

Abstract
Germline TSC1 or TSC2 mutations cause Tuberous Sclerosis Complex (TSC), a hamartoma syndrome with lung involvement. To explore the potential interaction between TSC1 and KRAS activation in lung cancer, mice were generated in which Tsc1 loss and KrasG12D expression occur in a small fraction of lung epithelial cells. Mice with combined Tsc1-KrasG12D mutation had dramatically reduced tumor latency (median survival 11.6 – 15.6 weeks) in comparison to KrasG12D alone mutant mice (median survival 27.5 weeks). Tsc1-Kras G12D tumors showed consistent activation of mTORC1, and responded to treatment with rapamycin leading to significantly improved survival, while rapamycin had minor effects on cancers in KrasG12D alone mice. Loss of heterozygosity for TSC1 or TSC2 was found in 22% of 86 human lung cancer specimens. However, none of 80 lung cancer lines studied showed evidence of lack of expression of either TSC1 or TSC2 or a signaling pattern corresponding to complete loss. These data indicate Tsc1 loss synergizes with Kras mutation to enhance lung tumorigenesis in the mouse, but that this is a rare event in human lung cancer. Rapamycin may have unique benefit for lung cancer patients in which TSC1/TSC2 function is limited.
Keywords: lung cancer, TSC1, KRAS, TSC2, mTOR
Introduction
Lung cancer is the leading cause of cancer-related deaths both in the USA and worldwide (Jemal et al., 2008; Molina et al., 2008). Non-small cell lung cancer (NSCLC) accounts for 80% of all cases. In NSCLC, several proto-oncogenes, such as KRAS and EGFR are known to be mutated at significant frequency (Thomas et al., 2007; Ding et al., 2008; Molina et al., 2008). In addition, loss of tumor suppressor gene function is known to occur in NSCLC (Weir et al., 2007; Ding et al., 2008). To dissect the role of tumor suppressor genes in lung tumorigenesis, we have generated a series of murine models utilizing an activatable KrasG12D allele (Jackson et al., 2001), and conditional alleles of several different tumor suppressor genes: p53, p16Ink4a, Ink4a/arf, and Lkb1 (Ji et al., 2007). Among these, loss of Lkb1 had the most potent effect in accelerating lung tumorigenesis, and led to several different histologic subtypes as well as invasion and metastasis (Ji et al., 2007). LKB1 inactivation also occurs in up to 35% of human lung cancer (Ji et al., 2007; Sanchez-Cespedes, 2007; Ding et al., 2008). LKB1 is a serine/threonine kinase that has multiple targets, including AMPK which phosphorylates and activates the TSC1/TSC2 complex (Corradetti et al., 2004; Shaw et al., 2004; Hardie & Sakamoto, 2006).
The TSC1/TSC2 complex is the only known GTPase for Rheb, serving to reduce Rheb-GTP levels, and thereby inhibit activation of mTORC1, a protein complex consisting of mTOR, RAPTOR, and mLST8 (Guertin & Sabatini, 2007; Huang & Manning, 2008). TSC1 and TSC2 are the targets of multiple kinases which regulate the GTPase activity of the complex, and thus they function as critical integrators of growth signals within the cell. Loss of either TSC1 or TSC2 prevents formation of a functional TSC1/TSC2 complex resulting in constitutive activation of mTORC1 and phosphorylation of its downstream targets S6K and 4E-BP1, with net effects of abnormal translational activation leading to cell growth and proliferation (Guertin & Sabatini, 2007; Huang & Manning, 2008).
Germline mutations of TSC1 or TSC2 result in Tuberous Sclerosis Complex (TSC), an autosomal dominant tumor suppressor gene syndrome that is characterized by widespread hamartoma development (Crino et al., 2006). The pulmonary manifestations of TSC include lymphangioleiomyomatosis and multifocal micronodular pneumocyte hyperplasia, although lung cancer is rare in TSC patients (Muir et al., 1998; McCormack, 2008).
Since Lkb1 loss synergized with Kras activation to accelerate tumorigenesis in the mouse (Ji et al., 2007), we hypothesized that part or all of this effect was due to loss of AMPK activation by LKB1, leading to functional inactivation of the TSC1/TSC2 complex and downstream mTORC1 activation (Corradetti et al., 2004; Shaw et al., 2004). To examine this hypothesis in vivo, we generated a novel lung adenocarcinoma murine model by intercrossing the conditional oncogenic LSL-KrasG12D allele (Jackson et al., 2001) and a conditional Tsc1 null allele (Kwiatkowski et al., 2002). Strikingly, homozygous or heterozygous loss of Tsc1 dramatically accelerated KrasG12D-driven lung cancer development. In addition, the mTORC1 inhibitor rapamycin appeared to have unique benefit in inducing tumor regression and extending survival in the Tsc1-KrasG12D mice. We then examined the potential role of the TSC1/TSC2 complex in human lung cancer development using both direct patient specimens, and a panel of 80 NSCLC cell lines. A significant fraction of human lung cancer samples showed evidence of TSC1 or TSC2 LOH, mainly TSC1 LOH. However, none of the cell lines showed evidence of complete loss of TSC1 or TSC2, suggesting that this event is rare in vivo in patients.
