

Abstract
Shape-memory materials (including polymers, metals, and ceramics) are those that are processed into a temporary shape and respond to some external stimuli (e.g., temperature) to undergo a transition back to a permanent shape.[1, 2] Shape memory polymers are finding use in a range of applications from aerospace to fabrics, to biomedical devices and microsystem components.[3–5] For many applications, it would be beneficial to initiate heating with an external trigger (e.g., transdermal light exposure). In this work, we formulated composites of gold nanorods (<1% by volume) and biodegradable networks, where exposure to infrared light induced heating and consequently, shape transitions. The heating is repeatable and tunable based on nanorod concentration and light intensity and the nanorods did not alter the cytotoxicity or in vivo tissue response to the networks.
Keywords: Nanorods, Polymers, Temperature, Biomaterials
The temperature at which shape transitions occur in polymers can be controlled through properties such as the glass transition temperature (Tg) or the melting temperature (e.g., through crystallization). In polymer networks the permanent shape is typically governed by covalent bonds, whereas the temporary shape is controlled by the thermal transition.[1, 2] For many implantable biodevices, the Tg of the material is defined to be slightly below body temperature so that implantation in vivo induces the desired shape change. These materials are being applied in applications such as implantable stents, self-tying sutures, and for securing materials to tissues.[6, 7] Beyond a shape-change, the transition from a glassy to rubbery network or transition through a lower critical solution temperature may also lead to changes in molecule diffusion through the network.[8, 9] However, one drawback to the use of these materials is that the shape change is not completely controllable, in that the change happens nearly instantaneously when implanted.
Typically, the temperature change is induced by direct heating of the polymer (e.g., when implanted) and there have been few examples where external triggers (e.g., light) are used to control the heating process.[10] Light has been used in systems where photoactive moieties are incorporated; however, the molecular rearrangement that occurs is often not reversible or the chemistry is not widely applicable to all polymer systems.[11–14] Yet, there are advantages to using light in that there is spatial control to heating, heating can be defined to the implant/device and not the surroundings, and that the heating can be performed indirectly (e.g., transdermally). For transdermal light exposure, wavelengths are limited in that many do not penetrate tissues significantly; yet, near-infrared wavelengths penetrate tissues well[15] and have been used extensively for the heating of nanoparticles in vivo. Specifically, gold nanoparticles have found use in biomedical applications, particularly for thermal ablation of tumors,[16–21] where the particles are injected and a laser is directed at the tissue for heating. Thus, the combination of nanoparticles (e.g., nanorods) with polymers would provide a facile system where thermal transitions can be induced indirectly with light exposure for a range of polymers.
Temperature transitions (e.g., Tg) in polymer networks are readily controlled through a range of structural parameters, including the crosslinking density of the material.[22, 23] In most instances, non-degradable crosslinkers are used (e.g., diacrylated ethylene glycols); however, there are now many applicable biodegradable polymers for use in these applications.[7, 24, 25] One group of biodegradable and crosslinkable polymers, poly(β-amino ester)s (PBAEs), were recently synthesized with acrylate end groups for use in biomedical applications.[26] There are a range of PBAEs available with varied degradation and mechanics based on the reagents used, in addition to the macromer molecular weight and branching.[26–28] One macromer (termed A6) has previously been characterized extensively as a homopolymer and for cytotoxicity[29] and can be used to crosslink monofunctional molecules such as tert-butyl acrylate (tBA), which has been used extensively in shape-memory applications.[22, 23] The chemical structures of tBA and A6 are shown in Figure 1A. A6 acts as a crosslinker and degrades through hydrolysis of ester units.
Figure 1.

