

Abstract
MicroRNAs (miRNAs) have recently emerged as an important new class of cellular regulators that control various cellular processes and are implicated in human diseases, including cancer. Here, we show that loss of let-7 function enhances lung tumor formation in vivo, strongly supporting the hypothesis that let-7 is a tumor suppressor. Moreover, we report that exogenous delivery of let-7 to established tumors in mouse models of non-small cell lung cancer (NSCLC) significantly reduces tumor burden. These results demonstrate the therapeutic potential of let-7 in NSCLC and point to miRNA replacement therapy as a promising approach in cancer treatment.
Keywords: Let-7, microRNAs, lung cancer, K-ras
Introduction
MicroRNAs (miRNAs) control various cellular processes such as cell death, differentiation, and development and are implicated in human diseases, including cancer (Esquela-Kerscher and Slack, 2006). Aberrant expression of specific miRNAs is associated with tumorigenesis and miRNA coding genes are often found in fragile genomic regions gained and lost in cancers (Calin et al., 2004b; Sevignani et al., 2007). Many members of one miRNA family in particular, let-7, map to chromosomal regions frequently deleted in lung cancer (Calin et al., 2004b), and reduced let-7 expression in non-small cell lung cancer (NSCLC) patients is correlated with poor prognosis (Takamizawa et al., 2004; Yanaihara et al., 2006). Moreover, let-7 miRNAs are thought to function as tumor suppressors through their negative regulation of multiple oncogenes, such as RAS, MYC, HMGA2, and promoters of cell cycle progression, such as CDC25A, CDK6, and Cyclin D2 (Johnson et al., 2007; Johnson et al., 2005; Lee and Dutta, 2007; Mayr et al., 2007; Park et al., 2007; Sampson et al., 2007; Yu et al., 2007). Administration of let-7 blocks the growth of cultured lung cancer cells and also prevents the onset of tumor formation in a mouse model of NSCLC (Esquela-Kerscher et al., 2008; Johnson et al., 2007; Kumar et al., 2008).
Materials and Methods
Lung cancer xenografts
Human H460 non-small cell lung carcinoma cells were cultured in RPMI media (Invitrogen, Carlsbad, CA) following standard tissue culture procedures. H460 cells were trypsinized, counted and subcutaneously injected into the lower back of 6–8 week old NOD/SCID mice (Jackson Laboratories, Bar Harbor, MA) using 3 × 106 cells in 100 μl RPMI with 50% matrigel (BD Biosciences, San Jose, CA) per injection. Once cancer cells have developed palpable tumors caliper measurements were taken daily and tumor volume was calculated using the formula V = length × width2/2, in which the length is greater than the width. When tumors reached an average volume of 150 mm3, 50 μl synthetic miRNA complexed with the siPORTamine transfection reagent (Ambion) was delivered intratumorally in 3-day intervals. Synthetic miRNAs are double-stranded and ready-to-use miRNA mimics and were purchased from Ambion, Life Technologies, Austin, TX (pre-miR; cat. no. AM17100). For each injection, 6.25 ug miRNA was complexed with 1.6 μl siPORTamine (Ambion; cat. no. AM4502) reagent in 50 μl phosphate-buffered saline. Mice were sacrificed by CO2 inhalation either one or three days after the last treatment, and tumors were collected and prepared for histology and RNA isolation. All animal experiments were performed under an IACUC approved animal study protocol.
Quantitative real-time PCR
Total RNA from H460 tumors was isolated using the mirVANA PARIS RNA isolation kit (Ambion, Austin, TX) following manufacturer’s instructions. For RT-PCR detection of let-7 mRNA targets in H460 xenografts, 500 ng purified RNA was reverse transcribed with random decamers using MMLV-RT (cat. no. 28025-021, Invitrogen, Carlsbad, CA) with the following incubations: 42°C for 60 min; 85°C for 5 min. For RT-PCR detection of the let-7b oligonucleotide, 10 ng purified RNA was heat-denatured at 70 °C for 2 min and reverse transcribed using the let-7b TaqMan miRNA Assay (Applied Biosystems, Foster City, CA) with the following conditions: 16°C for 30 min; 42°C for 30 min; 85°C for 5 min and MMLV-RT (Invitrogen). Gene and let-7b expression levels were determined by real-time PCR using Platinum Taq Polymerase reagents (Invitrogen) on the ABI Prism 7900 SDS (Applied Biosystems). TaqMan Gene Expression Assays (Applied Biosystems) were used with the following cycling conditions: 95°C for 1 min (initial denature); then 50 cycles of 95°C for 5 sec, 60°C for 30 sec. The 18S rRNA was amplified as an internal reference to adjust for well-to-well variances in amount of starting template. The let-7b TaqMan miRNA Assay (Applied Biosystems) was used with the following cycling conditions: 95°C for 1 min (initial denature); then 50 cycles of 95°C for 15 sec, 60°C for 1 min. Total copy numbers of let-7b molecules in tumor tissues were calculated using a standard curve generated with 103–1012 let-7b copies amplified on the same plate. Quantification of levels of let-7a, was performed using the Taqman microRNA PCR system (ABI, per standard protocol). Levels were normalized to mice treated with Ad-Cre alone (baseline) to determine changes in expression levels 4 weeks post lentivirus infection.