Materials and Methods
Mouse cohorts
Mice bearing the Lox-Stop-Lox-KrasG12D allele were provided by Tyler Jacks, Massachusetts Institute of Technology (Jackson et al., 2001). Tsc1L/L mice were generated by floxing exons 17 and 18 of the Tsc1 gene, as described previously (Kwiatkowski et al., 2002). To generate LSL-KrasG12D Tsc1L/L mice, LSL-KrasG12D mice were first crossed with Tsc1L/L mice and the progeny LSL-KrasG12D Tsc1L/+ mice were backcrossed to Tsc1L/L mice. Due to this breeding scheme, the strain background was mixed for all mice studied. Mice were treated with adenoviral Cre (AdCre) by nasal inhalation at 4-7 weeks of age to induce KrasG12D expression and/or inactivation of Tsc1L alleles by cleavage at the Lox sites in the infected respiratory epithelium. The animals were housed in a pathogen-free environment in a barrier facility at Harvard School of Public Health; all animal experiments performed were approved by the Institutional Animal Care and Use Committee at Harvard Medical School. Mice were terminated when severe dyspnea, weight loss, or other signs of morbidity were seen. The logrank test was used to compare the survival of different groups of mice.
Lung tissue preparation for histology and immunohistochemical studies
Lung tissue was prepared using methods described previously (Ji et al., 2007). In brief, mice were sacrificed, the left lung was removed and snap-frozen, while the right lung was inflated and fixed in buffered 10% formalin overnight. Paraffin sections were prepared, and cut at 5 microns for hematoxylin and eosin (H&E) staining.
Apoptosis was assessed in lung cancer sections by the terminal deoxynucleotidyl transferase dUTP-biotin nick end labeling (TUNEL) method using ApopTag Peroxide In Situ Apoptosis Detection Kit (Chemicon International, Inc.). To assess cell proliferation, a rabbit polyclonal antibody against Ki67 (1: 2500; Vector Lab) was used. An antibody against pS6(S240/244) (New England Biolabs) was also used for immunohistochemistry. TUNEL-positive and Ki67-positive cells were each counted as the number of positive cells per high power field (200x) in a blinded manner. pS6(S240/244) expression was assessed on a scale from 0 to 4 in tumors in a blinded manner, integrating both strength of the signal and uniformity among cells in the tumors into this score.
In vivo rapamycin treatment
Rapamycin treatment was given by intraperitoneal (IP) injection every other day at 6mg/kg (Meikle et al., 2007). Rapamycin was suspended in 5% (v/v) Tween-80 and 5% (v/v) PEG 400. In one cohort, magnetic resonance imaging (MRI) was used to assess lung tumor involvement before and after rapamycin or control vehicle treatments. MRI images were used to quantify the extent of tumor involvement using NIH ImageJ (version 1.33) as described previously (Li et al., 2007).
In a second survival cohort, treatment with rapamycin was initiated at 9 weeks following AdCre infection, and continued every other day until survival was terminated due to respiratory difficulties, or death.
Cell culture
Lung cancer cell lines were obtained from the ATCC by Dana-Farber Cancer Institute Lung Cancer Program. All cell lines were maintained in the growth medium recommended by the supplier.
Immunoblotting
Cell and mouse tumor lysates were prepared in lysis buffer (Cell Signaling Technology) supplemented with protease and phosphatase inhibitor I and II cocktails (Calbiochem), using a Dounce homogenizer for the tumor nodules, and clarified by centrifugation at 13,000 g for 10 minutes.
Protein concentrations were determined using the DC Protein Assay (Bio-Rad) and equivalent amounts (50 μg) were subjected to SDS-PAGE on 4-12% gradient gels (Invitrogen), transferred to PVDF membranes and detected by immunoblotting with antibodies indicated using SuperSignal West Pico Chemiluminescent substrate (Pierce Biotechnology). Antibodies used were against: TSC1, pAKT(S473), AKT, pS6(S240/244), pS6(S235/236), S6, pERK(T202/Y204), ERK (Cell Signaling Technology); TSC2, (Santa Cruz Biotechnology, Santa Cruz, CA).
Multiplex ligation-dependent probe assay (MLPA) for analysis of TSC1/TSC2 for LOH, and genotyping of the Tsc1 allele in mouse samples
MLPA analysis for LOH in TSC1 and TSC2 was performed using a set of 23 homemade probes, consisting of 10 probes in TSC1, 10 probes in TSC2, and 3 control probes from different chromosomes, as described (Kozlowski et al., 2007). MLPA products were quantified by capillary electrophoresis on an ABI 3100 Genetic Analyzer (Applied Biosystems, Inc.), using GeneMapper v3.5 (ABI). Excel programs were used to transform the peak height data to normalized values, such that the average signal from control samples after normalization was 1. We classified cases as demonstrating LOH for TSC1 or TSC2 when the average signal from probes for those genes was ≥ 0.2 lower than the average signal from other probes in the set, and a comparison of signal intensity was significant at a p < 0.001 in a two sided t-test.
