

Abstract
Inhibition of an initiating oncogene often leads to extensive tumor cell death, a phenomenon known as oncogene addiction1. This has led to the search for compounds that specifically target and inhibit oncogenes as anti-cancer agents. Whether chromosomal instability (CIN) generated as a result of deregulation of the mitotic checkpoint pathway2,3, a frequent characteristic of solid tumors, has any effect on oncogene addiction, however, has not been explored systematically. We show here that induction of chromosome instability by overexpression of the mitotic checkpoint gene Mad2 does not affect the regression of Kras driven lung tumors upon Kras inhibition. However, tumors that experience transient Mad2 overexpression and consequent chromosome instability recur at dramatically elevated rates. The recurrent tumors are highly aneuploid and have varied activation of pro-proliferative pathways. Thus, early CIN may be responsible for tumor relapse after seemingly effective anti-cancer treatments.
Chromosome instability (CIN) is commonly found in human solid tumors and extensive mouse modeling studies suggest that it is sufficient to initiate tumorigenesis in a variety of cellular contexts4-6. We have previously reported that ubiquitous overexpression of Mad2, which induces whole chromosome instability and rearrangements3, is sufficient for tumor initiation in multiple tissues including lung3. To determine whether CIN imparted by Mad2 overexpression could accelerate tumor formation driven by a classical oncogene, we generated cohorts of mice carrying a type II alveolar epithelial cell-specific doxycycline inducible (CCSP-rtTA) transactivator transgene7 in addition to rtTA-responsive Mad2 and/or murine oncogenic K-Ras4bG12D transgenes8. Resulting progeny were divided into groups maintained on normal or doxycycline diets at weaning. We refer to these mice as TI-K, TI-M, and TI-KM for “tetracycline inducible” Kras, Mad2 and Kras+Mad2, respectively. Eight weeks after induction, western blot analysis of lungs confirmed that only mice exposed to doxycycline expressed exogenous Mad2 protein, resulting in levels twice those of control animals (Fig. 1a). Endogenous levels of Mad2 were not upregulated upon KrasG12D activation (Fig.1a and Supplementary Fig. 1a).
Figure 1. Mad2 Overexpression Cooperates with KrasG12D in Lung Tumorigenesis.
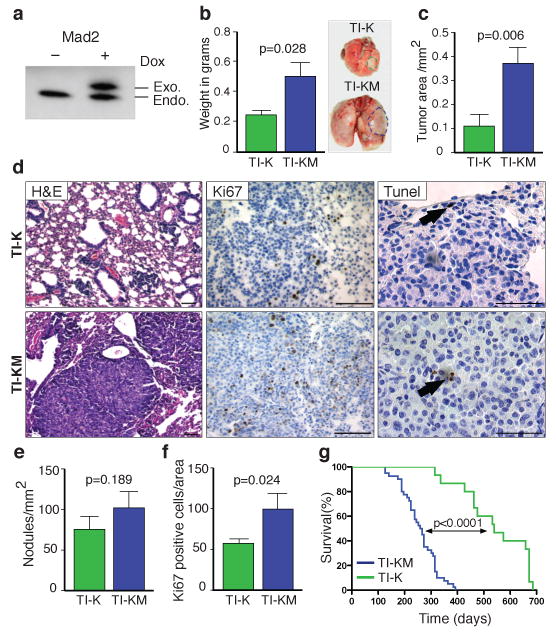
a, Mad2 western blot of TI-KM lungs maintained with (+) or without (−) doxycycline. b, Lung weights from TI-K and TI-KM mice on doxycycline for 8 weeks (Right). Representative macroscopic pictures show lung tumor-size (dotted lines). c, Total tumor area in TI-K and TI-KM lungs. d, H&E from TI-K or TI-KM mice after 8 weeks on doxycycline (left panel), Ki67 staining (middle panel) and TUNEL (right panel) (black bar: 100 μm). e, Tumor nodule/mm2 of lung tissue in TI-K versus TI-KM animals. f, Percentage of Ki67 positive cells. g, Kaplan-Meier curve of TI-K and TI-KM mice. Error bars represent mean and s.e.m. from at least 4 different mice. P values were determined by unpaired t test.