Tumor histologies and immunohistochemistries
Tumor tissues were fixed in formalin and embedded in paraffin using the Microm Tissue Embedding Center (Labequip, Ltd.; Markham, Ontario, Canada). 5 μm tissue sections were prepared and stained with hematoxylin and eosin (H&E) according to standard protocols. For immunohistochemistries, primary antibodies specific for Ki-67 (cat. no. M7249; DAKO, Carpinteria, CA), active Caspase-3 (cat. no. AF835; R&D Systems, Minneapolis, MN), N-Ras (cat. no. sc-20; Santa Cruz Biotechnology, Santa Cruz, CA) and CDC25A (cat. no. sc-97; Santa Cruz Biotechnology) were used. All antibodies were visualized by secondary horseradish peroxidase-conjugated immunoglobulins (cat. no. ab6721; Abcam, Cambridge, MA). Briefly, slides were washed in tris-buffered saline (TBS; 10 mM Tris, 150 mM NaCl, pH 7.6) and incubated in 3% (v/v) hydrogen peroxide for 10 min to suppress endogenous activity. Slides were washed with TBS and incubated in 5% (v/v) normal goat serum (cat. no. 005-000-001; Jackson ImmuResearch Laboratories, Inc., West Grove, PA) diluted in incubation buffer (0.1% [w/v] BSA in TBS) to reduce nonspecific binding of antibodies. Slides were incubated with primary antibodies in incubation buffer overnight at 4°C. After several washes in TBS, secondary antibodies were added and incubated for 60 min at room temperature. Secondary antibodies were visualized by adding 3,3′-diaminobenzidine (DAB; cat. no. K3465; DAKO) for 2–5 minutes, followed by several washing steps. Negative controls were performed by omitting the primary antibody. The slides were counterstained with hematoxylin for 10 seconds and mounted on cover slips with mounting medium (cat. no. 4112; Richard-Allan Scientific, Kalamazoo, MI). For determining apoptotic bodies by TUNEL assay, ApopTag Plus Peroxidase In Situ Apoptosis Kit (Chemicon, Temecula, CA, USA) was used according to the manufacturer’s instructions.
Anti-scr and anti-let-7 synthesis
The anti-miRs were synthesized by the Keck Facility (Yale University New Haven, CT, USA). To enhance their stability the anti-miRs were generated using 2′-O-methyl-modified nucleotides (italic) and phosphorothioate bonds (*) as follows:
anti-let-7g: 5′ A*C*UGUACAAACUACUACCU*C*A 3′;
anti-scramble: 5′C*U*CAACAACGUAUAUUCAG*C*A 3′.
The anti-scramble RNA was designed using the Scramble RNA software to have same GC% than anti-let-7g but no homology with the mouse genome (Levenkova et al., 2004). The anti-miR-reporter has a fluorescein amidite (FAM) attached at 5′ of anti-let-7g to follow their delivery using a fluorescent microscope with GFP detection wavelengths.
In vivo adenoviral infection and anti-miR delivery to LSL-K-ras G12D mice
Approximately 6-week-old LSL-K-ras G12D animals were anesthetized with an isoflurane/propylene glycol mixture and intranasally co-inoculated with adenovirus and anti-miRs (anti-let-7g or scrambled). The protocol was adapted from (Bitko and Barik, 2008; Bitko et al., 2005; Esquela-Kerscher et al., 2008; Jackson et al., 2001): 5×108 PFU of Ad-Cre, 60 μg of anti-miR and 0.01M CaCl2 were mixed in a final volume of 125 μl of MEM and inoculated to the mice in two 62.5 μL instillations as described (Jackson et al., 2001). Ad-Cre, used to induce tumor formation in LSL-K-ras G12D mice, was purchased from the Gene Transfer Vector Core Facility at the University of Iowa. Mice were sacrificed 7 weeks post-infection, and the PBS-perfused lungs were retained for analysis. Lungs were prepared for histological analysis to examine tumor burden by fixing the tissue in 4% paraformaldehyde overnight, embedded in paraffin and stained with hematoxylin and eosin. The delivery of anti-miRs in the lung was tested using anti-miR-5′ FAM oligonucleotides using a fluorescent microscope. Ad-Cre recombination was tested using a PCR assay to verify that K-ras G12D mice had undergone Cre-mediated removal of the stop element at the mutant K-ras locus as previously described (Jackson et al., 2001). Lung and tumor areas were quantified using ImageJ software in manual measurement mode as described previously (Esquela-Kerscher et al., 2008). The overall tumor burden was measured as a ratio of total tumor area to total lung area by taking the average of every seventh slide section throughout the lung. Nomenclature based on that of Nikitin et al.(Nikitin et al., 2004).
