

Abstract
Autism spectrum disorders (ASD’S) are highly heritable. Consequently, gene discovery promises to help illuminate the pathophysiology of these syndromes, yielding important opportunities for the development of novel treatments and a more nuanced understanding of the natural history of these disorders. Although the underlying genetic architecture of ASD’s is not yet known, the literature demonstrates that it is not, writ large, a monogenic disorder with Mendelian inheritance, but rather a group of complex genetic syndromes with risk deriving from genetic variations in multiple genes. The widely accepted “Common Disease-Common Variant” hypothesis predicts that the risk alleles in ASD’s and other complex disorders will be common in the general population. However, recent evidence from gene discovery efforts in a wide range of diseases raises important questions regarding the overall applicability of the theory and the extent of its usefulness in explaining individual genetic liability. In contrast, considerable evidence points to the importance of rare alleles both with regard to their value in providing a foothold into the molecular mechanisms of ASD and their overall contribution to the population-wide risk. This chapter reviews the origins of the common versus rare variant debate, highlights recent findings in the field, and addresses the clinical implications of both common and rare variant discoveries.
Keywords: Autism Genetics, Rare Variants, Copy Number Variation, Association
Introduction
Autism is an often-debilitating disorder of development with a world-wide prevalence of approximately 0.1 percent.[1] The clinical hallmarks include fundamental deficits in social functioning and language development as well as the presence of narrowed and/or repetitive interests and behaviors.[2] Autism is the most prevalent syndrome among a spectrum of disorders that are currently grouped together in the Diagnostic and Statistical Manual of Mental Disorders Fourth Edition Text Revision (DSM IV-TR) under the rubric of Pervasive Developmental Disorders (PDD); these include Pervasive Developmental Disorder Not Otherwise Specified (PDD-NOS, 0.15 percent prevalence), Rett’s Disorder (0.006 percent prevalence), Asperger’s Disorder (ASP) (0.025 percent prevalence) and Childhood Disintegrative Disorder (CDD) (0.001 percent prevalence). [1, 3] In addition, it is has become commonplace to refer to PDDs as Autism Spectrum Disorders (ASD) and within this group to include individuals with so-called “not-quite Autism,” (NQA) that is, persons who fall just below the threshold for diagnosis in one of the key domains.
Autism affects predominately males with a male-to-female ratio of approximately 4.3:1. [1] The male predominance identified in Asperger’s syndrome (ASP) may be as high as 14:1.[4] The oft-cited prevalence in the lay literature of 1/166 individuals includes the entire spectrum of disorders, which makes accurate comparisons with rates determined prior to the development of diagnostic criteria for PDD-NOS and ASP highly problematic. The question of whether, using consistent diagnostic approaches, there is an increasing prevalence of ASD remains a subject of debate, and a detailed treatment is beyond the scope of this review. It is important for the sake of this discussion, however, to note that the best epidemiological evidence to date suggests that there may be as much as a 3 fold increase in the prevalence of individuals meeting full diagnostic criteria for autism over the past four decades. [1] If this reflects a true increase in incidence, it would have implications for our understanding of potential genetic mechanisms, which will be discussed in more depth below.
Whether the prevalence or the sensitivity of detection of ASD (or both) has increased over the past 40 or so years, the numbers of individuals seeking care has certainly grown markedly. This increase has and will continue to pose significant societal challenges: disorders within the spectrum typically result in marked social, cognitive, and behavioral impairments that are life-long, and current treatment approaches are palliative at best. All told, the public health burden, reflected in annual costs, was recently estimated at 35 billion dollars in the United States alone.[5]
While there is currently intense interest in ASD among the public as well as the scientific community and dramatic progress has been made in a variety of areas of research, the underlying pathophysiology of these syndromes remains largely a mystery. [6,7] As ASDs are thought to be among the most heritable of all developmental neuropsychiatric conditions, the identification of susceptibility genes would seem to hold tremendous promise for elucidating the underlying cellular and molecular mechanisms of disease and to pave the way for improvements in diagnosis and the development of novel therapeutic strategies. At the same time, despite very strong evidence (reviewed below) for a genetic contribution and some notable recent findings[8, 9] the rate of progress in gene discovery has not been as rapid as anyone would have hoped. As a result, there has been mounting skepticism in some quarters regarding the wisdom of continued investment in genetic approaches and the ability of investigators in this area to make tangible contributions to the lives of affected individuals and their families.
Such concerns warrant serious consideration. There is little doubt that the promise of genetic investigation of social disability has not yet been fully realized. However, it is also the case that studies of the genetics of polygenic disorders in general, including ASD, have just begun to reach maturity. The slope of the discovery curve over the past 4–5 years has become increasingly steep, and it is no longer hyperbole to suggest that recent findings are offering the first glimpses of the biology underlying common conditions. Indeed, there is an extraordinary convergence of factors that is driving a rapidly accelerating rate of return on investment in the area of ASD. The combination of public and private interest in supporting research efforts, highly effective advocacy groups, the rapid evolution of genomic tools and methodologies, and a long-term investment in developing large DNA collections has already resulted in key findings and promises a flurry of important discoveries over the next several years.
The ensuing chapter will review the genetics of ASD with a particular focus on the “steep part” of the discovery curve, that is, studies published over the past 5 years. The discussion will be divided into four sections: the first will address broad conceptual issues that help explain the early difficulties with gene discovery in ASD; the second will turn to a discussion of key controversies regarding the nature of the genetic contribution to ASD which continue to enliven debate in the field; third, major recent advances will be highlighted, and finally, the chapter will address the clinical implications of recent findings, including recommendations for the genetic evaluation of newly presenting patients with ASD.
The Heritability of Autism Spectrum Disorders
Autism is one of the most familial of all psychiatric disorders, with heritability estimated to be approximately 90%.[10] The twin studies on which such estimates are based note that the concordance rate for monozygotic twins is between 70–90% compared to the corresponding value for dizygotic twins of no more than 10%.[10, 11] The spread of concordance estimates for a given twin type reflects a degree of diagnostic uncertainty, with the lower bound determined based on strict diagnostic criteria and the upper bound reflecting siblings that both fall within the PDD spectrum. Given evolving diagnostic nosology and methods, it would not be surprising if ongoing twin studies lead to some modest downward adjustment of heritability estimates. Of note, the risk to siblings of autistic individuals is at least 20 times higher than among the general population,[12] which is similar to findings for ASD’s in general.[13]
The Allelic Architecture of ASDs
While both twin and family studies demonstrate the important contribution of genes to ASDs, they do not address some of the most important questions for researchers interested in discovering the specific character and identity of these risks. These include key questions such as 1) how many genes may be involved both in the individual and among the affected population at large; 2) whether variations in the genetic code need occur only within a single gene (i.e., simple/Mendelian inheritance) in a given individual to dramatically increase the risk for ASD or whether simultaneous variations in multiple genes must occur alone or in combination with non-genetic factors (complex/non-Mendelian inheritance) to result in pathology; 3) what the magnitude of the risk carried by individual transcripts is, and 4) whether the sequence variations associated with ASDs will be common or rare in the general population. Indeed, these questions, which center on the underlying “allelic architecture” of ASDs, are critically important for study design. Consequently, a brief review of the topics is an important prelude to a discussion of recent findings.
Over the past decade, large-scale gene discovery efforts have clearly shown that autism is not a simple/Mendelian disorder converging on a single gene at the level of the population, as is reviewed elsewhere.[14] However, this fact should not be mistakenly taken to suggest certainty regarding the other issues raised above which, in reality, remain far less clear. In fact, at present, it seems ASD may, indeed, be transmitted in a Mendelian fashion within a single individual or family[15–17] However, whether this applies to only a tiny fraction of affected individuals or a larger proportion of ASD families remains unclear. Moreover, recent studies have clearly suggested that common genetic variation in the population may contribute to ASD in a complex/non-Mendelian manner.[8] Whether this suggests oligo or polygenic inheritance in the individual and the affected population and what proportion of the overall genetic risk is transmitted in this fashion has not yet been clarified.