Eight weeks after transgene induction, the lungs of TI-KM mice were nearly twice as large as those of TI-K mice (Fig. 1b) and total tumor area was significantly increased (Fig. 1c). However, tumor nodule number was not increased (Fig. 1e), indicating that tumor initiation is not affected by Mad2 overexpression. Adenocarcinomas in TI-KM mice exhibited a more aggressive morphology, with invasion into the pleura and higher proliferation rates (Fig. 1d-f) but no significant differences in apoptosis (Fig. 1d). Moreover, a statistically significant decrease in survival was seen in TI-KM versus TI-K mice. Whereas TI-K mice survived for 543 ± 32 days, TI-KM mice survived for 258 ± 11 days (P < 0.0001) (Fig. 1g). All mice died of lung tumors, as determined by postmortem examination. Although overexpression of Mad2 by itself under the CMV promoter induces lung tumors in 35% of animals3, only 1/40 Mad2 overexpressing mice (TI-M) in a Kras wild type background harbored a lung adenoma after 15 months on doxycycline (data not shown). This may be the result of Mad2 transgene expression being restricted to type II pneumocytes in the CCSP-rtTA model compared with ubiquitous overexpression in CMV-rtTA mice. Importantly, neither total nor exogenous levels of Mad2 in TI-KM animals are higher than in TI-M mice arguing that the increase in tumor burden is dependent on combined Kras and Mad2 overexpression (Supplementary Fig. 1a-c).
In both TI-K and TI-KM mice lung adenocarcinomas were SP-C positive, consistent with a type II-like classification (Supplementary Fig. 2a-b), and negative for CCSP8 (Supplementary Fig. 2c-d), which was seen in normal cells lining the airways (Supplementary Fig 2e-f). Hence, combined lung specific Mad2 and Kras overexpression leads to the formation of lung adenocarcinomas that are more aggressive than, though histologically similar to, those seen with Kras alone.
We have previously reported that ubiquitous overexpression of Mad2 leads to tumor initiation through the acquisition of a CIN phenotype3. To determine if the CIN generated by high levels of Mad2 could be contributing to the more severe phenotype in TI-KM mice, FISH analysis was performed on lung tissue using chromosome 12, 16 and 17 specific probes. TI-K lung tumors showed higher levels of aneuploidy (13%) compared to normal lung (2%, not shown). However, TI-KM tumors showed a three fold increase in aneuploid cells over TI-K (38% vs. 13%, p<0.0001) (Fig. 2a-b). TI-M animals developed very low levels of aneuploidy (5,7%), consistent with the observed lack of tumor formation. Despite high rates of aneuploidy seen in TI-KM primary tumors, array CGH analysis failed to detect any gross abnormalities (data not shown). This most likely reflects the absence of stable, clonally propagated events that significantly contribute to the CGH profile.
Figure 2. Mad2 Overexpression Increases Aneuploidy but is not Enough to Overcome Oncogene Addiction.