To assess the genotype of mouse DNA samples from mice bearing combinations of the floxed (L), null (K), and wild type (W) alleles, a panel of 9 MLPA probes was generated to interrogate these three alleles, including 4 control probes and probes specific for each allele. MLPA analysis was performed on DNA samples from mice of known genotypes as controls, and then performed on tumor nodules to determine percent of each allele present, after normalization and comparison with control samples. Oligonucleotide sequences for the probe sets and details are available on request.
Results
KrasG12D mutation synergizes with Tsc1 loss in the bronchial epithelium to lead to accelerated lung cancer development and early mortality
To generate mice with both expression of KrasG12D and loss of Tsc1 in the lung epithelium, we crossed mice bearing the Lox-Stop-Lox KrasG12D (hereafter LSL-KrasG12D) allele (Jackson et al., 2001) to Tsc1L/L mice (Kwiatkowski et al., 2002). Mice bearing different combinations of both alleles were then treated with an adenovirus encoding the cre recombinase (AdCre) by nasal inhalation to induce Cre-mediated recombination in the lung epithelium at age 4 – 7 weeks (Figure 1A). This treatment is known to infect a small percent of lung bronchial epithelial cells. Following AdCre treatment, the median survival was 11.6 weeks for LSL-KrasG12D Tsc1L/L mice, in contrast to 27.5 weeks for LSL-KrasG12D (alone) mice (Logrank test, p = 0.002). In addition, AdCre-treated LSL-KrasG12D Tsc1L/+ mice had a median survival of 15.6 weeks, which was also significantly different from both the LSL-KrasG12D mice (p = 0.04), and the LSL-KrasG12D Tsc1L/L mice (p = 0.009). All AdCre-treated Tsc1L/L mice survived over 50 weeks (Figure 1A).
Figure 1. Loss of Tsc1 markedly accelerates oncogenic Kras-driven lung tumorigenesis in the mouse, with activation of mTORC1.
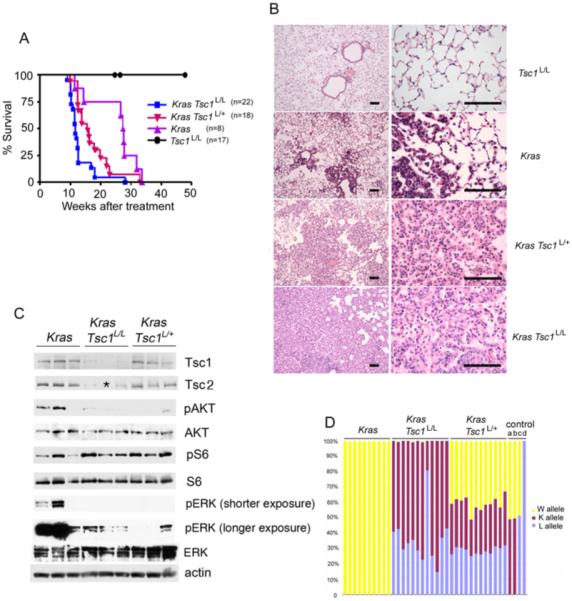
(A) LSL-KrasG12D Tsc1L/L mice display accelerated lung cancer development and early mortality. Kaplan-Meier survival curves of mouse cohorts following nasal inhalation of 5 × 106 PFU of AdCre. The median survival was 11.6 weeks for LSL-KrasG12D Tsc1L/L mice, 15.6 weeks for LSL-KrasG12D Tsc1L/+ mice, 27.5 weeks for LSL-KrasG12D mice, and over a year for Tsc1L/L mice.
(B) H&E-stained sections of lung from mice 12.5 weeks following AdCre inoculation shows extensive tumor in both LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mice. Low power (left) and high power (right) views are shown. Bar, 100μm.
(C) Immunoblot analysis of protein expression and mTOR signaling in tumors from these mice shows that Tsc1 and Tsc2 expression is reduced in the lung tumor nodules of both LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mice in comparison to tumors from LSL-KrasG12D mice. pS6(S240/244) expression is increased and pAKT(S473) expression is reduced, consistent with mTORC1 activation in these tumors. pERK levels are higher in the LSL-KrasG12D mice, compared to other genotypes. AKT, S6, ERK, and actin are all controls. Note that one lung tumor sample was depleted, and was not included in the Tsc2 blot, indicated by *.
(D) MLPA analyses of Tsc1 alleles demonstrate conversion of L to K alleles in both LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ tumor nodules. Yellow, red, and blue bars indicate the relative amount of wild type (W), knockout (K), and conditional allele (L), respectively. The four samples at right (a-d) are control samples from mice with genotypes KW, KW, LW, and LL, respectively. Note that the W allele persists in LSL-KrasG12D Tsc1L/+ tumor nodules.