As noted, in addition to the issue of “how many,” a second fundamental question regarding allelic architecture revolves around the question of “how big”. As a general proposition, genetic variations that have very large effects on early-onset disabling conditions tend to be rare in the population and are often observed to be transmitted in Mendelian fashion. Conversely, common genetic variations associated with disease tend to carry relatively small risks (an issue which will be discussed at greater length below). Given these relationships, the question of the effect size of a genetic variation closely relates to the rate at which it is likely to observed within the population (see Figure 1). Traditionally, rare disease alleles were defined as having a frequency of less than 1%. However, it is now common in the literature to define those with a frequency of 5% or less as rare and those with a frequency of 1% or less as “very rare.” Conversely, of course, common alleles would be those found in the population at a frequency of greater than 5%.
Figure 1.
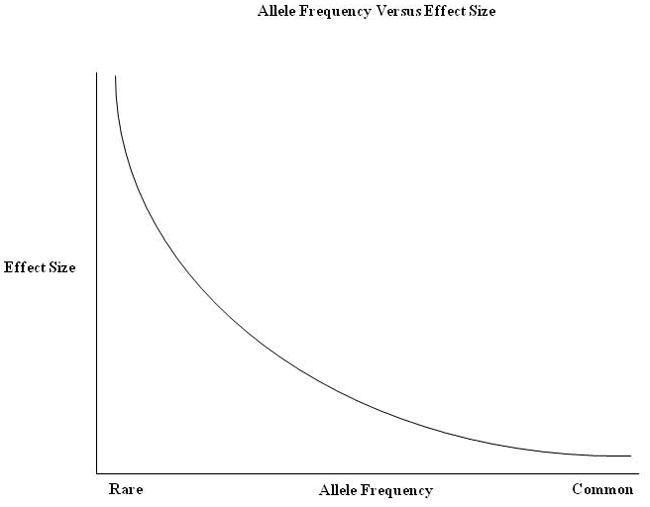
This graph illustrates the anticipated relationship between allele frequency and effect size for both common and rare disorders. Mendelian (or simple genetic) syndromes fall to the left-most aspect of the x-axis. To date, most genome-wide association studies across all of medicine have demonstrated that common alleles carry very small risks (far right side of the graph). Recent findings with regard to common risk variants in ASD have followed this pattern. Many of the rare mutations so far identified in ASD, appear to fall somewhere in the middle of this figure, showing low allele frequency and effect sizes that are not as great as those found in single gene disorders.
The foregoing brief discussion outlines some of the key basic issues that remain not only for autism genetics but also for many other common, complex disorders. Determining whether disease-related alleles are common or rare in the population, whether they are transmitted in a Mendelian or complex fashion, and the degree to which a syndrome or disease displays locus heterogeneity (that is genetic variations in multiple different genes leading to the same phenotype) are not simply points of academic interest. Since available genetic and genomic tools and analytic approaches tend to be specialized in the sense that they are optimal for studying rare or common variation, but typically not both, study design currently must take into account hypotheses about the allelic architecture of ASD, and the success or failure of an investigation may rest heavily on these assumptions.
The Common Disease, Common Variant Hypothesis
Over the past decade, one of the leading theories regarding the allelic architecture of common disorders (by definition those that affect more than 1% of the population) is the so-called “Common Disease, Common Variant (CDCV) Hypothesis.” Building on previous work by Chakravarti,[18] an influential article by Reich and Lander published in 2001[19], nicely summarizes the underlying reasoning: archeological data and evolutionary genetics supports the “Out of Africa” theory of evolution which posits that all humans descended from a small population over a relatively brief period of time. Specifically, it is thought that approximately 10,000 individuals grew into the current world population in roughly 100,000 years. The original, ancestral population, owing to its small size, could not have supported a large variety of disease alleles at any particular locus. Thus, by definition, disease alleles for common diseases in that original population must have been common, and disease alleles for rare diseases must have been rare. The hypothesis goes on to posit that due to rapid population expansion, common disease alleles massively proliferated. Rare disease alleles also spread, but as they were rare in the original population, not as widely as their common counterparts.
In addition to the dynamics of human population expansion, a second key aspect of the CDCV hypothesis rests on the rate of new mutation in the genome: new alleles are constantly introduced into the population with the result that novel variations will “dilute out” both rare and common disease alleles. However, the rate of introduction of new mutations is slow relative to the very dramatic expansion described by the Out of Africa theory. Thus, the fraction of new mutation in the population is predicted to be small relative to widely proliferated common variants but may represent a considerable proportion of the disease burden for initially rare disorders. In short, it is supposed that in today’s population alleles causing rare diseases will still be individually rare and that new mutations have not yet had time to “dilute out” common variants sufficiently, such that common diseases are predicted still to be mostly due to common variation in today’s population.
The proponents of the theory cite empiric evidence from rare monogenic disorders, pointing out that for rare diseases, such as Wilson’s disease (prevalence 1/30,000), there are greater than 50 disease-causing alleles so far identified. Moreover, as expected based on the CDCV theory, each allele is individually rare, with the most common variant explaining only 11% of the population risk[19] Moreover, based on similar logic one would predict that the rarer the disease, the greater the number of disease-causing alleles would be present and the lower the percentage of population risk would be explained by the most common allele. This appears to hold true in many cases, including with regard to Anaridia (prevalence of 1/100,000) with greater than 250 disease-causing alleles, the most common of which explains only 5% of the population risk.[19]
A handful of early findings from the study of common diseases also lent support to the theory. For example, APOE4 is a common allele found in approximately 15% of individuals of European descent and explains a significant amount of both the inter-individual genetic variation and roughly 50% of the population risk for Alzheimer’s disease.[20] Moreover, it has been determined to be an “ancient allele” dating back at least as far as the original population described in the CDCV hypothesis.[20] A similar situation has been identified with respect to macular degeneration.[21, 22]
These findings engendered confidence that similar common alleles of relatively large effect would explain much of the genetic variation in all or nearly all common complex diseases. However, despite both very significant recent methodological advances in the study of common variation (see below) and studies of hundreds of disorders, alleles similar to APOE 4 have been by far the exception rather than the rule, with most discoveries identifying alleles with effects that are an order of magnitude less than anticipated and accounting for a relatively small fraction of inter-individual genetic risk.
Type 2 diabetes provides a case in point. Multiple studies of this common disorder have identified a variety of common alleles. However, the effect sizes of these alleles have been very small compared with APOE4, explaining only a small percentage of the inter-individual and individually, a very small portion of the overall variance – 0.04%–0.5%.[23] In one of the earliest large scale studies employing what have turned out to be quite powerful genome-wide methods to study common variants, eight disease-related risk alleles explained only 2.3% of the overall variance. [24] Moreover, the majority of the alleles discovered have been in regions of DNA that are intergenic making the transition from gene discovery to an understanding of pathophysiology relatively difficult.