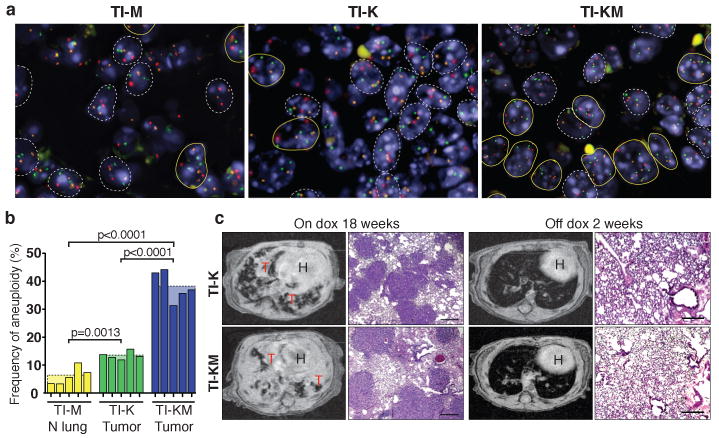
a, FISH images from TI-M normal lung, TI-K and TI-KM lung tumor showing aneuploid cells in yellow and normal cells in white. DNA: blue, chromosome 12 probe (green), 16 (red) and 17 (orange). b, Frequency of aneuploidy in 5 individual mice from each group. c, MR images and histological sections of lung tumors from TI-K and TI-KM mice after 18 weeks on doxycycline (left). After 2 weeks of doxycycline withdrawal, lung opacities disappeared from MR images, and tumors regressed histologically. H: heart, T: tumor. Scale bar: 200 μm.
RNA microarray analysis confirmed the diversity of tumor subtypes in the Kras and Mad2 tumors. While the majority of Kras tumors clustered together, the TI-KM primary tumors were markedly different from each other and from the cluster of mutant Kras overexpressors. These results suggest that Mad2 overexpression induces a wide spectrum of chromosomal aberrations and expression changes which are likely contributing to enhanced tumorigenesis. The precise causative events have not been identified (Supplementary Fig. 3).
Continued expression of mutant Kras is required for tumor maintenance8. In contrast to what is observed with classical oncogene overexpression, continuous Mad2 overexpression is not required for tumor maintenance, as primary tumors induced after Mad2 overexpression in two different ubiquitous inducible model systems do not regress upon transgene de-induction3. To determine whether the CIN imparted by Mad2 overexpression affected Kras oncogene addiction, TI-K or TI-KM mice with lesions evident by Magnetic Resonance Imaging (MRI) were identified (Supplementary Table 1) and then switched to a regular diet. MRI 2 weeks after de-induction revealed a striking reduction in tumor volume both in TI-K and TI-KM mice (Fig. 2c and Supplementary Fig. 4). Examination of lungs from all mice confirmed normal histology with areas of pleural and parenchymal fibrosis. Thus, CIN associated with Mad2 overexpression is not sufficient to prevent tumor regression following abrogation of Kras and Mad2 expression. It is likely that these tumors regress in response to mutant Kras withdrawal and not Mad2, since, as stated above, de-induction of Mad2 in the CMV-rtTA and tTA systems never leads to tumor regression by itself3.
Next, we sought to determine whether CIN would affect the rate of tumor recurrence. 25 TI-K and 24 TI-KM mice maintained on doxycycline were monitored for tumor appearance by MRI (Supplementary Table 2). When solid tumors were identified (between 12-28 weeks), doxycycline was removed from the diet and the mice periodically screened for tumor re-growth. While TI-K tumors never relapsed after de-induction of the oncogene (even up to one year off doxycycline), tumors recurred in 11 out of 24 TI-KM mice sacrificed 4 to 11 months after doxycycline withdrawal (Fig. 3a-b). These nodules were solid or well defined proliferating papillary adenocarcinomas morphologically indistinguishable from the primary tumors (Fig. 3e). The presence of recurrences in TI-KM animals is not a result of the larger primary tumor size since even in TI-KM animals selected for a small tumor volume, 50% recurrence was observed (Fig. 3c and Supplementary Table 3). In fact, the average volume of primary KM tumors that went on to recur in this selected population was significantly smaller than that of K and of KM primaries that never recurred (Fig. 3c). Mad2 overexpression alone led only to a single tumor in 1 out of 40 animals after 15 months of induction, arguing that the KM relapses are dependent on having expressed both transgenes.
Figure 3. Chromosomal Instability Increases the Likelihood of Recurrence in Kras Tumors.