Pathologic analysis of lung tissue demonstrated that AdCre-treated LSL-KrasG12D Tsc1L/L mice at 12.5 weeks post-treatment had developed multifocal adenocarcinoma with extensive involvement of the lung parenchyma, consistent with their progressive respiratory insufficiency and death (Figure 1B). Similarly, AdCre-treated LSL-KrasG12D Tsc1L/+ mice also showed multifocal adenocarcinoma of the lung on histological analysis at that age, but this was somewhat less extensive. At 12.5 weeks, AdCre-treated LSL-KrasG12D mice (with wild type Tsc1) had multiple microscopic foci of adenocarcinoma, that was much less developed, similar to previous reports (Jackson et al., 2001; Ji et al., 2007). By standard pathologic analysis, the lung adenocarcinomas from mice of these three genotypes were indistinguishable. Consistent with their extended survival, AdCre-treated Tsc1L/L mice had no lung tumors or other pathology when sampled at 1 year post treatment. These results indicate that both complete and haploid loss of Tsc1 cooperate significantly with activating Kras mutation to accelerate lung tumorigenesis.
Signaling analyses indicate that LSL-KrasG12D Tsc1L/L mouse lung cancers have activated mTORC1
Immunoblot analysis of lung tumor lysates demonstrated that Tsc1 protein levels were reduced in lung cancers developing in both LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mice in comparison to tumors from LSL-KrasG12D mice (Figure 1C). Tsc2 protein levels were also correspondingly reduced, consistent with the chaperone-like effect of Tsc1 on Tsc2 levels (Harrington et al. 2004).
Phospho-S6-S240/244 (pS6(S240/244)) levels were increased in the tumor lysates from both LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mice, consistent with activation of mTORC1 due to loss of functional Tsc1/Tsc2 in those tumors, again in contrast to tumor lysates from LSL-KrasG12D mice (Figure 1C). In addition, phospho-AKT-S473 (pAKT(S473)) levels were also reduced, on average, in both LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mouse lung cancers when compared to LSL-KrasG12D alone lung tumors. This effect is well-known in a variety of cell types and systems, as a mechanism of feedback regulation consequent to loss of Tsc1/Tsc2 and activation of mTORC1 (Zhang et al., 2003; Harrington et al., 2004; Huang et al., 2008). ERK activation appeared to be higher in the LSL-KrasG12D alone lung tumors, as assessed by levels of pERK(T202/Y204) (Figure 1C).
To examine the possibility that lung tumors developing in LSL-KrasG12D Tsc1L/+ mice had undergone genomic deletion or homologous recombination leading to loss of the Tsc1+ allele, we used multiplex ligation-dependent probe assay (MLPA) to investigate the copy number of the various alleles of Tsc1 in these tumors (Kozlowski et al., 2007). Lung tumors from LSL-KrasG12D Tsc1L/L mice showed a 65% (median) level of conversion of the conditional to the null allele, consistent with recombination at both conditional alleles in tumor cells, and contamination by inflammatory, stromal, and vascular cells (Fig. 1D). Similarly, LSL-KrasG12D Tsc1L/+ mice displayed 52% (median) conversion of the single conditional Tsc1 allele to the null allele. They also showed no major reduction in the copy number of the wild type (+) allele of Tsc1 (median 41%), suggesting that these tumors developed through a haploinsufficiency mechanism.
Haploid loss of TSC1 is common in human lung cancer specimens
As these results suggested that the Tsc1 gene could function as a tumor suppressor gene for lung cancer development in the mouse, we explored its involvement in human lung cancer. MLPA was used to assess TSC1 and TSC2 genomic copy number in DNA prepared directly from frozen lung cancer samples from 86 NSCLC patients (Figure 2) (Kozlowski et al., 2007). Genomic loss was seen in 19% and 5% of patients, respectively, for TSC1 and TSC2 (Figure 2A). In addition, one patient’s cancer displayed a copy number for TSC1 that was 40%, consistent with complete deletion of TSC1 in tumor cells from this patient, considering stromal and inflammatory cell contamination (Figure 2B).
Figure 2. MLPA analyses of human lung cancers identifies TSC1 and TSC2 genomic loss.

(A) Summary of MLPA analyses on 86 human NSCLC specimens demonstrates the proportion of samples with genetic events affecting TSC1 or TSC2.
(B) MLPA graphs. a, NSCLC sample without TSC1 or TSC2 loss; b, sample with TSC1 genomic loss, with 60% signal for all TSC1 probes; c, sample consistent with complete genomic deletion of TSC1, with 40% signal for 8 of 10 probes within TSC1; d, MLPA analysis of a control sample from a TSC patient with a heterozygous germline deletion involving exons 8 - 23.
In summary, 22% of 86 lung cancer specimens showed evidence of TSC1 and/or TSC2 genomic deletion by MLPA. However, the frequency of TSC2 LOH was so low, that this may represent genomic background copy number change, rather than a specific deletion of TSC2.