In short, while recent studies have been strikingly successful in providing reproducible evidence for the contribution of common variants to common disease, the findings so far have simultaneously raised important questions as to how much of the allelic architecture of these disorders will be explained by the CDCV hypothesis. This has led to a glass half-full situation in which those interested in common variants rightfully tout the tremendous recent progress in gene discovery in complex genetics, while many others in the scientific community have viewed the same results as evidence of the potentially flawed theoretical underpinnings of the theory. For example, the CDCV hypothesis relies heavily on the “Out of Africa” theory of evolution, which while widely accepted, is still challenged by a competing theory, the Multi-Modal theory of evolution which, if correct, would result in markedly different predictions regarding the dynamics of disease-allele proliferation.[25] Perhaps a more important point is that the CDCV hypothesis also assumes that conditions that were common in the original population did not negatively impact reproductive fitness, such that associated disease alleles were not selected out of the population.[25]
The very small effect sizes of the genetic variants so far identified for most common disorders may be viewed as calling this last assumption into question. The inability to identify risk alleles with more than modest effects raises the possibility that this observation is a signature of natural selection at work. Indeed, there is increasing speculation that the findings in Alzheimer’s disease and macular degeneration may be special cases, as these diseases affect the truly “aged,” and, consequently, are relatively immune from purifying selection. This point is further supported by evidence from early onset Alzheimer’s where all familial forms of the disease so far identified are monogenic disorders cause by heterogeneous and rare alleles.[26]
The reliance of the CDCV hypothesis on risk alleles not impacting reproductive fitness warrants particular attention in the case of autism. While it is certainly possible that individual alleles in a highly polygenic disorder might carry such small risks as to escape purifying selection, or that so-called balancing selection may be operating (i.e., a risk allele results in reproductive advantages in one context or environment while leading to a negative impact on fitness in another), it also seems logical that an allele carrying relatively large risks for a syndrome that fundamentally impacts social communication might sufficiently reduce reproductive fitness as to drive the frequency of the mutation down to low levels in the population. And if purifying selection has been at work with regard to autism genes from early in human history, this would suggest that there would be a greater proportion of rare alleles contributing to autism than would be predicted by the CDCV hypothesis. Finally, there are other reasons to question the applicability of the CDCV hypothesis with regard to ASD. Given the concern over the increasing prevalence of ASD, there is a tendency to consider this a spectrum of common diseases with a single underlying biology. However ASD might well reflect a collection of rare disorders resulting from hundreds of different genetic defects but leading to a shared phenotype, similar to the case of mental retardation.[27]
Studying Rare Variation
While the CDCV hypothesis has been a leading school of thought, particularly in psychiatric genetics, there have nonetheless been strongly held alternate views of the likely genetic architecture of common diseases and autism in particular. Not surprisingly, these have focused on the potential contribution of rare variation. With respect to ASD, gene discovery efforts focusing on low frequency alleles can be conceptualized as falling into three broad categories: 1) studies aimed directly at the CDCV hypothesis, namely investigation of whether rare as opposed to common variation accounts for the lion’s share of population risk for PDD; 2) studies aimed at investigating “extreme outliers,” that is, presumed unusual families that transmit the phenotype in a Mendelian fashion. These are of interest regardless of whether they represent only a small fraction of cases of so-called idiopathic autism; and 3) studies of known rare monogenic syndromes that share features with ASD.
The first of these alternatives, the rare variant common disease approach supposes that common disease and autism in particular may reflect the convergence of multiple, rare variations in the same gene (allelic heterogeneity) or multiple genes (locus heterogeneity) leading to a common/shared phenotype. Given a sufficiently large number of genomic “targets,” individually rare mutations could accumulate in the population so as to account for a significant proportion of a commonly occurring disorder.[28, 29] Such variation could either be transmitted from generation-to-generation or de novo. In the latter case, one would expect a considerable number of apparently sporadic cases (in which only the proband in the family was affected) and a very high rate of monozygotic (MZ) concordance versus dizygotic (DZ) concordance (both of which have been suggested with regard to ASD – see below).
Regardless of whether rare variants account for the majority of genetic risk for ASD, a focus on identifying low frequency alleles nonetheless represents an important avenue of study. The investigation of extreme outliers has played a central role in illuminating the pathophysiology of a range of common complex disorders, from hypercholesterolemia[30] to hypertension.[31] In these cases, the importance of identifying rare mutations has less to do with accounting for population risk and rather focuses on the importance of gaining a foothold in the molecular and cellular mechanisms of disease. Rare Mendelian mutations may be particularly valuable, even if extremely rare, both because the methods of gene discovery for this type of variation are quite powerful and well elaborated and because the identified alleles are likely to carry large effects and correspond to coding regions of the genome, changes in the genetic code that are, at present, typically far easier to investigate in the laboratory than those variations that correspond to non-coding, regulatory or intergenic and intragenic regions.
Finally, a particularly relevant rare variant approach with regard to social disability involves the study of monogenic syndromes, such as Fragile X, neurofibromatosis, and tuberous sclerosis that show phenotypic overlap with ASD. Such examples of so-called syndromic autism have often been relegated to the sidelines in the study of the genetics of ASD by those interested in “pure” social disability. However, recent findings in the study of these conditions have provided remarkable insights into the developing central nervous system, and promise to transform our understanding of the pathophysiology and treatment of developmental delay and ASD.[32–34]
Recent Findings in Autism Genetics
The foregoing has outlined important conceptual issues in the study of the genetics of common disease in general and autism in particular. With this in mind, we now turn to a consideration of the past five years in the genetics of ASD, and outline recent progress both with regard to rare and common variants. As noted below, the weight of the empirical evidence highlights the critical role already played by the discovery of rare variation in ASD and suggests that both common and rare variant approaches have been and will continue to be highly relevant to the understanding of this potentially debilitating spectrum of syndromes.
Studies Geared to Finding Common Autism Alleles
Several study designs have been geared largely to investigating the contribution of common disease alleles to autism. The first of these types of studies are non-parametric linkage studies, the most common of which is the Affected Sib-Pair design (ASP). (See O’Roak and State, 2008 for a more in depth description of this method.[6] Briefly, this approach studies the transmission of genetic variation from one generation to the other in an effort to identify regions of the genome carrying disease risk. The analysis specifically avoids specifying the mode of transmission of a disorder, an approach that is intended to increase the ability to identify alleles contributing to risk in a complex fashion. While theoretically the ASP approach may identify either common or rare variations, it is not particularly robust in the face of high locus heterogeneity, making its application most useful in a practical sense to studying common risk alleles.
So far, more than a dozen such studies have been completed using genome-wide non-parametric approaches. The largest and most recent of these involved 1,181 multiplex families and 10,000 markers.[35] Taking all of these into account, nearly every chromosome has shown some evidence in favor of linkage, but no single region has been found to be highly significant, and no disease related variation/mutation has been identified yet within any of the most promising intervals. Nonetheless, attempts at further replication, studies of endophenotypes and intensive fine mapping of some of these intervals have yielded some quite interesting findings (discussed in more detail below). [36, 37]
Candidate gene association has also been a mainstay of methods aimed at identifying common alleles contributing to ASD. In these studies, variations in or near a gene or genes of interest are examined. Unlike linkage studies that evaluate the transmission of genomic segments from generation to generation, this type of analysis typically relies on evaluation of the frequency of previously identified common variation in cases versus controls. Such studies are extremely practical in that evaluation of known common alleles is inexpensive compared to rare variant discovery and many of the relevant study designs allow for the use of all probands whether or not additional family members are available, affected, and/or willing to participate in genetic research. The latter considerations are relevant to the feasibility of recruiting large numbers of patients for study, something that has been increasingly appreciated to be an important aspect of common variant studies.
While these approaches are attractive due to their relative ease of implementation and their utility in hypothesis-driven investigations, they have by and large not been reliable. Indeed, a comprehensive review several years ago looking across all of medicine, including in psychiatry, found that of 603 different reported gene-disease associations, of which 166 had been studied 3 or more times, only 6 were consistently replicated, with none in autism or other psychiatric disorders (with the exception APOE4).[38]
With the development of genome-wide as opposed to candidate gene based association studies and a related explosion of reproducible findings, several explanations for the poor track record of candidate approaches have emerged. These include (but are not limited to) a tendency to underestimate the sample size needed to identify risk (based on an initial overestimation of effect size of common risk alleles), often a failure to account sufficiently for the confound of ethnic variation (aka population stratification), the low prior probability of picking the right variation(s) to study, and overly permissive statistical thresholds.
While these potential flaws are found in many studies of ASD (as they are across all of medicine), several recent candidate gene investigations have employed more rigorous methodologies and have provided some evidence for replication, which ultimately is the gold standard for genetic findings. While not an exhaustive list, the genes EN2, MET and CNTNAP2 have emerged as relatively strong candidates from these recent single locus association studies.
Engrailed 2 (EN2) is a homeobox transcription factor that maps to the long arm of chromosome 7 and plays a key role in the development of the midbrain and cerebellum. Benayed and Gharani et al. reported significant association of this gene with autism in an initial sample to which the Millonig lab has subsequently added additional support by showing that mis-expression of mouse En2 in primary cortical cultures impairs neuronal differentiation.[39]
While the initial study was notable for the use of an internal replication sample prior to publication, the results of subsequent genetic studies have not been as clear. Zhong et al. found no association between autism and SNPs in the EN2 region,[40] and a second small study in the Chinese population was not able to replicate the initially reported SNP but did find some evidence for association of haplotypes in the region[41] Given these results, EN2 must still be considered a candidate for involvement in ASD, though so far it has not been independently replicated in a fashion that confers it clear disease risk status.
