

Sequence alignment of the US9 protein homologs of HSV-1(F), HSV-2(G), simian B virus, equine herpes viruses 1 and 4, bovine herpes virus 1, and feline herpes virus 1, simian varicella, varicella-zoster, and pseudorabies viruses generated by the lineup and pretty programs of the Genetics Computer Group software. The last line shows the consensus sequence. Residues conserved among at least five of the sequences are shown as capitalized letters, whereas those conserved among at least seven of the sequences are shown as bold letters. Dots indicate gaps inserted by the program to create the best alignment.
(ii) Ubiquitination usually, but not necessarily, leads to the degradation of the ubiquitin conjugate in an ATP-dependent process that involves the large 26 S proteinase complex and its catalytic core, the proteasome. Ubiquitination has been shown to play a role in several cellular processes such as regulation of the cell cycle and gene expression, generation of antigenic peptides for MHC class 1-dependent presentation on cell surfaces, cellular stress response, cell surface receptor modification, DNA repair, import of proteins into mitochondria, uptake of precursors of neurotransmitters into synaptosomes, biogenesis of peroxisomes, assembly of ribosomes, programmed cell death, etc. The expected result of ubiquitination is that the targeted protein is rapidly degraded, modulating the final outcome of the process in which it is involved. Contrary to the expectation that it would be rapidly degraded, US9 protein seemed to be relatively stable, both immediately after entry of the virus into cells and late in infection after its synthesis. Other examples of ubiquitinated proteins with a relative long half-life have been reported in the literature (e.g., histone H2A, refs. 26 and 27). The function of this type of ubiquitination is not well understood.
(iii) HSV has evolved two functions designed to reduce the effects of degradation of proteins by the ubiquitin pathway. Thus, ICP0 binds both cyclin D3 and the ubiquitin-specific protease, and at least one outcome of the interaction is the stabilization of cyclin D3 (14). In addition, HSV-1 ICP47 blocks the translocation into the endoplasmic reticulum of antigenic peptides generated by the ubiquitin pathway for presentation at the cell surface (16–18). The discovery that US9 protein is ubiquitinated but remains stable suggested the possibility that the protein is involved in some aspect of cellular machinery for degrading unwanted proteins and led to the demonstration that it is associated with proteasomes. This association of US9 protein together with the observation that US9 coding sequence is very well conserved among the herpes viruses comprising the alphaherpesvirinae subfamily, suggests that one of its functions may be to perturb ubiquitin-mediated protein degradation.
We conceive of three possibilities. First, US9 protein could be turning off all ubiquitin-mediated protein degradation. Such a step would reduce availability of virus-derived antigenic peptides for presentation to the immune system and would inhibit the turnover of cellular proteins whose accumulation is cell cycle dependent and that are required for efficient viral replication. Cellular inhibitors of proteasome function have been described, and some of them associate with the proteasome (28–30). Examples of viral products reported to inhibit the proteasome-mediated protein degradation include HIV-1 tat (31) and the X protein of hepatitis B virus (32, 33).
The second possibility is that US9 protein blocks the degradation of specific proteins. As noted above, HSV ICP0 seems to interfere with the ubiquitin-mediated proteolytic pathway, possibly to prevent cyclin D3 degradation. US9 protein could represent an additional viral mechanism to regulate specific cellular functions.
A third possibility is that US9 protein mediates the specific degradation of a protein noxious for viral replication. A similar mechanism has been proposed to explain the induction of NF-κB transactivating activity by the human T-cell leukemia virus (HTLV-1) tax protein. In resting T lymphocytes, NF-κB is sequestered to the cytoplasm by members of the I-κB family of inhibitors. HTLV-1 tax promotes I-κB degradation (34, 35) and thereby activates NF-κB.
Studies in progress should define the significance and consequences of the ubiquitination of US9 protein and of its interaction with proteasomes.
Acknowledgments
We thank Mark Hochstrasser and Klavs B. Hendil for invaluable advice and for gifts of antibodies to ubiquitin and subunit 12 of proteasome. These studies were aided by National Cancer Institute Grant CA47451 and the U.S. Public Health Service.