TSC1/TSC2 loss in human lung cancer cell lines
To explore the loss of TSC1/TSC2 in human lung cancer in greater detail, we surveyed Akt-mTOR signaling in 80 lung cancer cell lines (Supplemental Table 1). None of the 80 cell lines were found to display evidence of complete loss of TSC1 or TSC2 by immunoblotting (data not shown). However, 9 (11%) and 2 (3%) of the 80 cell lines showed evidence of LOH for TSC1 or TSC2, respectively, by MLPA analysis (Supplemental Table 1). In addition, some cell lines showed high levels of phospho-S6(S240/244) in the absence of serum. However, those same lines generally showed elevated activation of pAKT (S473), consistent with this effect being due to upstream activation of PI3K, PTEN loss, or other mutation.
Rapamycin effects in the Kras - Tsc1 mouse lung cancer model
Since rapamycin is a highly specific therapy which blocks mTORC1 through interaction with FKBP12 (Sabers et al., 1995), we assessed the potential benefit of rapamycin as a treatment for the LSL-KrasG12D Tsc1L/L mouse model. Magnetic resonance imaging (MRI) was used to assess tumor extent before and after a 2 week period of treatment with rapamycin at 6 mg/kg IP every other day (Figure 3A). LSL-KrasG12D Tsc1L/L mice showed a clear response to rapamycin with an average 54% reduction in tumor extent, as assessed by MRI (Figure 3A, 4A). LSL-KrasG12D Tsc1L/+ tumors showed a smaller response with an overall 13% average reduction in tumor volume, though several mice showed a much greater response. LSL-KrasG12D mice showed on average stable disease (2% increase) during this period of treatment, while LSL-KrasG12D Lkb1L/L tumors in general showed major progression during rapamycin treatment (average 107% increase). Note that placebo-treated control mice of the LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Lkb1L/L genotypes (Figure 4A squares) showed a variable but generally marked increase in tumor extent during two weeks of observation while a much slower growth rate was seen in placebo-treated LSL-KrasG12D mice, consistent with their markedly different survivals (Figure 4A, 1A).
Figure 3. Rapamycin decreases tumor burden and causes apoptosis in LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mice.

(A) MRI images (left and middle columns) of mice before and after 2 weeks of rapamycin treatment (6 mg/kg IP every other day). H indicates the heart. H&E stained lung tissue sections (right column) after 2-5 weeks of rapamycin treatment show evidence of regression in the LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mice. The top three H&E sections were from mice treated for 2 weeks, the bottom mouse was treated for 5 weeks. Bar, 100μm.
(B) Representative photographs of cancers from the LSL-KrasG12D Tsc1L/L and LSL-KrasG12D mice showing apoptosis (TUNEL staining) without treatment, and after two doses of rapamycin. Bar, 100μm.
Figure 4. Quantitative tumor response, apoptosis, proliferation, and pS6 expression in response to rapamycin in LSL-KrasG12D Tsc1L/L, LSL-KrasG12D, and other mice.

(A) Relative change in tumor burden in mice treated with rapamycin for 2 weeks (circles). MRI scans from before and after treatment were assessed for tumor volume in mm3 by a blinded observer, and the ratio between after treatment and before treatment volumes was plotted. Six mice of various genotypes were placebo-treated controls in this experiment, shown as squares. The lines indicate average change among the treated mice.
(B) Scatter plots of number of positive cells per high power field (HPF) in lung tumor nodules stained to show evidence of apoptosis (TUNEL). The mice had genotypes LSL-KrasG12D Tsc1L/L or LSL-KrasG12D, and were treated with rapamycin 6mg/kg every other day for 0, 1, 2, or 4 doses, as indicated on the x axis.
(C) Scatter plots of number of positive cells per HPF in lung tumor nodules stained to show evidence of proliferation (antibodies against Ki67), as in B.
(D) Scatter plots of relative staining intensity for pS6(S240/244) in tumor nodules from rapamycin-treated mice, as in B.
For B-D, all pairwise comparisons between LSL-KrasG12D Tsc1L/L and LSL-KrasG12D tumors after the same number of doses are not significant (P > 0.05 by Mann Whitney test), except for the comparison between TUNEL positivity (B) after 2 and 4 doses, p=0.028 and 0.048 respectively.
Analysis of tumor sections from the mice after 1 to 4 doses of rapamycin showed that both LSL-KrasG12D Tsc1L/L and LSL-KrasG12D tumors showed increased TUNEL positivity consistent with apoptosis after rapamycin treatment (Figure 3B, 4B). However, this was more pronounced in the LSL-KrasG12D Tsc1L/L mice than in the LSL-KrasG12D mice after 2 and 4 doses of rapamycin (p=0.028 and 0.048, respectively, Figure 4B). In addition, LSL-KrasG12D Tsc1L/L mice showed evidence of a sustained reduction in proliferation following 2 and 4 doses of rapamycin, in contrast to LSL-KrasG12D mice, although this did not meet statistical significance (Figure 4C). In both types of mice there was marked reduction in pS6(S240/244) levels, as assessed by immunohistochemistry. pS6 (S240/244) levels were somewhat higher in the LSL-KrasG12D Tsc1L/L mice without treatment, but this did not meet statistical significance (Figure 4D).