Of course, the potential explanations for this are myriad. It is possible that the initial result represents Type 1 error despite the authors’ best efforts to avoid this. Alternatively, the initial study may have simply overestimated the effect size of the risk allele, suggesting that the aforementioned efforts at replication have been underpowered. As will be discussed in more detail subsequently, the failure to identify this or any of the other candidate genes discussed in this section in the first genome wide association study (GWAS) of autism would tend to support either alternative. To the extent that the effect sizes have been overestimated, one would have to conclude that the total sample sizes studied to date with regard to ASD have not been nearly sufficient to rule out the contribution of these transcripts.
A similar story has evolved with regards to the MET oncogene, also located on chromosome 7 in a region that was found to show suggestive linkage to ASD in an ASP study [42]. Campbell et al. performed a rigorous analysis that included an internal replication sample and functional assays leading to the identification of a significant association between a regulatory SNP upstream of the MET gene and autism.[43] Additional genetic and biological studies have lent support to this initial observation, including work from the same laboratory.[44] In addition, in an independent study of 185 cases and 88 controls, Soussa found significant association, but to different markers than those implicated in the Campbell study.[45]
A third gene also on chromosome 7 (7q35), Contactin Associated Protein 2 (CNTNAP2), has emerged recently as a candidate for involvement in a range of developmental disordes including autism, language development, and seizure, based both on common and rare variant findings. The common variant findings will be discussed in this section and the rare variant research will be discussed subsequently.
In 2002 Alarcon and colleagues in Dan Geschwind’s lab implicated the 7q35 region in two non-parametric linkage studies focusing on an “age at first word” language phenotype among individuals with ASD [36, 47]. In 2008 these authors reported a follow-up fine mapping association study using the same measures and evaluating 1,172 trios from AGRE. This was a two stage study in which genes meeting an initial nominally significant cutoff were investigated in a second independent set of 304 AGRE trios.[46] The only marker that remained significant through both rounds of analysis corresponded to CNTNAP2.
At the same time Arking and colleagues in Aravinda Charkravarti’s lab conducted an analysis of linkage among 72 multiplex families and identified a suggestive peak at 7q35. Subsequently, they performed a follow-up TDT in this interval showing association with a single SNP at CNTNAP2 (permutation P<0.006). This was a different allele from that reported by Alarcon et al [46]. However, an internal replication using TDT analysis on an additional set of 1,295 trios supported the identified association based on the broader autism diagnosis.
Vernes and colleagues subsequently studied CNTNAP2 in relation to specific language impairment.[48] Using chromatin immunoprecipitation, they first demonstrated that the protein product of FOXP2, a gene causing a monogenic form of speech and language disorder, binds to CNTNAP2 and regulates its expression[48]. They then went on to demonstrate a positive association between SNPs in CNTNAP2 and an endophenotype of specific language disorder among a relatively small sample [48] but in the same region identified as associated in the Alarcon study described above.[46]
The aforementioned analyses are among the most rigorous contemporary candidate gene association studies to date. Importantly, the first large-scale genome-wide association study (GWAS) of autism was recently completed and identified significant association of ASD to an intergenic region on chromosome 5 – 5p14.1, mapping between the neuronal adhesion molecules Cadherin 9 and Cadherin 10.[49] As suggested above, the transition from candidate gene to genome-wide approaches in general has represented a key methodological shift in common variant studies. The latter have been shown to have considerable advantages with regard to reproducibility. Most likely this has resulted from the unbiased nature of the initial investigation (essentially all genes are queried simultaneously), the tendency to study somewhat larger sample sizes which have allowed identification of common variants carrying small risks, the use of genome-wide genotyping data and sophisticated methods to guard against population stratification, and a general agreement among the scientific community with regard to an appropriate statistical threshold for genome-wide significance that account for the very large number of tests inherent in such studies.
This recent finding must be viewed as a major success for the common variant approach to ASD. At the same time, it is consistent with studies of other complex conditions that have suggested that such variation is likely to be only a part of the overall story. For example, despite a sample size of 10,000 individuals, only a single significant locus was identified. Moreover, the most significantly associated SNP showed an odds ratio of 1.19, again consistent with the general findings of very modest effects for common variants in other complex disorders.[49] Finally, the identification of association with a marker mapping approximately 1 million base-pairs from either Cadherin 9 or Cadherin 10 (either or both of which are plausible candidates) presents a significant challenge for follow-up studies aimed at understanding the biology of this variation.[50–54]
In sum, with regard to common variants, the results of the non-parametric linkage studies remain uncertain as do the majority of promising candidate gene associations. The GWAS finding provides additional support for the contribution of common alleles, but makes the future identification of a common variant carrying even moderate risks for autism unlikely.
Rare Alleles and ASD
Cytogenetic Studies
The study of abnormalities in chromosomal structure, cytogenetics, has proven to be an important source of rare variant findings in a variety of disorders, including ASD. Traditionally, these abnormalities have been detected via microscopic examination of chromosomes. More recently, sub-microscopic structural changes have been detectable using the analysis of copy number variation (CNV).[55]
Rare microscopic chromosomal abnormalities occur at a mean rate of up to 7.4% in autism versus less than 1% in the general population.[56] Moreover, multiple studies have converged on particular chromosomal abnormalities in autism, the most common of which are maternally inherited duplications at 15q11-13. These duplications are found in as many as 1–3% of patients diagnosed with idiopathic autism. [57, 58]
Several studies have originated from or been strongly supported by traditional cytogenetic evidence leading to a number of key findings. For example, Thomas et al. in 1999 reported de novo deletions or translocations that affected the same region on the X chromosome at Xp22.3 in three girls with autism confirmed by the Autism Diagnostic Interview (ADI).[59]
Jamain and colleagues in 2003 looked for rare deleterious mutations in genes mapping to this interval and came across the first example of a clear functional mutation in a case of otherwise idiopathic autism which corresponded to the gene NLGN4X, a neuronal adhesion molecule subsequently found to be important for the specification of excitatory versus inhibitory synapses.[60, 61] The identified frameshift mutation in NLGN4X led to a premature termination of the protein with the loss of the critical transmembrane domain. This was found in two affected brothers (one with autism and one with Aspergers). The mutation was also found to be de novo in the unaffected mother, a finding that is not unexpected for a deleterious X-linked mutation. As expected, the mutation was not found in an unaffected brother, nor was it present in 350 unrelated controls.[60]
Jamain et al. also screened NLGN3 and NLGN4Y in 158 subjects with Autism or Aspergers and found a single suspect missense mutation in NLGN3.[60] NLGN3 is a homologue of NLGN4 but is located on a different region of the X chromosome at Xq13. The finding was again identified in two affected brothers, one with autism and the other with Aspergers.[60] While this substitution was present in a highly conserved region of the protein and was later found to alter synaptic function in the mouse, its relationship to disease was less clear initially, and, unlike the mutation in NLGN4X, has not been replicated in other human genetic studies.[62]
Shortly after publication of the Jamain et al paper, Laumonier and colleagues reported on the study of a large family with X-linked mental retardation.[63] Three of the 13 children with MR also had autism or PDD. Parametric analysis clearly supported linkage to the Xp22.3 region corresponding to the NLGN4X, and subsequent sequencing of this transcript in affected individuals revealed a two base-pair deletion leading to a frameshift and premature stop codon. Similar to the independent mutation observed by Jamain and colleagues this resulted in loss of the transmembrane domain and was not found in several hundred healthy male controls.[63]
Several mutation screenings of modest samples of patients (several hundred individuals) have not identified additional mutations in the genes NLGN4X or NLGN3 clearly carrying risks for [64–67] ASD. However, in addition to the convergence of findings from the Lammonier and Jamain studies cited above, both neurobiological and molecular evidence has accumulated supporting the importance of NLGN4X. Most importantly, genes coding for molecules that interact with NLGN4X, including SHANK3 and NRXN1 have both been strongly implicated in ASD. Given the expectation of a high rate of genetic heterogeneity. This type of evidence showing multiple mutations in a relevant molecular pathway as opposed to just a single gene is an important avenue for confirmation of rare variant findings and autism. [17, 68, 69]
In this regard the evidence for the involvement of SHANK3 is particularly compelling. Thomas Bourgeron’s lab, initially responsible for the first NLGN4X finding, subsequently identified de novo and transmitted structural and sequence variations in this transcript that codes for an important post synaptic binding partner of NLGN4. In an independent subsequent study [69], four de novo abnormities and nine inherited missense variants were identified among 400 families. Three of the de novo events were large-scale deletions, encompassing 277 kb, 3.2 Mb, and 4.36 Mb, while the fourth was a missense variant. The high rate of de novo mutations involving coding segments of SHANK3 in individuals with idiopathic ASD identified in independent studies, the finding of developmental delay and autistic features in patients with the 22q13 deletion syndrome (the genomic segment corresponding to where SHANK3 resides), and the interaction with NLGN4X provide strong convergent evidence for the importance of rare mutations in this transcript for ASD.