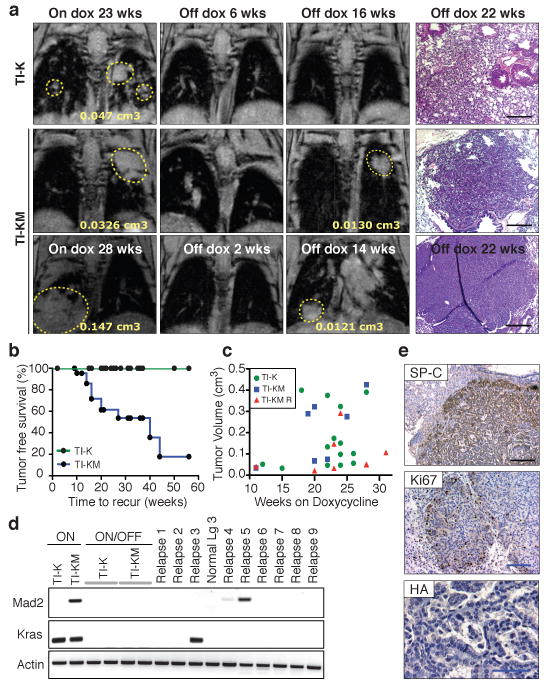
a, MRI from TI-K and TI-KM mice on doxycycline showing lung tumors (yellow circles) (left panel), after 2-6 weeks of doxycycline withdrawal showing complete regression (middle panel) and after 14-16 weeks off doxycycline (right panel) showing recurrent tumors in TI-KM mice at the same anatomical site as the primary tumor. Histological sections: right panel. b, Relapse-free survival curve of TI-K and TI-KM mice; censured events indicated by circles. c, Initial tumor volume of TI-K and TI-KM mice that never recur and TI-KM that developed a relapse. d, RT-PCR from normal, TI-K and TI-KM lungs on doxycycline showing the corresponding transgenes and from TI-K, TI-KM and relapses off doxycycline. e, SP-C, Ki67 and HA immunostaining of a relapse. (Black bar: 200 μm, blue bar: 100 μm).
It is, however, formally possible that these recurrences are de novo tumors derived from cells that had expressed Kras and Mad2 but were not part of the initial tumor mass, a possibility that we consider unlikely for the following reasons: no such events have been reported in other inducible mouse models of oncogene addiction9,10; TI-KM animals maintained off doxycycline never develop lung tumors in this model system; TI-KM mice that were on doxycycline for 5 weeks, during which time no tumors developed, and switched to a regular diet for 35 additional weeks did not develop tumors (Supplementary Fig. 5); and in 3 out of 3 informative cases where primary tumors were small enough to localize by MRI, recurrences appeared at the same anatomical site as the primary tumor (Fig. 3a).
Tumor relapse has been observed in several tetracycline inducible mouse models of oncogene addiction11. Although many of these recurring tumors show doxycycline independent re-expression of the transgene, additional mechanisms responsible for tumor recurrence have also been identified. We performed HA specific immunohistochemistry and/or RT-PCR analysis to detect the Mad2 and Kras transgenes in the primary tumors, recurrent lesions and tumor free lung samples from TI-K and TI-KM mice (Fig. 3d-e and Supplementary Fig. 6b). None of the non-tumor-bearing lungs after doxycycline withdrawal re-expressed the inducible transgenes. Surprisingly, only one of the recurrences re-expressed the Kras transgene and 2/11 re-expressed the Mad2 transgene (which by itself is insufficient to induce tumor formation), suggesting modes of escape that are independent of the initiating oncogene. RT-PCR and sequencing of commonly mutated exons revealed that the recurrences were not a result of secondary point mutations in the endogenous Hras, Kras or Nras loci (data not shown), a common mechanism of acquired resistance to targeted therapies in other inducible mouse models12 and in human cancer13-15.