Both LSL-KrasG12D Tsc1L/L mice and LSL-KrasG12D Tsc1L/+ mice treated with rapamycin at 6 mg/kg IP every other day showed a significant improvement in survival (Figure 5). A striking improvement in survival was seen in the LSL-KrasG12D Tsc1L/L mice (median survival 2.9 vs. 18.6 weeks, p = 0.0018). The improvement in survival in the LSL-KrasG12D Tsc1L/+ mice was somewhat less dramatic but still highly significant (6.9 vs. 16.3 weeks, p = 0.002), likely due in part to the slower tumor onset in LSL-KrasG12D Tsc1L/+ mice.
Figure 5. Rapamycin improves survival of LSL-KrasG12D Tsc1L/L and LSL-KrasG12D Tsc1L/+ mice.
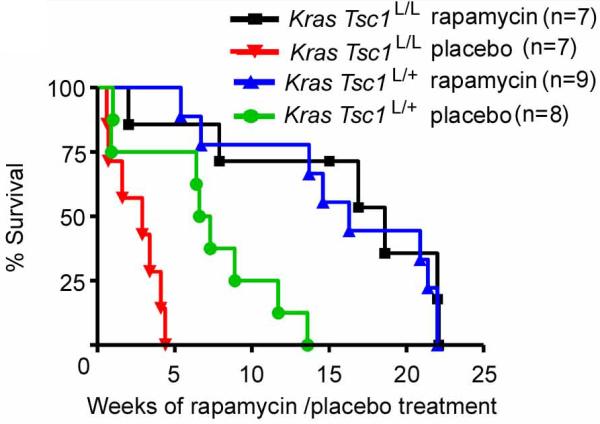
Kaplan-Meier survival curves of mice of two genotypes treated with either rapamycin (6 mg/kg IP every other day) or placebo, beginning 9 weeks following AdCre inhalation. Rapamycin significantly extended the survival of both LSL-KrasG12D Tsc1L/L (p=0.0018) and LSL-KrasG12D Tsc1L/+ mice (p=0.002).
Discussion
Lung cancer is the most common cause of cancer death for both females and males in the US population (Jemal et al., 2008; Molina et al., 2008). In the past 10 years, the greatest progress in lung cancer treatment research has been in the identification of a strong correlation between the effectiveness of molecularly targeted therapies (erlotinib, gefitinib) against the EGFR tyrosine kinase and tumor genotype (activating mutation in EGFR) (Ciardiello & Tortora, 2008). This experience lends hope to the concept that a detailed molecular characterization of each lung cancer may lead to the identification of unique molecularly targeted treatments designed to exploit the critical oncogenic signaling in each tumor. Here we show that lung cancers in the mouse in which TSC1/TSC2 function is eliminated or reduced are uniquely sensitive to a molecularly targeted therapy (rapamycin, RAD001, CCI-779, etc.). However, we observed that complete loss of TSC1/TSC2 is very rare in the human lung cancer cell lines we studied, consistent with other recent results (Ding et al., 2008).
We observed major in vivo synergism between activation of Kras and haploid or complete loss of Tsc1 in the development of lung cancer in the mouse. Mice that were either Tsc1L/L or Tsc1L/+, as well as LSL-KrasG12D, developed lung cancer on exposure to AdCre at a rate that was 1.8-2.4-fold faster than that seen in mice with LSL-KrasG12D alone (Figure 1A). Although there were distinct differences in histology and metastatic behavior, their survival was similar to that of AdCre-treated LSL-KrasG12D Lkb1L/L mice (Ji et al., 2007). In addition, it was more limited than that of AdCre-treated mice bearing the LSL-KrasG12D allele, and conditional alleles of either p16INK4a, p53, or Ink4a/Arf (Ji et al., 2007). Both Tsc1L/L LSL-KrasG12D and Tsc1L/+ LSL-KrasG12D mice demonstrated unique sensitivity to treatment with rapamycin as a single agent, in contrast to mice with Kras activating mutation alone, or LSL-KrasG12D Lkb1L/L mice. Survival of both genotypes of Kras-Tsc1 mice was significantly prolonged in response to rapamycin treatment (Figure 5). Our observations that rapamycin treatment was able to cause stable disease (neither growth of tumor nor significant reduction) in the LSL-KrasG12D only mice is similar to a previous report in a different Kras lung model in which CCI-779 was given as an mTORC1 inhibitor (Wislez et al., 2005). In that study, high dose CCI-779 (20 mg/kg daily for 4 weeks) led to stabilization of lung tumor extent, with prevention of the appearance of new lung tumors, and a small reduction in tumor size, but mainly due to induction of apoptosis in tumor-infiltrating macrophages (Wislez et al., 2005).