Several studies have also implicated Neurexin 1 (NRXN1), a trans-synaptic binding partner for Neuroligins. The Autism Genome Project Consortium reported a combined linkage and copy number analysis involving 1168 subjects.[35] This investigation, one of the first to address the issue of CNVs in autism, was confined to an analysis of only large-scale variations because it used first-generation arrays with 10,000 probes. Despite this, a family was identified in which two affected siblings shared the same 300 kb deletion, encompassing the coding region of NRXN1. This was not present in the parents, highlighting the phenomena of germline mosaicism. Rare missense variants and balanced chromosomal abnormalities disrupting NRXN1 in ASD patients have been reported as well. [70, 71]
The convergence of findings suggesting an ASD related NRXN/NLGN/SHANK3 pathway may also point more broadly to the importance of other cell-adhesion molecules in ASD. In 2005, Fernandez et al. studied a child with features of a rare deletion syndrome on the short arm of chromosome 3 who presented with social disability.[72] Using cytogenetic techniques, they identified and mapped a balanced translocation that disrupted the coding segment of the transcript Contactin 4, suggesting a potential role for these neuronal adhesion molecules in ASD. Two subsequent studies have provided additional evidence for the role of rare variation and particularly CNVs, in Contactin 4 [9, 73]
As noted above, subsequent studies involving both common and rare variant findings have implicated a similar molecular, Contactin-Associated Protein-Like 2 (CNTNAP2) in ASD. As a general proposition, contactins bind to contactin-associated proteins to mediate their functions, at least in the peripheral nervous system. CNTNAP2 was first and, so far most convincingly, tied to ASD by Strauss et al who mapped a homozygous recessive mutation in CNTNAP2 leading to intractable epilepsy, developmental delay and autistic features.[16] Moreover, the range of phenotypic expression of recessive mutations in this transcript have recently been expanded to include periventricular leukomalacia and hepatosplenomegaly.[75]
Subsequently, Bakkaloglu et al. mapped a de novo chromosomal abnormality in the only affected member of a pedigree and found the rearrangement disrupted CNTNAP2.[74] The authors comprehensively re-sequenced this molecule in 635 patients and 942 controls. This relatively large-scale re-sequencing effort demonstrated a two-fold increase in the burden of rare variants in cases versus controls and identified a single rare variant associated with affected status. While the cytogenetic and rare variant association findings were interesting in light of the other evidence implicating rare variants in CNTNAP2 to developmental delay, the authors pointed out that 1) the increase in the burden of rare mutations in cases and controls did not reach statistical significance, and 2) based on the methodology used in their study, they could not rule out the confound of population stratification with respect to the rare associated allele.
Parametric Linkage Analysis
The use of parametric linkage to study consanguineous families, as exemplified by the Strauss study noted above, represents another important alternative to cytogenetic approaches to the identification of extreme outlier families. Subsequent to the Strauss paper described above, Morrow et al. conducted a large-scale homozygosity mapping study in consanguineous Middle Eastern families.[76] They identified multiple, mostly non-overlapping regions of homozygosity, that is, regions of the genome in which a single identical chromosomal segment is inherited from both mother and father due to a recent common shared ancestor. Although the study did not reach genome-wide statistical significance, several large, rare, inherited homozygous deletions were found that disrupted either the coding or potential regulatory regions of brain-expressed transcripts, including DIA1 (Deleted in Autism-1) (c3orf58), NHE9 (Sodium/Proton Exhanger9), PCDH10 (protocadherin 10) and CNTN3 (Contactin 3). The authors found additional strong evidence supporting a role for the gene NHE9, the (Na+, K+)/H+ exchanger, in ASD through the identification of a rare nonsense mutation in two male siblings with autism, one of whom has epilepsy and the other probable seizures, in a non-consanguineous family. They found rare amino acid changes in NHE9 in nearly 6% of patients with both autism and epilepsy versus only 0.63% of controls. Further, based on an independent set of studies reported in the same publication, three of the genes located within or closest to the two largest deletions (DIA1, NHE9, PCDH10) were found to either be regulated by neural activity and/or were the targets of activity-induced transcription factors. This suggests that changes in activity-regulated gene expression during brain development may contribute to ASD.
CNVs and ASD
The development of new technology has now expanded the scope of cytogenetic studies. In addition to potentially identifying smaller “abnormalities” of the type that has led to the important outlier findings discussed above, CNV analysis now offers researchers the first truly cost-effective tools for scanning the entire genome for rare variants, allowing for an assessment of the Rare Variant, Common Disease (RVCD) hypothesis. The earliest studies in this regard seemed to support an overall contribution of rare variation to the population risk of ASD, consistent with the RVCD hypothesis. The first study to suggest this found that 7–10% of simplex autism families, 2 to 3% of multiplex families but only 1% of control families carried rare de novo CNVs.[55] Subsequent studies have supported this overall pattern.[77, 78] Moreover, the finding of an increased rate of de novo variation is consistent both with the MZ and DZ data as well as research suggesting that increasing paternal age at the time of conception is a risk factor for autism.[79]
Interestingly, these studies initially focused on relatively large CNVs, and used samples that included patients with significant developmental disability in addition to ASD. Whether the same overall increase in mutation burden will be as evident as the resolution of CNV analysis increases and more diverse samples (with, for example, higher IQs and less dysmorphology) are included, is not yet clear.
In addition to identifying an increase in very large de novo CNVs on a population basis, several recent CNV studies have identified specific regions of the genome that appear to carry substantial risk for ASD. For example, both de novo deletions and duplications at 16p11.2 have been identified in patients with ASD[80, 81], and, when evaluated together, they have been found to increase the risk of autism. [81] Weiss et al. found them in 1% of autism case samples versus in less than 0.1% of the general population. Statistical significance was achieved independently in three distinct populations and 15 brain-expressed genes in the region are being explored.[81]. Marshall et al. conducted a similar CNV analysis that further supported the association of CNVs at 16p.11.[77] However, a more recent study did not replicate the 16p11 findings due to an increased frequency of these variants seen in controls.[9] This disparity may suggest sample heterogeneity or potentially the confound of population stratification, something that was not explicitly explored in the initial Weiss report. Additional studies with well-characterized and fastidiously matched controls, as is now common in GWAS studies, will be required to further evaluate this locus. Moreover, additional biological studies will be tremendously helpful in determining whether the inclusion of both deletions and duplications in this and other CNV association analyses makes biological sense. Certainly, recent findings in other pervasive developmental disorders support this hypothesis. [82]
Several other recent CNV studies have supported the role of this class of variation to ASD risk. For example, Marshall et al., in addition to finding support for the 16p.11 locus as mentioned above, identified CNVs (either deletions or duplications) that were over-represented in cases versus controls for SHANK3, NLGN4, and NRXN1 genes [77] They also identified several new candidates, including DPP6 and DPP10, [77].