Viral constructs and in vivo infection
Adenovirus expressing Cre-recombinase was obtained as described previously (Jackson et al., 2001). Lentivirus expressing let-7a was purchased from Select Biosciences (Mountain View, CA). LSL-K-ras G12D (Jackson et al., 2001) (strain number 01XJ6) mice were obtained from the NCI-Frederick Mouse Repository and treated with Adenovirus and Lentivirus as described previously (Esquela-Kerscher et al., 2008).
Results
In vivo anti-let-7 delivery to lungs leads to increased tumor burden
Available data suggest that the let-7 miRNA functions as a tumor suppressor that is frequently lost in non-small cell lung cancer (NSCLC) (Johnson et al., 2005; Takamizawa et al., 2004; Yanaihara et al., 2006), yet no loss of function assays have been reported to formally test this hypothesis. The mouse let-7 miRNA family consists of multiple members, encoded by 12 genes in the mouse genome, nine of which code for unique mature let-7 molecules, and three that code for redundant sequences (Roush and Slack, 2008). We previously showed that five of the let-7 family members, let-7a, let-7b, let-7c, let-7d, and let-7g consistently reduced the number of proliferating A549 and HepG2 cells by similar levels (Johnson et al., 2007). Because we have not yet detected a difference with any particular let-7 family member, this suggests functional redundancy. Since it is likely that their biological functions overlap, in vivo loss of functional genetic tests with the let-7 family are technically difficult. To circumvent this difficulty, we developed a novel protocol to test the role of let-7 family members as tumor suppressors in a well-characterized K-ras autochthonous NSCLC mouse model lung (Jackson et al., 2001), LSL-K-ras G12D, containing a Cre recombinase dependent allele of activated Kras with endogenous 5′ and 3′ regions. We combined a successful protocol for delivery of small RNAs by intranasal passage (Bitko and Barik, 2008; Bitko et al., 2005) with established in vivo antisense technology (Krutzfeldt et al., 2005). We designed an anti-miR molecule targeting the let-7g sequence (anti-let-7g) and a control with same GC percentage but no homology in the mouse genome (anti-scrambled (scr)) (See Materials and Methods for details). We chose let-7g for this experiment because let-7g maps to 3p21 which has been implicated in the initiation of human lung cancers (Calin et al., 2004a) and also because there is evidence suggesting that let-7g mediated tumor suppression is especially potent in tumors harboring oncogenic K-ras mutations (Kumar et al., 2008). However, we did not rule out the possibility that our anti-let-7g molecule cross-reacts with other let-7 family members due to their high similarity.
To monitor the delivery of anti-miRs to the lung, we labeled the anti-miR with a FAM molecule attached at its 5′OH. We inoculated this labeled anti-miR into mouse lungs by intranasal passage and harvested their lungs 2 hours (h) and 16h later. Both time points showed similar fluorescence signal in the lung (Suppl Fig. 1), demonstrating efficient and quick up-take of the modified RNAs. These observations match previously reported data from Bitko et al.(Bitko and Barik, 2008) showing that naked siRNAs can be successfully delivered to the lung via intranasal passage and remain stable.
We transiently delivered non-labeled anti-miRs to mice carrying a knock-in activated K-rasG12D allele, that we had simultaneously inoculated with an adenovirus expressing Cre recombinase (Ad-Cre) to activate LSL-K-ras G12D (Suppl. Fig 2) and initiate lung tumors (as described in Esquela-Kerscher et al. (Esquela-Kerscher et al., 2008)). In both cases, the lungs show a range of proliferative lesions including bronchiolar papillary hyperplasia, adenoma, atypical adenomatous hyperplasia and focal low grade adenocarcinoma. However, mice treated with anti-let-7g (n=5) displayed more extensive (p<0.01) tumor burden 7 weeks after intranasal inoculation relative to anti-scr control (n=5) with more prominent bronchiolar papillary hyperplasia (Fig. 1). We speculate that transient knock-down of let-7g (and potentially other let-7 family members) function enhances tumorgenicity by elevating levels of let-7 target genes soon after delivery. This could act synergistically with the K-ras mutation to form a more aggressive NSCLC phenotype similar to the case of the double mutant mouse model K-ras G12D; Trp-53flox/flox (Jackson et al., 2005). This experiment demonstrates a role for let-7 in the suppression of lung tumors via K-ras (Johnson et al., 2005) in vivo and is strong evidence that let-7 functions as a tumor suppressor in the lung.