In parallel to what was seen with the primary tumors, recurrent tumors in TI-KM mice were highly aneuploid (39,4%). Lung tissue from TI-K animals after tumor regression showed very low levels of aneuploidy (2,7%), similar to wild type lung, suggesting that this baseline aneuploid level is insufficient to drive tumor recurrence. In contrast, elevated rates of aneuploidy were observed both in the TI-KM primary tumors (38%) and TI-KM lungs after regression (12,9 %) prior to tumor relapse. This suggests that high levels of aneuploidy in the primary tumor and in cells remaining after de-induction of Kras and Mad2 enable survival after oncogene withdrawal thereby facilitating relapse (Fig. 4a-b).
Figure 4. Recurrent Tumors from TI-KM Mice are Heterogeneous and Highly Aneuploid.
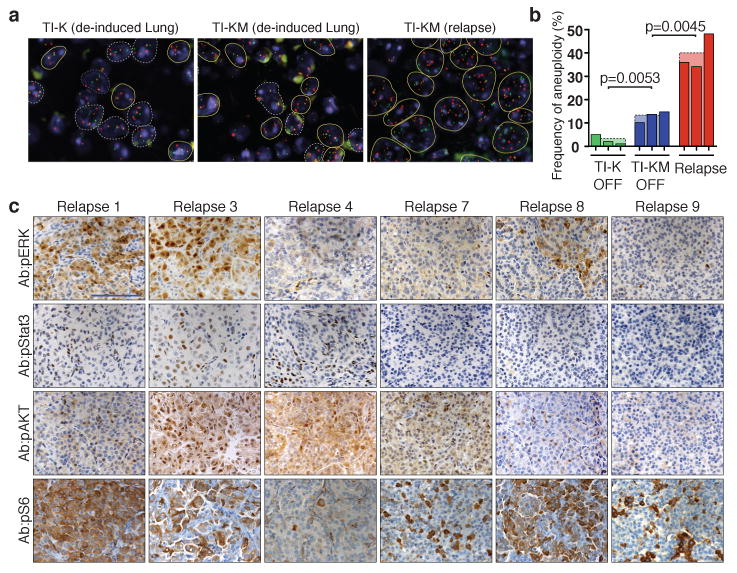
a, FISH images from a TI-K and TI-KM regressed lung and a TI-KM tumor relapse showing aneuploid cells in yellow lines and normal cells in white. b, Frequency of aneuploidy in 3 independent TI-K and 3 TI-KM regressed lungs and 3 recurrent tumors of TI-KM mice showing an increase in aneuploid cells in the recurrent tumors. P values were determined by unpaired t test. c, Immunohistochemistry of pERK, pStat3, pAKT and pS6 in tumor relapses showing positive staining for all 3 pathways in relapses 1,3 and 8 while relapses 4 and 7 are only positive for pAKT and pS6. Scale bar: 100 μm.
KrasG12D lung tumors show positive staining for pErk, pStat3, pAkt and pS6, indicating that the MAPK, STAT3 and PI3K pathways are all stimulated by oncogenic Kras 16 (Supplementary Fig. 7). Only 5 out of 10 KM recurrences retained MAPK pathway activation and 4 out of 10 showed low level pStat3 activity (Fig. 4c and Supplementary Fig. 8). 10/10 recurrences scored positive for AKT, though the different levels and patterns seen by IHC make this unlikely to be the sole determinant of recurrence. To further characterize the signaling pathways implicated in tumor relapse, RNA microarray analysis was performed in 6 TI-KM recurrent tumors. Consistent (Supplementary Fig. 9) with the IHC analysis, recurrent tumors were very heterogeneous, different from each other and from the K or KM primary tumors. Thus, recurrent tumors employ alternative signaling pathways to escape oncogene addiction, a result consistent with the notion that Mad2-mediated CIN in the primary tumor provides extensive diversity and therefore an evolutionary advantage to oncogene driven solid tumors.