Bucan and colleagues conducted a similar study.[78] using multiple independent samples of cases as well as controls. They identified 14 deletions present at least once in both groups of probands, but in neither control set (N= 2539).[78] As in previous studies, some of these supported previous candidate genes, including NRXN1. Newly identified genes included BZRAP1 at 17q22 and MDGA2 at 14q21.3. The former codes for an adaptor molecule thought to regulate synaptic transmission by linking vesicular release machinery to voltage gated Ca2+ channels.[78] The latter is less well-characterized but the researchers noted that the protein structure as predicted by BLASTP is unexpectedly similar to that of Contactin 4, discussed above.[78]
Finally, a very recent large-scale study supported prior rare variant discoveries, including the relevance of CNTN4 and Neurexin 1 in ASD. [9] As in the case of 16p11, the authors have chosen at times to combine duplications and deletions in considering replication: for example, previous findings have pointed to a role for deletions or disrupting translocations in CNTN4, while the report of replication in this case focused on duplications present in cases and absent in controls. In addition to providing additional evidence for previously mapped regions, this recent study suggested an entirely new molecular mechanism: four of the candidate genes identified were related to the ubiquitin pathway. The authors point out that “the ubiquitin system operates both pre and post-synaptically to regulate the range of synaptic attributes including endo and exocytosis, dendritic elaboration and the formation of the post-synaptic density.”[9]
Syndromic Autism
As mentioned above, another line of evidence implicating the contribution of rare alleles to autism genetics is the overlap in phenotype between autism and rare monogenic diseases. For example, “ASD may be diagnosed in 30% of males with FXS and likewise Fragile X mutations may be found among as many as 7–8% of individuals with idiopathic ASD.”[83] Similarly, mutations in MECP2, the Rett Syndrome gene, have been found among cases of “idiopathic autism” without the Rett phenotype. For example, 2 of 69 females diagnosed as having “idiopathic autism” were found to have MECP2 mutations in one case series.[84] Likewise, autistic patients have an increased risk for neurofibromatosis (100 fold) and other rare monogenic diseases like tuberous sclerosis and Joubert’s Syndrome, and patients with these disorders are reported to have an increased risk for having autism. [85, 86] The association between autism and these neuropsychiatric syndromes is reviewed in further detail elsewhere.[87–89]
While the above-mentioned evidence (and related biological studies) underscores that the study of rare monogenic syndromes may be extraordinarily useful in understanding idiopathic autism, the reported rates of phenotypic overlap need to be viewed with a modicum of caution. First, a distinction must be drawn between evidence that derives from the finding of rare syndromic mutations in children who are thought to represent cases of idiopathic autism versus evidence that derives from the identification of ASD features in individuals with developmental delay syndromes. With regard to the latter, it is important to note that many studies focusing on this question are limited in their ability to blind diagnostic assessments owing to both the nature of patient recruitment and the often pathognomonic physical features of affected individuals. An additional consideration is that ASD diagnoses are not uniformly made using state-of-the-art instruments, and that there may be a considerable degree of diagnostic uncertainty arising in cases of severe developmental delay.[90]
Clinical Implications
What are the clinical implications of the genetic findings to date (summarized in Table 1)? It is worthwhile to start by re-stating the obvious: there is not a single gene or genetic test that definitively diagnoses autism. The diagnosis of autism remains a clinical/syndromic one. However, this does not preclude the usefulness of genetic testing in aiding in diagnosis, family planning or prognosis, the importance of which should not be underestimated.[91]
Table 1.
Summary Evidence for Selected ASD Candidate Genes
| Gene/Region | Chr | Evidence |
|---|---|---|
| Neurexin 1 (NRXN1) | 2 | |
| Contactin 4 | 3 | |
| Sodium/Proton Exchanger 9 (NHE9) | 3 |
|
| 5p14.1 | 5 |
|
| Contactin Associated Protein 2 (CNTNAP2) | 7 |
|
| Engrailed 2 (EN2) | 7 | |
| MET oncogene | 7 |
|
| 16p11.2 | 16 | |
| SHANK3 | 22 |
|
| Neuroligan 3 (NLGN3) | X | |
| Neuroligan 4X (NLGN4X) | X |
|
There are several institutional practice parameters and guidelines that provide recommendations regarding genetic testing in autism. In addition, there are scholarly articles that review the subject. On certain recommendations, there is near complete agreement. Others remain debated, and practice often differs from clinic to clinic. Moreover, it is important to note that many of the formal practice parameters were developed prior to the recent explosion of data from CNV analyses and consequently tend to underestimate the now demonstrated yield of these approaches.
The American Academy of Child and Adolescent Psychiatry’s most recent Practice Parameter from 1999 offers broad recommendations regarding the need for genetic testing after the diagnosis of Autism has been made. They state that the presence of dysmorphic features or a family history of intellectual disability may “suggest” obtaining genetic screening for metabolic disorders or chromosomal analysis and/or genetics consultation.[92]. An updated version of these recommendations with more detailed recommendations based on current literature will soon be forthcoming.
In 2000, The American Academy of Neurology guidelines made the following “Level 2” evidence-based recommendation: “Genetic testing in children with autism, specifically high resolution chromosome studies (karyotype) and DNA analysis for [Fragile X], should be performed in the presence of mental retardation (or if mental retardation cannot be excluded), if there is a family history of [Fragile X] or undiagnosed mental retardation, or if dysmorphic features are present. However, there is little likelihood of positive karyotype or [Fragile X] testing in the presence of high-functioning autism.”[93]. The latter conclusion is currently a matter of some debate as ongoing studies focusing on higher functioning ASD continue to identify cases of previously undiagnosed Fragile X syndrome.
The 2007 American Academy of Pediatrics Clinical Report states that genetic testing, such as chromosomal analysis, subtelomeric FISH, and specific Fragile X testing, may be indicated in children with ASDs only if they have either coexisting global developmental delay or intellectual disability. The say, however, that newer techniques such as comparative genomic hybridization-microarray analysis (CGH) that can detect submicroscopic chromosomal abnormalities may become standard of care in the future but has not been sufficiently evaluated in children with ASDs to date.[94]
Thus, amongst the institutional guidelines, the consensus is that routine genetic screening or referral to a geneticist is not indicated for every patient diagnosed with idiopathic autism. Rather, screening or referral should be triggered only when suspicion is raised by the history or presentation. All agree that one such trigger is the presence of intellectual disability in the patient or a history of it in the family. Two of the guidelines suggest that another trigger should be dysmorphic features in the patient. All recommend chromosomal analysis and Fragile X testing at a minimum and/or referral to a geneticist. Again, we highlight here that the most recent of these recommendations were published in 2007 and could not take into account the widespread dissemination of high resolution CNV analyses.
In fact, there has been an increasing appreciation in the primary literature that more extensive genetic screening may be valuable on a more routine basis. Using a three-tiered protocol of neurogenetic evaluation of patients diagnosed with idiopathic autism, a 2006 study reported a 40% yield.[95] The first tier included the dysmorphology criteria found in the clinical guidelines above with these findings resulting in targeted genetics workups. If a diagnosis was not found in each tier, a subsequent panel of tests was undertaken. The subsequent tiers included karyotyping, Fragile X testing, MECP-2 testing, 22q11 FISH, 15 interphase FISH, Prader Willi/Angelman testing, 17p11 FISH, and sub-telomeric FISH if IQ was less than 50. The authors felt that the yield was sufficiently high to warrant more routine evaluation and testing by clinical geneticists.[95]
An excellent 2009 review of the subject also goes beyond the institutional guidelines in its recommendations. For example, it recommends karyotype and Fragile X testing for all patients with ASDs and MECP2 testing in all girls with autism and intellectual disability even in the absence of Rett symptoms.[91] It also points to the “near future “ (which has now arrived) when the cost of array based CGH testing for the detection of CNVs will be inexpensive enough that it should not be restricted only to patients with dysmorphology or history of intellectual disability.[91]
The definite trend in the literature over the past decade is for increased genetic testing and a decreasing threshold for obtaining such tests. This is well-justified as more is learned about the genetic causes of autism and as tests become more accurate and less expensive. These changes are occurring very rapidly, and it is difficult for general clinicians such as neurologists, pediatricians, and psychiatrists to keep abreast of the changes. Further, while there is some training in observing dysmorphologies in these fields, for most, it will fall far short of that which is routine for clinical geneticists. Thus, using dysmorphology as a criteria for work-up may lead to missed opportunities for more specific diagnosis. The cost of this is not simply academic. For example, missing a 22q11 deletion syndrome diagnosis may keep a patient with a treatable cardiac condition from receiving adequate care. Finally, increased testing, especially with high resolution microarrays, could lead to the identification of further syndromes. Owing to these issues, we favor a standard workup that includes Fragile X testing, and screening chromosomes with a high-resolution array, along with referral to a clinical geneticist for counseling in the case of positive results, and further consultation for those who screen negative, but have a history of developmental delay, regression or evidence of dysmorphology, including macrocephaly.