Figure 1. Anti-let-7 enhances tumor formation in activated K-ras lungs.

Macroscopic observation of the K-ras G12D mice lungs seven weeks after infection with Ad-Cre. The lung surface of the mice treated with anti-let-7 (A, C) shows higher levels of precancerous lesions in comparison with the lungs treated with anti-scr (B, D). This observation correlates with a higher load of neoplastic development observed histologically (C, D). Slides stained by H&E x100 amplified show advanced tumor development in lungs treated with anti-let-7 ((E) extended adenoma with acinar and papillar structures) in comparison with the lungs treated with anti-scr ((F) earlier neoplastic lesions such as adenomatous and epithelial hyperplasia shown with arrow). G. Quantification of total tumor area. The ratios of tumor area versus normal lung area are presented as a box-and-whisker plot. Boxes represent interquartile ranges (between the 25th and 75th quartiles) and the two-tailed p-value is indicated. The total range, mean (◇), and median (blank bar) are shown.
Intratumoral delivery of let-7 reduces tumor size in a lung cancer xenograft model
To study the anti-tumorigenic role of let-7 in lung cancer, we had previously examined two murine models of human lung cancer, a xenograft model using human lung cancer cells and the LSL-K-ras-G12D model. In the latter mouse model, tumorigenesis was initiated by the activation of a gain of function K-ras G12D gene through the inhalation of Ad-Cre (Jackson et al., 2001) as described above. Using these models, we showed that let-7 suppresses lung tumor initiation, resulting in a 66% reduction of tumor burden when comparing K-ras G12D mice treated with let-7 miRNA to those that were treated with a control miRNA, respectively (Esquela-Kerscher et al., 2008). Similar findings were made by the Jack’s lab (Kumar et al., 2008). Since in both studies the let-7 miRNA was delivered at the same time as tumorigenesis was initiated, these two studies showed that let-7 may be useful as a preventive therapy against lung cancer in the LSL-K-ras G12D mice. While these results revealed an inherent anti-oncogenic activity of let-7 during tumorigenesis, the therapeutic potential of let-7 on established tumors remained unknown.
Because most newly diagnosed cases of NSCLC are locally advanced or metastatic at presentation and refractory to most currently applied therapies (Isobe et al., 2005), we explored the therapeutic potential of let-7 in established tumors. Immunodeficient NOD/SCID mice were subcutaneously inoculated with human H460 lung cancer cells and housed until the tumor cells – initially loosely distributed in matrigel – had formed solid and palpable tumors with an average volume of 100–150 mm3. We chose the H460 cell line for this study because it is among the most aggressively growing NSCLC xenografts and is among the cell lines most resistant to other targeted therapies (Hohla et al., 2007; Siegfried et al., 1999). Beginning on day 11 following inoculation, let-7b or a negative control miRNA (miR-NC) were repeatedly administered by intratumoral injections every 3 days. These synthetic miRNAs were complexed with siPORTamine (siPORT), a lipid-based transfection reagent that enhances cellular uptake of the oligonucleotide. All mice were sacrificed once control animals developed tumors that exceeded an average volume of 600 mm3 (day 21). As shown in Figure 2A, mice that received miR-NC developed tumors at a pace similar to those treated with siPORT alone or phosphate-buffered saline (PBS). In contrast, local delivery of synthetic let-7b induced a specific inhibitory response and robustly interfered with tumor growth. Images of tumor-bearing mice on the day of sacrifice are shown in Figure 2B.
Figure 2. Growth inhibition of established lung tumor xenografts by synthetic let-7 oligonucleotides.