The role of aneuploidy in the initiation of human tumors has been modeled extensively in mice5,17,18. Overexpression of the mitotic checkpoint gene Mad2 is a very common occurrence in human tumors likely due to its direct activation by E2F upon inhibition of the Retinoblastoma tumor suppressor pathway2. Indeed, inducible overexpression of Mad2 in mice leads to modest levels of aneuploidy and robust tumor induction in multiple tissue types3. Extremely high levels of aneuploidy are observed in cells with complete loss of mitotic checkpoint function19-21 but these cells are non-viable, a fact reflected in the observation that no mitotic checkpoint null tumors have ever been reported, regardless of p53 status. While loss of mitotic checkpoint function in some settings can suppress tumor formation in vivo22-24, whether this is due to intolerable levels of aneuploidy and subsequent cell death or perhaps even non-cell autonomous effects on the tumor microenvironment has not yet been examined.
The requirement for continued expression of an initiating oncogenic stimulus for tumor cell survival (oncogene addiction1) is a puzzling phenomenon since withdrawal of a deregulated activity might be predicted to lead to reversion of the cell to a normal state. Nonetheless, its occurrence suggests that targeting oncogenes therapeutically might lead to profound tumor regression. Indeed, inhibition of the deregulated kinase activity of BCR-ABL with the drug Imatinib (Gleevec) in chronic myelogenous leukemia is an extremely effective targeted therapy25. Remarkably, patients in the accelerated phase of the disease, in which multiple chromosomal anomalies are observed, still show a strong clinical response albeit with significantly higher relapse rates26-28. Here we show that a similar phenomenon might take place in solid tumors. CIN induced by overexpression of the mitotic checkpoint gene Mad2 (observed in a wide variety of human tumors) does not alleviate addiction to Kras in a lung tumor model, since profound tumor regression takes place when Kras expression is inhibited whether or not CIN is present in the primary lesion. However, a dramatic increase in tumor recurrence occurs in the chromosomally unstable setting with emergent independence from the original oncogenic stimulus. While targeted therapy resistance is often initially due to point mutations or amplifications of the intended target gene, CIN might provide an escape route even after sequential, successful inhibition of the altered target. Moreover, CIN in the primary tumor may accelerate disruption of DNA repair pathways thereby facilitating the generation of point mutations. Thus, early CIN in tumors, which in some cases directly results from the loss of the Rb tumor suppressor pathway and overactivation of the mitotic checkpoint, may provide the evolutionary fuel responsible for high relapse rates in human cancers that initially respond to targeted or perhaps even conventional chemotherapeutic regimens. Mouse models of cancer which often show benign levels of CIN relative to human cancers may thereby overestimate the efficacy of clinical drug candidates.
Methods Summary
All mice (TI-K, TI-KM, TI-M, CCSP-rtTA) were of mixed 129/Bl6 background. Doxycycline was administered by impregnated food pellets (625ppm; Harlan-Teklad). For MRI analysis, mice were anaesthetized with isofluorane and images obtained by low resolution axial scout followed by high-spatial resolution T2 weighted scan. FISH analysis was carried out with 3 pericentromeric probes from chromosomes 12, 16 and 17 amplified from corresponding BAC clones and labeled by nick translation with SpectrumGreen, SpectrumRed and SpectrumOrange fluorophores. Quantification of aneuploidy was carried out in a minimum of 150 interphase nuclei counterstained with DAPI. Microarray expression analysis was carried out on MOE430A 2.0 Affymetrix arrays with RNA extracted using the Qiagen RNeasy kit. For CGH, DNA was extracted using a Qiagen DNeay kit, whole genome amplified and hybridized to normal genome controls on an Agilent mouse 244K array. RT-PCR was carried out using SuperScript III (Invitrogen) according to manufacturer's instructions and amplified with transgene specific primers. Protein expression was assessed by western blot of 40ug of tissue lysates incubated with antibodies against Mad2 (BD) and Actin (Amersham). Histopathology and immunohistochemistry were carried out on paraffin sections of formalin fixed tissue with Ki67 (Novocastra), anti-prosurfactant protein C (Chemicon), anti-Clara cell protein (Chemicon), anti-phospho-AKT (Cell Signalling Technology), anti-phospho-stat3 (Cell Signalling Technology), anti-phospho-S6 (Cell Signalling Technology) and anti-HA (Roche).