ABBREVIATIONS
- HSV-1
herpes simplex virus 1
- GST
glutathione S-transferase
- pfu
plaque-forming units
- ICP0
viral-infected cell protein no. 0
- TLCK
7-amino-1-chloro-3-tosylamido-2-heptanone
- TPCK
tosylamido-2-phenylethyl chloromethyl ketone
- PMSF
phenylmethylsulfonyl fluoride
References
- 1.McGeoch D J, Dolan A, Donald S, Rixon F J. J Mol Biol. 1985;181:1–13. doi: 10.1016/0022-2836(85)90320-1. [DOI] [PubMed] [Google Scholar]
- 2.Frame M C, McGeoch D J, Rixon F J, Orr A C, Marsden H S. Virology. 1986;150:321–332. doi: 10.1016/0042-6822(86)90297-7. [DOI] [PubMed] [Google Scholar]
- 3.Roizman B, Sears A E. In: Fields’ Virology. 3rd Ed. Fields B N, Knipe D M, Howley P, Chanock R M, Hirsch M S, Melnick J L, Monath T P, Roizman B, editors. New York: Raven; 1996. pp. 2231–2295. [Google Scholar]
- 4.Longnecker R, Roizman B. J Virol. 1986;58:583–591. doi: 10.1128/jvi.58.2.583-591.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jentsch F. Annu Rev Genet. 1992;26:179–207. doi: 10.1146/annurev.ge.26.120192.001143. [DOI] [PubMed] [Google Scholar]
- 6.Hershko A, Ciechanover A. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- 7.Ganot D, Leshinsky E, Eytan E, Hershko A. J Biol Chem. 1988;263:12412–12419. [PubMed] [Google Scholar]
- 8.Tanaka K, Tamura T, Yoshimura T, Ichihara A. New Biol. 1992;4:173–187. [PubMed] [Google Scholar]
- 9.Jennissen H P. Eur J Biochem. 1995;231:1–30. [PubMed] [Google Scholar]
- 10.Weissman A M. Immunol Today. 1997;18:189–198. doi: 10.1016/s0167-5699(97)84666-x. [DOI] [PubMed] [Google Scholar]
- 11.Ciechanover A. Biol Chem Hoppe-seyler. 1994;375:565–581. doi: 10.1515/bchm3.1994.375.9.565. [DOI] [PubMed] [Google Scholar]
- 12.Monaco J J. J Leukocyte Biol. 1995;57:543–547. doi: 10.1002/jlb.57.4.543. [DOI] [PubMed] [Google Scholar]
- 13.Finley D, Chau V. Annu Rev Cell Biol. 1991;7:25–69. doi: 10.1146/annurev.cb.07.110191.000325. [DOI] [PubMed] [Google Scholar]
- 14.Kawaguchi Y, Roizman B J. Virology. 1997;71:7328–7336. doi: 10.1128/jvi.71.10.7328-7336.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett R D, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. EMBO J. 1997;16:566–577. doi: 10.1093/emboj/16.3.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.York I A, Roop C, Andrews D W, Riddell S R, Graham F L, Johnson D C. Cell. 1994;77:525–535. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 17.Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Nature (London) 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 18.Fruh K, Ahn K, Djaballah H, Sempe P, van Endert P M, Tampe R, Peterson P A, Yang Y. Nature (London) 1995;375:415–418. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- 19.Ejercito P, Kieff E D, Roizman B. J Gen Virol. 1968;2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 20.Hendil K B, Kristensen P, Uerkvitz W. Biochem J. 1995;305:245–252. doi: 10.1042/bj3050245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward P L, Ogle W O, Roizman B. J Virol. 1996;70:4623–4631. doi: 10.1128/jvi.70.7.4623-4631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haas A L, Bright P M. J Biol Chem. 1985;260:12464–12473. [PubMed] [Google Scholar]
- 23.Spear P G, Roizman B. J Virol. 1972;9:143–159. doi: 10.1128/jvi.9.1.143-159.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hochstrasser M. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 25.Hough R, Pratt G, Rechsteiner M. J Biol Chem. 1987;262:8303–8313. [PubMed] [Google Scholar]
- 26.Hunt L T, Dayhoff M O. Biochem Biophys Res Commun. 1977;74:650–655. doi: 10.1016/0006-291x(77)90352-7. [DOI] [PubMed] [Google Scholar]
- 27.Levinger L, Varshavsky A. Cell. 1992;28:375–385. doi: 10.1016/0092-8674(82)90355-5. [DOI] [PubMed] [Google Scholar]
- 28.Driscoll J, Frydman J, Goldberg A L. Proc Natl Acad Sci USA. 1992;89:4986–4990. doi: 10.1073/pnas.89.11.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X S, Etlinger J D. Biochemistry. 1992;31:11963–11967. [Google Scholar]
- 30.Chu-Ping M, Slaughter C A, DeMartino G N. Biochim Biophys Acta. 1992;1119:303–311. doi: 10.1016/0167-4838(92)90218-3. [DOI] [PubMed] [Google Scholar]
- 31.Seeger M, Ferrel K, Frank R, Dubiel W. J Biol Chem. 1997;272:8145–8148. doi: 10.1074/jbc.272.13.8145. [DOI] [PubMed] [Google Scholar]
- 32.Huang J, Kwong J, Sun E C, Liang T J. J Virol. 1996;70:5582–5591. doi: 10.1128/jvi.70.8.5582-5591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher M, Runkel L, Shaller H. Virus Genes. 1995;10:99–102. doi: 10.1007/BF01724303. [DOI] [PubMed] [Google Scholar]
- 34.Maggirwarm S B, Harhaj E, Sun S C. Oncogene. 1995;11:993–998. [PubMed] [Google Scholar]
- 35.Beraud, C. & Greene, W. C. J. (1996) Acquired Immune Defic. Syndr. Hum. Retrovir. 13, Suppl. 1, S76–S84. [DOI] [PubMed]