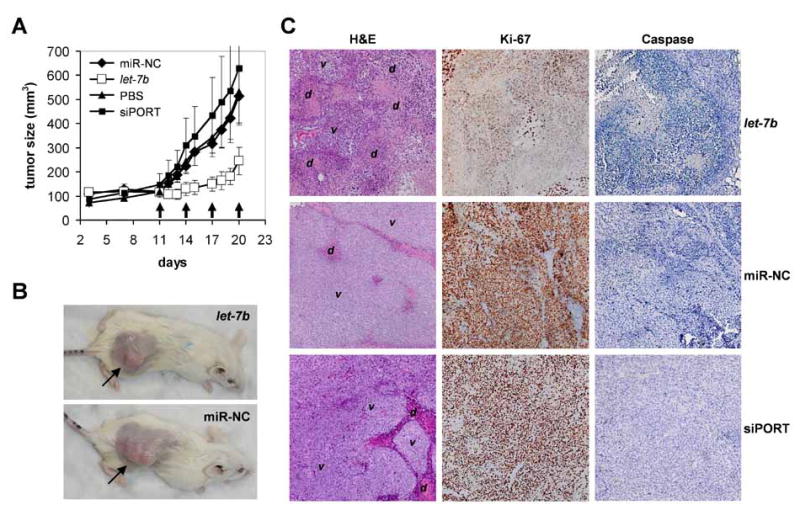
(A) Three × 106 H460 lung cancer cells in 50% matrigel were subcutaneously injected into immunodeficient NOD/SCID mice. On days 11, 14, 17 and 20, synthetic let-7b or miR-NC double-stranded and ready-to-use miRNAs conjugated with the siPORTamine transfection reagent were intratumorally delivered into groups of six animals. As additional negative controls, tumor-bearing mice received intratumoral injections of PBS (n=5) or the lipid formulation lacking any oligonucleotide (siPORT; n=6). Caliper measurements were taken to determine the length and width of each tumor and to calculate total tumor volumes. Standard deviations are shown in the graph. Arrows indicate days of treatment. (B) Images of mice carrying H460 tumors after sacrifice on day 21. Subcutaneous lung tumors are indicated by arrows. (C) Tumor histologies and immunohistochemistry staining specific for Ki-67 and active caspase 3 are shown. v, area containing viable H460 tumor cells; d, area containing dead cells, cell debris or cells undergoing cell death. No difference in caspase activity was detected between experiment and controls.
A histological analysis revealed that tumors treated with miR-NC or siPORT were densely packed with viable H460 cells and only occasionally contained small necrotic areas (Fig. 2C, left panel, center and bottom). These pockets of dead cells are likely due to the aggressive nature of H460 cells that rapidly outgrow other cancer cells and compete for blood supply and nutrients (Edinger and Thompson, 2004). Tumors that received let-7b, however, contained large areas filled with cell debris (Fig. 2C, left panel, top). These tumors also had enlarged necrotic cores in agreement with the supposition that intratumoral injections preferentially deliver the therapeutic to the center of the tumor. To get better insight into the mechanism of action, we performed immunohistochemistry stains using antibodies specific for the Ki-67 and caspase 3 proteins, markers for cellular proliferation and apoptosis, respectively. As shown in Figure 2C (right panel), H460 cells displayed reduced levels of Ki-67 in response to let-7b treatment. In contrast, caspase 3 levels remained unaltered. It is possible that let-7 does not specifically induce the apoptotic cascade or alternatively, apoptosis induced by let-7 occurred immediately after administration and is no longer detectable at the time of the analysis. This observation is in accord with our previous observations and indicates a role for let-7 over-expression in interfering with cell cycle progression rather than a major role in directly inducing programmed cell death (Esquela-Kerscher et al., 2008; Johnson et al., 2007).
To better correlate the therapeutic response with the delivery of let-7b, we determined the presence of let-7b oligonucleotides in tumor tissues 24 hours after the final administration. Total RNA from H460 tumors treated with let-7b, miR-NC, siPORT or PBS were analyzed by quantitative real-time PCR (qRT-PCR) using let-7b specific primers. Tumors injected with the let-7b oligo contained ~14-fold more let-7b miRNA compared to the endogenous levels that are present in the controls (Fig. 3A). Since crude tumor tissue was used for this analysis, these levels include extracellular as well as intracellular let-7b. To confirm that the synthetic let-7b has successfully been delivered into the cytoplasm of H460 tumor cells and actively engages in the RNA-induced silencing pathway, we evaluated the mRNA levels of NRAS and CDK6, both of which are directly targeted by the let-7 miRNA(Esquela-Kerscher et al., 2005; Johnson et al., 2007). We chose N-ras as a readout for let-7 activity because unlike the K-ras mRNA, N-ras mRNA seems to undergo degradation in response to let-7 treatment (Johnson et al., 2005). As shown in Figure 3B, both transcripts were strongly repressed in tumors that have been injected with the let-7b miRNA. Hence, these mRNA levels inversely correlate with increased accumulation of let-7b in these tumors, illustrating the successful cellular uptake of the synthetic let-7 mimetic (see Fig. 3A). In addition, the down-regulation of let-7 targets was confirmed on the protein level by immunohistochemistry directed against proteins for which suitable antibodies were available (Fig. 3C). Similar to the reduction of the mRNA, tumors treated with let-7b lacked expression of NRAS protein relative to tumors that received either miR-NC or siPORT. CDC25A protein, another let-7 target that was less responsive on the mRNA level, also showed reduced protein expression in tumors that were injected with let-7b (Johnson et al., 2007). Thus, the synthetic let-7b miRNA successfully entered H460 tumor cells and activated the let-7 silencing pathway to suppress its targets and reduce tumor growth.