We found that none of 80 NSCLC lines examined had a signaling pattern similar to that of TSC1 null or TSC2 null MEFs (Zhang et al., 2003; Harrington et al., 2004), and that all expressed both TSC1 and TSC2 to some extent. However, we did observe that 22% of human lung cancers and 14% of lung cancer cell lines displayed loss of a single allele of either TSC1 or TSC2 (Figure 2), similar to a previous report (Takamochi et al., 2001). This lack of occurrence of homozygous loss of either TSC1 or TSC2 in human lung cancer is also consistent with a larger body of data indicating that neither gene is a common targets for mutation/inactivation in human cancer. This is in striking contrast to other tumor suppressor genes that regulate mTOR signaling, including LKB1 which is commonly involved in lung cancer (Ji et al., 2007; Sanchez-Cespedes, 2007; Ding et al., 2008), and PTEN, less commonly involved in lung cancer, but much more so in colon cancer and glioblastoma. Indeed the only common adult malignancy in which TSC1 or TSC2 loss has been reported to occur at appreciable frequency is bladder cancer (Hornigold et al., 1999; Adachi et al., 2003; Platt et al., 2009). LKB1 and PTEN have broader effects beyond activation of mTORC1, and this may partially explain their more common involvement in adult cancers.
The reason for this discordance between lack of involvement of TSC1/TSC2 in human lung cancer, and a clear pathogenic role in this mouse model of lung cancer is not clear. One possibility is that due to their location in the genome, TSC1 and TSC2 are uncommon targets for genomic and mutational events. However, second hit events occur commonly in these genes in the tumors of TSC patients (Henske et al., 1996), arguing against a model of ‘relative genomic protection’. A second possibility is the observation that AKT activation is suppressed in tumors from TSC patients, Tsc mouse model tumors, and cell lines in which TSC1/TSC2 is completely lost (Zhang et al., 2003; Harrington et al., 2004). This has been attributed to a variety of feedback mechanisms, including suppression of IRS function, reduced PDGFR expression, and most recently elucidation of a functional role for the TSC1/TSC2 complex as a co-factor for the kinase activity of mTORC2, the AKT S473 kinase (Zhang et al., 2003; Harrington et al., 2004; Huang et al., 2008). Although this explanation is plausible and may contribute, one wonders why this does not occur in the mouse lung model studied here, in which loss of Tsc1 has a clear growth promoting effect, accelerating development of lung cancer. It is possible that the combination with Kras activating mutation and/or the timing of this combination is critical in leading to the accelerated lung tumor development in these mice.
The observation that haploid loss of Tsc1 in this mouse model had similar effects in accelerating lung tumorigenesis as complete loss (Figure 1A), is consistent with a model in which attenuated TSC1/TSC2 function leads to some activation of mTORC1 and complements KRAS mutational activation nearly as well as complete loss. The observation that TSC1 LOH is fairly common in NSCLC patient samples (Takamochi et al., 2001), is also consistent with a potential growth promoting effect of haploid expression. Haploinsufficiency is well-known as a growth promoting effect for a number of tumor suppressor genes, including PTEN, NF1, and that encoding p27Kip1 (Santarosa & Ashworth, 2004; Smilenov, 2006). In this mouse model, it is possible that ERK-mediated phosphorylation of TSC2, due to activated KRAS, synergizes with haploid loss of TSC1 to lead to inactivation of the TSC1/TSC2 protein complex, and mTORC1 activation (Ma et al., 2005).
Two mTORC1 inhibitors have been used in Phase I-II clinical trials in NSCLC patients. Both RAD001 and CC-779 were seen to have overall a 3-5% response rate, though in one trial in which CCI-779 was used, this reached 8% (Gridelli et al., 2008). Two small Phase I-II combination trials of RAD001 and gefitinib have been completed, with higher response rates (Milton et al., 2007), and several more are ongoing (Gridelli et al., 2008). Our data suggest the possibility that patients showing response might be those with haploid expression of TSC1/TSC2 in their lung cancers, but further study is required. Single agent rapamycin was quite dramatically effective in reducing tumor size (Figure 3A), and extending survival (Figure 5) of the LSL-KrasG12D Tsc1L/L mice, in contrast to limited or no effect on the tumors of mice with Kras mutation alone or combined with Lkb1 loss. This is consistent with the multiple downstream targets that are influenced by Lkb1 loss (Alessi et al., 2006). In addition, rapamycin as a single agent has clear activity in both the renal epithelial tumors that develop in mouse models of TSC (Kenerson et al., 2005; Lee et al., 2005; Pollizzi et al., 2009), and some activity in kidney angiomyolipomas that occur in TSC patients (Bissler et al., 2008). Thus, this additional experience in both preclinical models and patients supports the concept that rapamycin has particular effectiveness in tumors lacking functional TSC1/TSC2. Although evidently rare, lung cancers which completely lack TSC1/TSC2 are also likely to be very sensitive to rapamycin.