Full Online Methods
Animal Husbandry and Genotyping
TetO-Mad2 transgenic mice, TetO-Kras and CCSP-rtTA mice were kept in pathogen-free housing under guidelines approved by the MSKCC Institutional Animal Care and Use Committee and Research Animal Resource Center. Doxycycline was administered by feeding mice with doxycycline-impregnated food pellets (625ppm; Harlan-Teklad). Tail DNA was isolated using Qiaprep Tail DNeasy isolation kit (QIAGEN) according to the manufacturer's protocol. TetO-Mad2, TetO-Kras and CCSP-rtTA transgenic mice were genotyped as described in 3 and 8.
Magnetic Resonance Imaging
Individual mice were subjected to MRI for detection of tumors. In brief, mice were anesthetized with 2% isofluorane and images were obtained from a Bruker 4.7T 40cm bore magnet with a commercial 7-cm inner diameter birdcage coil in the Animal Imaging MRCore Facility at MSKCC. Low-resolution axial scout images were obtained initially, followed by high-spatial-resolution T2-weighted axial images (repetition interval (TR)=3,800ms, effective echo time (TE)=35ms, eight echoes per phase encoding step, spatial resolution=1.0mm slice thickness × 112μm×112μm in plane resolution and four repetitions of data acquisition for 8-9 min of imaging time.
FISH
Probes were synthesized from 3 pericentromeric BAC clones of chromosome 12, Chr 12-RP23-54G4,RP23-41E22 & RP23 16809 5a; Chr 16-RP23-290E4, RP23-356A24 & RP24 258J4 4a and Chr 17-RP23-354J18-6c, RP23-73N16 & RP23-202G20 and labeled with SpectrumGreen-dUTP, SpectrumRed-dUTP and SpectrumOrange-dUTP (Vysis) respectively. The BAC DNAs were labeled by nick translation according to standard procedures. The number of hybridization signals for these probes was assessed in a minimum of 150 interphase nuclei with strong and well-delineated contours.
RNA Expression Data
raw data (CEL files) was normalized and quantified to give log2 expression levels for each probe set using the gcRMA method available in the Bioconductor library for the R statistical analysis system. To compute differential expression, the LIMMA package (also from Bioconductor) was used for a variant of the standard t-test, which adds a correction term to the group variance. The False Discovery Rate (FDR) method was used to correct for multiple testing. Hierarchical clustering was carried out using a distance function derived from the correlation between samples (dist=(1-r)/2 were r is the Pearson correlation).
RNA and Protein Analysis
RNA was isolated using the RNeasy kit (Qiagen, Valencia, CA) and treated with DNaseI (Ambion) to eliminate any contaminating DNA. RT reactions were performed with SuperScript III (Invitrogen) according to the manufacturer's instructions, followed by PCR with primers specific for transgene amplification. Protein expression was assessed by immunoblotting using 40μg of total tissue lysates. Blots were probed with antibodies directed against Mad2 (BD Transduction Laboratories). HRP conjugated anti-mouse (Amersham) was used as a secondary antibody and proteins were visualized using the ECL detection system (Amersham). For quantitative western blots fluorochrome conjugated secondary antibodies (LICOR) were used in place of HRP and signals detected using LICOR Odyssey.
Histopathology
For immunohistochemistry analysis, representative sections were deparaffinized, rehydrated in graded alcohols and processed using the avidin-biotin immunoperoxidase method. Sections were subjected to antigen retrieval by microwave oven treatment using standard procedures. Diaminobenzidine was used as the chromogen and hematoxylin to counterstain nuclei. The primary antibodies used in immunohistochemistry were Ki67 (NovoCastra), anti-prosurfactant protein C (1:400 dilution, AB3786 Chemicon), anti-Clara cell protein (1:1000, AB07623 Chemicon), anti-phospho-MAPK (1:100 dilution, #4376 Cell Signaling Technology), anti-phospho-Akt (1:100 dilution, #3787 Cell Signaling Technology), anti-phospho-Stat3 (1:100 dilution, #9145 Cell Signaling Technology), anti-phospho-S6 (1:100 dilution, #2211 Cell Signaling Technology) and anti-HA (1:100 dilution, clone 3F10 #11867423001 Roche).