Summary and Conclusions
Autism and related conditions are highly heritable disorders. Consequently, gene discovery promises to help elucidate the underlying pathophysiology of these syndromes and, it is hoped, eventually improve diagnosis, treatment, and prognosis. The genetic architecture of autism is not yet known. What can be said from the studies to date is that writ large, autism is not a monogenic disorder with Mendelian inheritance. In many, but clearly not all individual cases, it is likely to be a complex genetic disorder that results from simultaneous genetic variations in multiple genes. The CDCV hypothesis predicts that the risk alleles in Autism and other complex disorders will be common in the population. However, recent evidence both with regard to autism and other complex disorders, raises significant questions regarding the overall applicability of the theory and the extent of its usefulness in explaining individual genetic liability. In addition, considerable evidence points to the importance of rare alleles for the overall population of affected individuals as well as their role in providing a foothold into the molecular mechanisms of disease. Finally, there is debate regarding the clinical implications of autism genetic research to date. Most institutional guidelines recommend genetic testing or referral only for idiopathic autism if intellectual disability and dysmorphic features are present. However, recent advances suggest that the combination of several routine tests combined with a low threshold for referral is well-justified in cases of idiopathic autism.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord. 2003;33(4):365–82. doi: 10.1023/a:1025054610557. [DOI] [PubMed] [Google Scholar]
- 2.American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM-IV-TR) 2000. [Google Scholar]
- 3.Martin A, Volkmar FR, Lewis M. Lewis’s Child and Adolescent Psychiatry: A Comprehensive Textbook. Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 4.Schopler E, Mesibov GB, Kunce LJ. Asperger Syndrome Or High-Functioning Autism? Plenum Pub Corp; 1998. [Google Scholar]
- 5.Ganz ML. The lifetime distribution of the incremental societal costs of autism. Archives of Pediatrics and Adolescent Medicine. 2007;161(4):343. doi: 10.1001/archpedi.161.4.343. [DOI] [PubMed] [Google Scholar]
- 6.O’Roak BJ, State MW. Autism genetics: strategies, challenges, and opportunities. Autism Research. 2008;1(1) doi: 10.1002/aur.3. [DOI] [PubMed] [Google Scholar]
- 7.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nature Reviews Genetics. 2008;9(5):341–55. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14. 1 associate with autism spectrum disorders. Nature. 2009 doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–73. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Nyholt DR, Magnussen P, et al. A genomewide screen for autism susceptibility loci. The American Journal of Human Genetics. 2001;69(2):327–40. doi: 10.1086/321980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey A, Le Couteur A, Gottesman I, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25(1):63. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 12.Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. 2005;66:3. [PubMed] [Google Scholar]
- 13.Bolton P, Macdonald H, Pickles A, et al. A case-control family history study of autism. Journal of Child Psychology and Psychiatry. 1994;35(5):877–900. doi: 10.1111/j.1469-7610.1994.tb02300.x. [DOI] [PubMed] [Google Scholar]
- 14.Gupta AR, State MW. Recent advances in the genetics of autism. Biol Psychiatry. 2007;61(4):429–37. doi: 10.1016/j.biopsych.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 15.Morrow EM, Yoo SY, Flavell SW, et al. Identifying Autism Loci and Genes by Tracing Recent Shared Ancestry. Science. 2008;321(5886):218. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strauss KA, Puffenberger EG, Huentelman MJ, et al. Recessive Symptomatic Focal Epilepsy and Mutant Contactin-Associated Protein-like 2. N Engl J Med; N Engl J Med. 2006;354(13):1370–7. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 17.Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–27. doi: 10.1038/ng1933. Epub 2006 Dec 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakravarti A. Population genetics—making sense out of sequence. Nat Genet. 1999;21(1 Supplement):56–60. doi: 10.1038/4482. [DOI] [PubMed] [Google Scholar]
- 19.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends in Genetics. 2001;17(9):502–10. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 20.Farrer L, Cupples L, Haines J, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–56. [PubMed] [Google Scholar]
- 21.Klein RJ, Zeiss C, Chew EY, et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science. 2005;308(5720):385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards AO, Ritter R, III, Abel KJ, et al. Complement Factor H Polymorphism and Age-Related Macular Degeneration. Science. 2005;308(5720):421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 23.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PIW, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostroem KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney ASF, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jorgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CNA, Payne F, Perry JRB, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjogren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genet. 2008;40(5):638–45. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 25.Iyengar SK, Elston RC. The genetic basis of complex traits: rare variants or “common gene, common disease”? Methods Mol Biol. 2007;376:71–84. doi: 10.1007/978-1-59745-389-9_6. [DOI] [PubMed] [Google Scholar]
- 26.Clark R, Hutton M, Fuldner M, et al. The structure of the presenilin 1(S 182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995;11(2):219–22. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- 27.Inlow JK, Restifo LL. Molecular and Comparative Genetics of Mental Retardation. Genetics. 2004;166(2):835–81. doi: 10.1093/genetics/166.2.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen JC, Pertsemlidis A, Fahmi S, et al. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proceedings of the National Academy of Sciences. 2006;103(6):1810–5. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji W, Foo JN, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40(5):592–9. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown MS, Goldstein JL. Expression of the Familial Hypercholesterolemia Gene in Heterozygotes: Mechanism for a Dominant Disorder in Man. Science. 1974;185(4145):61–3. doi: 10.1126/science.185.4145.61. [DOI] [PubMed] [Google Scholar]
- 31.Lifton RP, Gharavi AG, Geller DS. Molecular Mechanisms of Human Hypertension. Cell. 2001;104(4):545–56. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 32.Bear MF, Dölen G, Osterweil E, et al. Fragile X: translation in action. Neuropsychopharmacology. 2007;33(1):84–7. doi: 10.1038/sj.npp.1301610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2/– mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843–8. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Cui Y, Kushner SA, et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Current Biology. 2005;15(21):1961–7. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 35.Szatmari P, Paterson AD, Zwaigenbaum L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–28. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alarcón M, Cantor RM, Liu J, et al. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. The American Journal of Human Genetics. 2002;70(1):60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arking DE, Cutler DJ, Brune CW, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. The American Journal of Human Genetics. 2008;82(1):160–4. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirschhorn JN, Lohmueller K, Byrne E, et al. A comprehensive review of genetic association studies. Gen Med. 2002;4;2(2):45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 39.Benayed R, Gharani N, Rossman I, et al. Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. The American Journal of Human Genetics. 2005;77(5):851–68. doi: 10.1086/497705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong H, Serajee F, Nabi R, et al. No association between the EN2 gene and autistic disorder. Br Med J. 2003;40(1) doi: 10.1136/jmg.40.1.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Jia M, Yue W, et al. Association of the ENGRAILED 2 (EN2) gene with autism in Chinese Han population. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(4):434–8. doi: 10.1002/ajmg.b.30623. [DOI] [PubMed] [Google Scholar]
- 42.IMGSAC A. A genomewide screen for autism: Strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001;69:570–81. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell DB, Sutcliffe JS, Ebert PJ, et al. A genetic variant that disrupts MET transcription is associated with autism. Proceedings of the National Academy of Sciences. 2006;103(45):16834. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campbell DB, Li C, Sutcliffe JS, et al. Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. Autism research: official journal of the International Society for Autism Research. 2008;1(3):159. doi: 10.1002/aur.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sousa I, Clark TG, Toma C, et al. MET and autism susceptibility: family and case–control studies. European Journal of Human Genetics. 2008;17(6):749–58. doi: 10.1038/ejhg.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alarcón M, Abrahams BS, Stone JL, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. The American Journal of Human Genetics. 2008;82(1):150–9. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alarcon M, Yonan A, Gilliam T, et al. Quantitative genome scan and ordered-subsets analysis of autism endophenotypes support language QTLs. Mol Psychiatry. 2005;10(8):747–57. doi: 10.1038/sj.mp.4001666. [DOI] [PubMed] [Google Scholar]