Figure 3. let-7 accumulation and activity in H460 tumor xenografts.
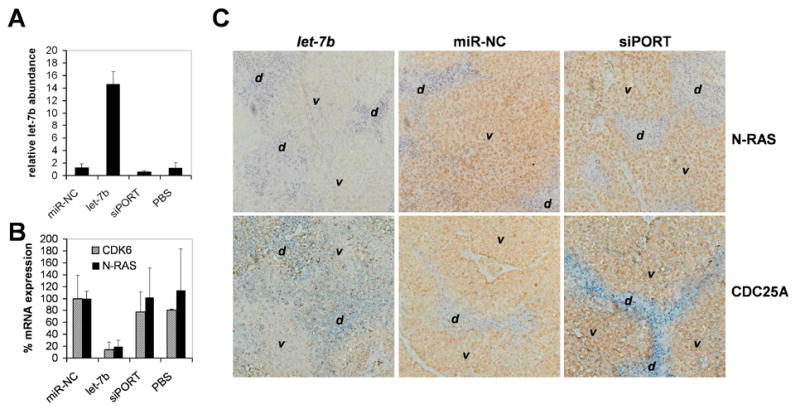
(A) Relative let-7b abundance in H460 tumors. qRT-PCR was performed using primers specific for let-7b and total RNA from tumors harvested 1 day post the final treatment. Ct values were used to calculate absolute let-7b copy numbers and expressed relative to the average expression in control tumors (1.0). Averages and standard deviations of 3 animals (miR-NC and let-7b) or 2 animals (siPORT, PBS) are shown. (B) qRT-PCR analysis measuring mRNA levels of let-7 targets. qRT-PCR was carried out using total RNA from H460 tumors harvested on day 21 and primers specific for the CDK6 and N-RAS transcripts. Raw Ct values were normalized to the ones of GAPDH, a house-keeping gene not regulated by let-7b, and graphed relative to CDK6 and N-RAS expression in miR-NC treated tumors (100%). Averages and standard deviations of 3 animals (miR-NC and let-7b) or 2 animals (siPORT, PBS) are shown. (C) Immunohistochemistries showing protein expression of the let-7 targets N-RAS and CDC25A. Staining of tumors treated with let-7b or of control tumors were carried out in parallel. v, area containing viable H460 tumor cells; d, area containing dead cells, cell debris or cells undergoing cell death.
Intranasal delivery of let-7 reduces established K-ras dependent lung tumors
To address whether let-7 miRNA can reduce tumor burden in late stage K-ras G12D animal lungs, 5 × 108 plaque-forming units (p.f.u) of Ad-Cre were administered intranasally to six-week-old LSL-Kras-G12D mice and the mice were maintained for 10 weeks. The 10 weeks post-infection group was split into 3 sets. One set acted as a baseline for the study (n=6) (Fig. 4A, bottom, left and center panels,). A second set was treated with 1× 106 infectious units (i.f.u) of a lentiviral vector expressing let-7a (lenti-let-7 that showed a three to four fold up-regulation of let-7 miRNA in A549 cells (Suppl Fig. 3)). The third set was treated with a control empty lentiviral vector (lenti-control). Baseline animals were sacrificed at 10 weeks post Ad-Cre infection. All other animals were incubated further and sacrificed either 2 or 4 weeks post lentivirus infection, and lung morphology, cell proliferation and apoptosis were assessed immunohistochemically. We observed similar lung morphology for both baseline mice and lenti-control mice that were sacrificed at 2 and 4 weeks post lentivirus infection. Baseline mice and lenti-controls showed bronchiolar papillary hyperplasia and extensive replacement of the parenchyma predominantly by papillary adenocarcinoma (Figure 4A lower two rows, Fig 4B, suppl Fig 4). In mice given lenti-let-7, we observed residual bronchiolar papillary hyperplasia but markedly reduced parenchymal tumor burden when compared to those treated with the lenti-control (75% reduction in tumor area) and the baseline animals (64% reduction), respectively (Fig. 4A, Top; Fig. 4B; Suppl Fig. 4). Also, a qRT-PCR analysis revealed that lungs from mice treated with lenti-let-7 contained ~5-fold more let-7 oligonucleotides compared to controls (Suppl. fig. 5A). These results show that let-7 can effectively cause remission of lung tumors in an advanced K-ras activated NSCLC mouse model.