Supplementary Material
Acknowledgments
We thank H. Varmus and W. Pao for the CCSP-rtTA and TetO-Kras mice; K. Politi, K. Podsypanina and M. Jechlinger for reagents, discussions and critical reading of the manuscript. We are grateful to C. Le and M. Lupu for MR imaging; M. Leversha, C. Kalyani and J. McGuire for FISH staining; K.M.D. La Perle, M. Jiao and M. Squatrito for immunohistochemistry and pathological analysis and A. Viale for CGH and Affymetrix. We thank Y. Chin, S. Curelariu and C. Coker for excellent technical assistance. Support: RS by the Charles H. Revson Foundation; JMS by a BCRP Predoctoral Traineeship Award from the US Department of Defense (CDMRP) and RB by the NIH.
Footnotes
Author Contributions: RS, JMS and RB designed the study, RS performed experiments, RS, JMS, NS and RB analyzed the data. RS, JMS and RB wrote the paper.
Author Information: The raw and processed microarray data have been deposited at GEO with the accession number GSE19753. Reprints and permissions information is available at www.nature.com/reprints.
Conflict of Interest: All authors declare no conflicts of interest.
References
- 1.Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3:448–457. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- 2.Hernando E, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- 3.Sotillo R, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 2008;24:457–466. doi: 10.1016/j.tig.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang S, Khoo C, DePinho RA. Modeling chromosomal instability and epithelial carcinogenesis in the telomerase-deficient mouse. Semin Cancer Biol. 2001;11:227–239. doi: 10.1006/scbi.2000.0374. [DOI] [PubMed] [Google Scholar]
- 7.Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275:11858–11864. doi: 10.1074/jbc.275.16.11858. [DOI] [PubMed] [Google Scholar]
- 8.Fisher GH, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes & development. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chin L, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 10.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 11.Giuriato S, Felsher DW. How cancers escape their oncogene habit. Cell Cycle. 2003;2:329–332. [PubMed] [Google Scholar]
- 12.Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6:577–586. doi: 10.1016/j.ccr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 13.Gorre ME, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science (New York, NY. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 15.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Politi K, et al. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes & development. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker DJ, Chen J, van Deursen JM. The mitotic checkpoint in cancer and aging: what have mice taught us? Curr Opin Cell Biol. 2005;17:583–589. doi: 10.1016/j.ceb.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Pérez de Castro I, de Cárcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28:899–912. doi: 10.1093/carcin/bgm019. [DOI] [PubMed] [Google Scholar]
- 19.Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–645. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- 20.Li M, York JP, Zhang P. Loss of Cdc20 Causes a Securin-Dependent Metaphase Arrest in Two-Cell Mouse Embryos. Mol Cell Biol. 2007;27:3481. doi: 10.1128/MCB.02088-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwon M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy Acts Both Oncogenically and as a Tumor Suppressor. Cancer Cell. 2006 doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Rao CV, et al. Colonic tumorigenesis in BubR1+/-ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 2005;102:4365–4370. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007;179:255–267. doi: 10.1083/jcb.200706015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 26.Cortes J, O'Dwyer ME. Clonal evolution in chronic myelogenous leukemia. Hematol Oncol Clin North Am. 2004;18:671–684. x. doi: 10.1016/j.hoc.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 27.Hochhaus A, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–2196. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 28.O'Dwyer ME, et al. The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood. 2002;100:1628–1633. doi: 10.1182/blood-2002-03-0777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.