Supplementary Material
Acknowledgements
We thank Kate McNamara and Sara Zaghlul (Dana-Farber Cancer Institute, Boston), and Christine Lam, June Goto, Iza Malinowska-Kolodziej, and Mei Zheng (Brigham and Women’s Hospital, Boston) for technical assistance; and Roderick T. Bronson (Harvard Medical School, Boston) and Robert Padera and Lucian Chirieac (Brigham and Women’s Hospital, Boston) for assistance with pathology review.
Financial support:
This work was supported in part by the NIH grants R01 AG2400401 (K.-K.W.), R01 CA122794 (K.-K.W.), K08 AG024004 (K.-K.W.), P01 CA120964 (D.J.K.), the Sidney Kimmel Foundation for Cancer Research (K.-K.W.), the Joan Scarangello Foundation to Conquer Lung Cancer (K.-K.W., D.J.K.), and the Flight Attendant Medical Research Institute (K.-K.W.).
Footnotes
Conflict of interest
There is no competing financial interest in relation to this work.
References
- Adachi H, Igawa M, Shiina H, Urakami S, Shigeno K, Hino O. Human bladder tumors with 2-hit mutations of tumor suppressor gene TSC1 and decreased expression of p27. J Urol. 2003;170:601–604. doi: 10.1097/01.ju.0000074621.74361.10. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridelli C, Maione P, Rossi A. The potential role of mTOR inhibitors in non-small cell lung cancer. Oncologist. 2008;13:139–147. doi: 10.1634/theoncologist.2007-0171. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda) 2006;21:48–60. doi: 10.1152/physiol.00044.2005. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, Hornigold N, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59:400–406. [PMC free article] [PubMed] [Google Scholar]
- Hornigold N, Devlin J, Davies AM, Aveyard JS, Habuchi T, Knowles MA. Mutation of the 9q34 gene TSC1 in sporadic bladder cancer. Oncogene. 1999;18:2657–2661. doi: 10.1038/sj.onc.1202854. [DOI] [PubMed] [Google Scholar]
- Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28:4104–4115. doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- Kenerson H, Dundon TA, Yeung RS. Effects of rapamycin in the Eker rat model of tuberous sclerosis complex. Pediatr Res. 2005;57:67–75. doi: 10.1203/01.PDR.0000147727.78571.07. [DOI] [PubMed] [Google Scholar]
- Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389–400. doi: 10.1007/s00439-006-0308-9. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- Lee L, Sudentas P, Donohue B, Asrican K, Worku A, Walker V, et al. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42:213–227. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- Li D, Shimamura T, Ji H, Chen L, Haringsma HJ, McNamara K, et al. Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell. 2007;12:81–93. doi: 10.1016/j.ccr.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133:507–516. doi: 10.1378/chest.07-0898. [DOI] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton DT, Riely GJ, Azzoli CG, Gomez JE, Heelan RT, Kris MG, et al. Phase 1 trial of everolimus and gefitinib in patients with advanced nonsmall-cell lung cancer. Cancer. 2007;110:599–605. doi: 10.1002/cncr.22816. [DOI] [PubMed] [Google Scholar]
- Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir TE, Leslie KO, Popper H, Kitaichi M, Gagne E, Emelin JK, et al. Micronodular pneumocyte hyperplasia. Am J Surg Pathol. 1998;22:465–472. doi: 10.1097/00000478-199804000-00012. [DOI] [PubMed] [Google Scholar]
- Pollizzi K, Malinowska-Kolodziej I, Stumm M, Lane H, Kwiatkowski D. Equivalent benefit of mTORC1 blockade and combined PI3K-mTOR blockade in a mouse model of tuberous sclerosis. Mol Cancer. 2009;8:38. doi: 10.1186/1476-4598-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt FM, Hurst CD, Taylor CF, Gregory WM, Harnden P, Knowles MA. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. 2009;15:6008–17. doi: 10.1158/1078-0432.CCR-09-0898. [DOI] [PubMed] [Google Scholar]
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270:815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007;26:7825–7832. doi: 10.1038/sj.onc.1210594. [DOI] [PubMed] [Google Scholar]
- Santarosa M, Ashworth A. Haploinsufficiency for tumour suppressor genes: when you don’t need to go all the way. Biochim Biophys Acta. 2004;1654:105–122. doi: 10.1016/j.bbcan.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Smilenov LB. Tumor development: haploinsufficiency and local network assembly. Cancer Lett. 2006;240:17–28. doi: 10.1016/j.canlet.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Takamochi K, Ogura T, Suzuki K, Kawasaki H, Kurashima Y, Yokose T, et al. Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am J Pathol. 2001;159:1941–1948. doi: 10.1016/S0002-9440(10)63041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wislez M, Spencer ML, Izzo JG, Juroske DM, Balhara K, Cody DD, et al. Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-ras. Cancer Res. 2005;65:3226–3235. doi: 10.1158/0008-5472.CAN-04-4420. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.