Figure 4. let-7 reduces lung tumor burden in an autochthonous NSCLC model.

Whole lungs, tumor histologies and immunohistochemistries specific for Ki-67 are shown. (A) (left and center panels, top) LSL-K-ras G12D mice administered with Ad-Cre (5×108) pfu for 10 weeks then lenti-let-7 (1×106 ifu) for four weeks display reduced lung lesions, hyperplasias and adenomas compared to (left and center panels, center) LSL-K-ras G12D mice treated with Ad-Cre (5×108) for 10 weeks then the empty lenti-control (1×106 ifu) for four weeks and (left and center panels, bottom) baseline animals, which were sacrificed at 10 weeks after Ad-Cre infection. (Right panel) Ki-67 staining of lungs. (B) Quantitative analysis of tumor burden in LSL-K-ras G12D animals treated with Ad-Cre and lenti-let-7 (n=6) versus LSL-K-ras G12D animals treated with Ad-Cre and lenti-control (n=6) and Ad-Cre treated baseline animals (n=6). The ratios of tumor area versus normal lung area are presented as a box-and-whisker plot. Boxes represent interquartile ranges (between the 25th and 75th quartiles) and the two-tailed p-value is indicated. The total range, mean (◇), and median (blank bar) are shown. pfu, plaque forming units; ifu, infectious units.
To evaluate the form of cell death in vivo, we performed TdT-mediated dUTP nick end labeling (TUNEL) assays and Ki67 staining to measure the level of apoptosis and proliferation, respectively. Consistent with the H460 xenograft model, let-7 treated lungs, the control and baseline lungs showed similar levels of TUNEL-positive cells (Suppl fig. 4B). In contrast, let-7 treated lungs lacked Ki67, suggesting that let-7 over-expression interfered with proliferation of tumor cells in lungs with activated K-ras G12D (Fig. 4A, top, right panel). Therefore, we conclude that let-7 blocks tumor growth by repressing proliferation and eliminating tumor cells potentially through a non-apoptotic mechanism. Apoptosis-unrelated cell death as a means to eliminate cancer cells is a common mechanism of many therapeutic agents and is reminiscent of the prevailing form of cell death in solid tumors after cytotoxic therapy (Brown and Wilson, 2003). Let-7 over-expression may lead to cytotoxic-type damage or may disrupt DNA repair pathways that would correct for naturally occurring damage in the quickly dividing cancer cells.
Discussion
While much work remains to be done to determine the tumor-suppressive mechanism of let-7 in in vivo lung tumors, studies from our lab, the Jack’s group, and others suggest that let-7 may act through direct repression of the Kras, HMGA2, and c-Myc oncogenes (Johnson et al., 2005; Kumar et al., 2008; Lee and Dutta, 2007; Sampson et al., 2007). Although Kras might seem to be a critical target in this mouse model, we speculate that other targets are also important, as a recent study showed that the sole inhibition of c-Myc resulted in significant inhibition of tumor formation in early stage, and reduction of tumors in late stage K-ras activated lungs (Soucek et al., 2008).
In conclusion, our studies show that let-7 is a tumor suppressor in lung cancer, and provide proof-of-concept for let-7 replacement therapy as a novel and promising modality to treat lung cancer. Similar to targeted therapies that tackle a gain-of-function in cancer, such as EGFR inhibitors, the reintroduction of the let-7 tumor suppressor miRNA interferes with the oncogenic properties of tumor cells and induces a therapeutic response. In contrast to an inhibitory approach, however, “miRNA replacement therapy” seeks to restore expression and function of a naturally occurring molecule. Hence, miRNA replacement therapy represents a unique therapeutic opportunity following a different strategy than small molecule inhibitors, siRNAs and miRNA antagonists, and deserves further exploration. This approach has been successfully demonstrated for liver and metastatic prostate cancer (Kota et al., 2009; Takeshita et al., 2009). While our results presented here suggest that let-7 miRNAs may prove useful in the treatment of human NSCLC, the challenge of a let-7 therapy will be the successful delivery of the miRNA to the tumor cells. Local delivery of synthetic let-7 by intratumoral injections resulted in strong inhibition of overall tumor growth. However, this delivery route might be inadequate in a clinical setting as peripheral tumor cells remained present and showed limited knock-down of let-7 targets by immunohistochemistry. Therefore, a delivery technology that facilitates universal access to all tumor cells, such as a systemic delivery route, might be needed to make a let-7 therapeutic more efficacious. Alternatively, adenoviral or retroviral delivery of let-7, as demonstrated here, could become useful tools in the treatment of respiratory malignancies.