- 48.Vernes SC, Newbury DF, Abrahams BS, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359(22):2337. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14. 1 associate with autism spectrum disorders. Nature. 2009 doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feuk L. ASHG 2008 Annual Meeting: from enormous cohorts to individual genomes. Genome Medicine. 2009;1:9. doi: 10.1186/gm9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldstein DB. Common Genetic Variation and Human Traits. N Engl J Med; N Engl J Med. 2009;360(17):1696–8. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 52.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316(5829):1341–1345. doi: 10.1126/science.1142382. Epub 2007 Apr 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445(7130):881–885. doi: 10.1038/nature05616. Epub 2007 Feb 11. [DOI] [PubMed] [Google Scholar]
- 54.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316(5829):1336–1341. doi: 10.1126/science.1142364. Epub 2007 Apr 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J, Zwaigenbaum L, Szatmari P, et al. Molecular Cytogenetics of Autism. Curr Genomics. 2004;5:347–64. [Google Scholar]
- 57.Cook E, Jr, Lindgren V, Leventhal B, et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60(4):928. [PMC free article] [PubMed] [Google Scholar]
- 58.Schroer RJ, Phelan MC, Michaelis RC, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet. 1998;76(4):327–36. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 59.Thomas NS, Sharp AJ, Browne CE, et al. Xp deletions associated with autism in three females. Hum Genet. 1999;104(1):43–8. doi: 10.1007/s004390050908. [DOI] [PubMed] [Google Scholar]
- 60.Jamain S, Quach H, Betancur C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–9. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chih B, Afridi SK, Clark L, et al. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet. 2004;13(14):1471–7. doi: 10.1093/hmg/ddh158. [DOI] [PubMed] [Google Scholar]
- 62.Tabuchi K, Blundell J, Etherton MR, et al. A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice. Science. 2007;318(5847):71–6. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laumonnier F, Bonnet-Brilhault F, Gomot M, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74(3):552–557. doi: 10.1086/382137. Epub 2004 Feb 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blasi F, Bacchelli E, Pesaresi G, et al. Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(3):220–1. doi: 10.1002/ajmg.b.30287. [DOI] [PubMed] [Google Scholar]
- 65.Gauthier J, Bonnel A, St-Onge J, et al. NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2005;(1) doi: 10.1002/ajmg.b.30066. [DOI] [PubMed] [Google Scholar]
- 66.Vincent JB, Kolozsvari D, Roberts WS, et al. Brief Research Communication: Mutation Screening of X-Chromosomal Neuroligin Genes: No Mutations in 196 Autism Probands. AMERICAN JOURNAL OF MEDICAL GENETICS PART B. 2004;129:82–4. doi: 10.1002/ajmg.b.30069. [DOI] [PubMed] [Google Scholar]
- 67.Yan J, Oliveira G, Coutinho A, et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry. 2004;10(4):329–32. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- 68.Meyer G, Varoqueaux F, Neeb A, et al. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology. 2004;47(5):724–33. doi: 10.1016/j.neuropharm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 69.Moessner R, Marshall CR, Sutcliffe JS, et al. Contribution of SHANK3 mutations to autism spectrum disorder. The American Journal of Human Genetics. 2007;81(6):1289–97. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feng J, Schroer R, Yan J, et al. High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409(1):10–3. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 71.Kim HG, Kishikawa S, Higgins AW, et al. Disruption of neurexin 1 associated with autism spectrum disorder. The American Journal of Human Genetics. 2008;82(1):199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fernandez T, Morgan T, Davis N, et al. Disruption of Contactin 4 (CNTN4) Results in Developmental Delay and Other Features of 3p Deletion Syndrome. The American Journal of Human Genetics. 2004;74(6):1286–93. doi: 10.1086/421474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roohi J, Montagna C, Tegay DH, et al. Disruption of contactin 4 in 3 subjects with autism spectrum disorder. Br Med J. 2008 doi: 10.1136/jmg.2008.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bakkaloglu B, O’Roak BJ, Louvi A, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. The American Journal of Human Genetics. 2008;82(1):165–73. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jackman C, Horn ND, Molleston JP, et al. Gene associated with seizures, autism, and hepatomegaly in an Amish girl. Pediatr Neurol. 2009;40(4):310–3. doi: 10.1016/j.pediatrneurol.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 76.Morrow EM, Yoo SY, Flavell SW, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321(5886):218. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. The American Journal of Human Genetics. 2008;82(2):477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bucan M, Abrahams BS, Wang K, et al. Genome-Wide Analyses of Exonic Copy Number Variants in a Family-Based Study Point to Novel Autism Susceptibility Genes. PLoS Genet. 2009;5(6):e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reichenberg A, Gross R, Weiser M, et al. Advancing Paternal Age and Autism. Arch Gen Psychiatry. 2006;63(9):1026. doi: 10.1001/archpsyc.63.9.1026. [DOI] [PubMed] [Google Scholar]
- 80.Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16 p 11.2 microdeletions in autism. Hum Mol Genet. 2008;17(4):628. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 81.Weiss LA, Shen Y, Korn JM, et al. Association Between Microdeletion and Microduplication at 16p11.2 and Autism. Obstet Gynecol Surv. 2008;63(6):361. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 82.Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. The American Journal of Human Genetics. 2005;77(3):442–53. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113(5) doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- 84.Carney RM, Wolpert CM, Ravan SA, et al. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol. 2003;28(3):205–11. doi: 10.1016/s0887-8994(02)00624-0. [DOI] [PubMed] [Google Scholar]
- 85.Marui T, Hashimoto O, Nanba E, et al. Association between the neurofibromatosis-1 (NF1) locus and autism in the Japanese population. AMERICAN JOURNAL OF MEDICAL GENETICS PART B. 2004;131:43–7. doi: 10.1002/ajmg.b.20119. [DOI] [PubMed] [Google Scholar]
- 86.Smalley SL. Autism and Tuberous Sclerosis. J Autism Dev Disord. 1998;28(5):407–14. doi: 10.1023/a:1026052421693. [DOI] [PubMed] [Google Scholar]
- 87.Zafeiriou DI, Ververi A, Vargiami E. Childhood autism and associated comorbidities. Brain and Development. 2007;29(5):257–72. doi: 10.1016/j.braindev.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 88.Kumar R, Christian S. Genetics of autism spectrum disorders. Current Neurology and Neuroscience Reports. 2009;9(3):188–97. doi: 10.1007/s11910-009-0029-2. [DOI] [PubMed] [Google Scholar]
- 89.Fombonne E, Du Mazaubrun C, Cans C, et al. Autism and associated medical disorders in a French epidemiological survey. Journal of Amer Academy of Child & Adolescent Psychiatry. 1997;36(11):1561. doi: 10.1016/S0890-8567(09)66566-7. [DOI] [PubMed] [Google Scholar]
- 90.Fred R. Volkmar, Matthew State, Ami Klin. Autism and autism spectrum disorders: diagnostic issues for the coming decade. Journal of Child Psychology and Psychiatry. 2009;50(1–2):108–15. doi: 10.1111/j.1469-7610.2008.02010.x. [DOI] [PubMed] [Google Scholar]
- 91.Lintas C, Persico A. Autistic phenotypes and genetic testing: state-of-the-art for the clinical geneticist. Br Med J. 2009;46(1):1. doi: 10.1136/jmg.2008.060871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.MDGR, Tanguay P, Valerie Arnold M, et al. Summary of the practice parameters for the assessment and treatment of children, adolescents, and adults with autism and other pervasive developmental disorders. J Am Acad Child Adolesc Psychiatry. 1999;38:1611. doi: 10.1097/00004583-199912000-00028. [DOI] [PubMed] [Google Scholar]
- 93.Filipek P, Accardo P, Ashwal S, et al. Practice parameter: Screening and diagnosis of autism Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Child Neurology Society. Neurology. 2000;55(4):468–79. doi: 10.1212/wnl.55.4.468. [DOI] [PubMed] [Google Scholar]
- 94.Johnson CP, Myers SM. Identification and evaluation of children with autism spectrum disorders. Pediatrics. 2007;120(5):1183. doi: 10.1542/peds.2007-2361. [DOI] [PubMed] [Google Scholar]
- 95.Schaefer GB, Lutz RE. Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genetics in Medicine. 2006;8(9):549. doi: 10.1097/01.gim.0000237789.98842.f1. [DOI] [PubMed] [Google Scholar]