Supplementary Material
Acknowledgments
We would like to acknowledge Xianping Liang for help with animal husbandry and genotyping, and Paul Lebourgeois for histopathological analyses. PT was supported by a NIH NRSA postdoctoral fellowship (F32CA130376). PM was supported by a postdoctoral fellowship from Ministry of Education and Science of Spain (MEC) and from the Hope Funds for Cancer Research. JBW was supported by the ASTRO Junior Faculty Career Research Training Award. FJS and JBW were supported by grants from the NIH (CA131301-01A1) and from the Connecticut Department of Public Health (RFP #2006-0913).
Footnotes
Conflict of interest
FS and JW are inventors on pending patent applications in the miRNA area. Part of this work was supported by a grant to FS from Asuragen.
References
- Bitko V, Barik S. Nasal delivery of siRNA. Methods Mol Biol. 2008;442:75–82. doi: 10.1007/978-1-59745-191-8_6. [DOI] [PubMed] [Google Scholar]
- Bitko V, Musiyenko A, Shulyayeva O, Barik S. Inhibition of respiratory viruses by nasally administered siRNA. Nat Med. 2005;11:50–5. doi: 10.1038/nm1164. [DOI] [PubMed] [Google Scholar]
- Brown JM, Wilson G. Apoptosis genes and resistance to cancer therapy: what does the experimental and clinical data tell us? Cancer Biol Ther. 2003;2:477–90. doi: 10.4161/cbt.2.5.450. [DOI] [PubMed] [Google Scholar]
- Calin GA, Liu C-G, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proceedings of the National Academy of Sciences. 2004a;101:11755–11760. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004b;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–9. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Johnson SM, Bai L, Saito K, Partridge J, Reinert KL, et al. Post-embryonic expression of C. elegans microRNAs belonging to the lin-4 and let-7 families in the hypodermis and the reproductive system. Dev Dyn. 2005;234:868–77. doi: 10.1002/dvdy.20572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–64. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- Hohla F, Schally AV, Kanashiro CA, Buchholz S, Baker B, Kannadka C, et al. Growth inhibition of non-small-cell lung carcinoma by BN/GRP antagonist is linked with suppression of K-Ras, COX-2, and pAkt. Proc Natl Acad Sci U S A. 2007;104:18671–6. doi: 10.1073/pnas.0709455104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe T, Herbst RS, Onn A. Current management of advanced non-small cell lung cancer: targeted therapy. Semin Oncol. 2005;32:315–28. doi: 10.1053/j.seminoncol.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–8. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–22. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–47. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–17. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci U S A. 2008;105:3903–8. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–30. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenkova N, Gu Q, Rux JJ. Gene specific siRNA selector. Bioinformatics. 2004;20:430–2. doi: 10.1093/bioinformatics/btg437. [DOI] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–9. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–16. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- Park SM, Shell S, Radjabi AR, Schickel R, Feig C, Boyerinas B, et al. Let-7 prevents early cancer progression by suppressing expression of the embryonic gene HMGA2. Cell Cycle. 2007;6:2585–90. doi: 10.4161/cc.6.21.4845. [DOI] [PubMed] [Google Scholar]
- Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–16. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–70. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- Sevignani C, Calin GA, Nnadi SC, Shimizu M, Davuluri RV, Hyslop T, et al. MicroRNA genes are frequently located near mouse cancer susceptibility loci. Proc Natl Acad Sci U S A. 2007;104:8017–22. doi: 10.1073/pnas.0702177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried JM, Krishnamachary N, Gaither Davis A, Gubish C, Hunt JD, Shriver SP. Evidence for autocrine actions of neuromedin B and gastrin-releasing peptide in non-small cell lung cancer. Pulm Pharmacol Ther. 1999;12:291–302. doi: 10.1006/pupt.1999.0210. [DOI] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–83. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–6. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- Takeshita F, Patrawala L, Osaki M, Takahashi RU, Yamamoto Y, Kosaka N, et al. Systemic Delivery of Synthetic MicroRNA-16 Inhibits the Growth of Metastatic Prostate Tumors via Downregulation of Multiple Cell-cycle Genes. Mol Ther. 2009 doi: 10.1038/mt.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–98. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.