(A) Structures of macromer A6 and monomer tBA used for network formation; (B) Reaction behavior (double bond conversion) with ultraviolet light exposure during the crosslinking of 15:85 A6:tBA networks without (solid) or with (dotted) nanorods (high concentration); (C) Bulk and TEM images of nanorods encapsulated in 15:85 A6:tBA networks (bar = 100 nm, inset bar = 20 nm); (D) Storage modulus (solid lines) and tan d (dotted lines) for 15:85 A6:tBA networks without (black) or with (grey) nanorods (mid concentration).
Gold nanorods were synthesized using a previously reported seed-mediated growth process[30] and were further pegylated to alter their solubility.[31] The nanorods were imaged on a transmission electron microscope (Figure S1A) and exhibited a characteristic morphology with an average length, width, and aspect ratio of 31.08 ± 4.52 nm, 9.24 ± 2.75 nm, and 3.64 ± 0.82, respectively, with the majority having elongated shapes and a small fraction that were rounded (aspect ratio ~1) (Figure S1B). The nanorods exhibited typical absorbances for both the longitudinal and transverse plasmon peaks (Figure S1C), with a slight shift based on pegylation. Using a previously determined molar extinction coefficient of 4.4×109 M−1cm−1,[31] the nanorod concentration was determined for defined loading into polymers.
The nanorods were then added to a solution of A6, tBA, and photoinitiator at a range of concentrations (Table 1) and polymerized with ultraviolet light exposure. Importantly, there were no changes in the polymerization behavior in samples without and with nanorods (Figure 1B); thus, there are no concerns with the particles interfering with the polymerization. This is due to the minimal absorption (Figure S1C) of the nanorods at the polymerization wavelength (365nm) and that the nanorods are loaded at relatively low concentrations (<1% by volume for all formulations). The samples exhibited a purple color dependent on the concentration of nanorods incorporated and TEM images indicate that the nanorods are dilute, that there are no visual changes in nanorod shape after encapsulation, and that there is no significant clustering of the nanorods in the networks (Figure 1C).
Table 1.
Properties of 15:85 A6:tBA networks containing a range of nanorod concentrations.
| Group | Nanorod Concentration (mol AuNR) | Tg (°C) |
|---|---|---|
| None | 0 | 40.7 ± 1.8 |
| Low | 1.8·10−13 | 44.0 ± 0.1 |
| Mid | 7.2·10−13 | 43.1 ± 0.8 |
| High | 3.3·10−12 | 53.3 ± 3.6 |
With respect to viscoelastic properties, samples exhibited a typical profile for crosslinked networks with a decrease in the elastic modulus with increased temperature (transition from glassy to rubbery network), as well as a broad tan δ curve (Figure 1D). The tan δ is the ratio of the loss modulus to the storage modulus and is indicative of the ability of a material to dissipate energy. The peak of the tan δ curve is one method used to determine the Tg of the material. An increase in the ratio of the crosslinker A6 to tBA in the networks increased the Tg of the polymers (Figure S2) by altering the crosslinking density, which has been observed by others.[32] Thus, the crosslinker concentration provides a simple technique to alter the thermo-mechanical properties of networks towards their intended use and desired Tg. The addition of the nanorods slightly increased the Tg for many networks (Figure S2 and Table 1) and the magnitude of increase was dependent on the nanorod concentration. A representative profile of the storage modulus and tan δ for a network without and one with nanorods (mid concentration) is shown in Figure 1C. The profiles for both of these parameters shifted slightly to the right with inclusion of nanorods in the networks. The influence of the addition of particles on the network properties is dependent on the interfacial interactions between the polymer and the particles,[33, 34] and it is not unexpected to see this change with addition of nanorods. For the remainder of the work reported, a formulation of 15:85 A6:tBA was used since it exhibits a Tg slightly higher than body temperature and is useful for potential implant applications. Although fluid uptake could alter these transition temperatures, there is very little swelling in these systems due to the hydrophobicity of the network. For example, there was less than a 1% increase in mass after swelling in water for 24 hours, regardless of the incorporation of nanorods.
The polymers without and with nanorods were assessed for cytotoxicity using both indirect (non-contact) and direct (surface adhesion) methods. As reported in Figure 2A, there were no differences in fluorescence (i.e., mitochondrial activity) at any time points for cells cultured alone or in the presence of the polymers (non-contact) without or with nanorods. This indicates that there are no toxic leachable components from the samples. When fibroblasts were cultured on the surfaces of composites, there was ample spreading and cytoskeletal organization (Figure 2B). When quantified, there were no statistical differences between groups without and with nanorods at any of the time points. For example, there were ~8900 and ~8600 cells on films without and with nanorods, respectively, after 48 hours. These preliminary cytotoxicity studies indicate that the polymer formulations are non-toxic at early time points and that the addition of nanorods does not increase toxicity.
Figure 2.

(A) Relative cellular viability (reported as the fluorescence from mitochondrial activity assay) of cells cultured in the presence of 15:85 A6:tBA networks without and with nanorods (mid concentration) and cultured for 1 (white), 2 (grey), and 3 (black) days and compared to control TCPS. There were no statistically significant differences between groups at each time point; (B) representative images of fibroblast adhesion (stained for actin fibers and nuclei) to the surface of 15:85 A6:tBA networks without (top) and with (bottom) nanorods (mid concentration) after 24 hours (bar = 30 um); (C) representative images of H&E stained histological sections of 15:85 A6:tBA networks without (top) and with (bottom) nanorods (mid concentration) 12 weeks after implantation subcutaneously. Anova with Tukey’s post-hoc test was used to determine significant differences among groups, with p < 0.05.
To further assess the cellular interactions, samples were implanted subcutaneously. At all time points, a thin fibrous capsule (~100 μm) is present around the implant with minimal associated chronic inflammation. There was no evidence of inflammation or necrosis within the adjacent subcutaneous fibroadipose tissue, skin adnexal structures or deep skeletal muscle (Figure 2C and Figure S3). This represents a typical host response to a non-toxic material and there were no differences observed between the samples without and with nanorods. Specifically, there were no differences in the cellular density, type of cells, or extent of fibrous encapsulation between groups. The lack of differences could be attributed to the low concentrations of nanorods and that the cells are interacting only with the surface of the material, rather than directly with the nanorods. As expected, there was degradation in the materials as the A6 crosslinks undergo hydrolysis and there were no significant differences in samples without and with nanorods; however, the process was slow (~5 wt% at 12 weeks) due to the hydrophobicity of the tBA, which comprises the majority of the samples. This may be advantageous since the transition temperatures could be altered with degradation. Depending on the application, slow degradation may be desirable, or other monomers may be used to expedite degradation by changing bulk hydrophilicity.
To examine the heating potential of the composites, slabs of the samples were held within a beam of infrared light (770 nm) for 60 seconds. Since the polymer and initiator absorb minimally (Figure S1C) at the excitation wavelength, the material is anticipated to be relatively transparent to this light. The samples all began at room temperature and the surface temperature change after light exposure for 60 seconds is reported in Figure 3A. Significant and rapid heating was observed in samples that contained nanorods and was dependent upon both the concentration of nanorods and the light intensity. For example, 15:85 A6:tBA networks with the high concentration of nanorods heated ~50°C when exposed to 0.3 W of infrared light. These values are probably lower than what is found at the top surface of the composites since the intensity is lowered through the sample. Essentially no change in temperature was observed for samples that did not contain nanorods. The transmittance of light (Figure S4) correlated to these findings, with increased temperatures corresponding to decreases in light transmittance through the sample due to absorption by the nanorods. The heating was also reversible with intermittent light exposure (Figure 3B), meaning the sample could be repeatedly heated and cooled with 5 minute intervals of light exposure and non-exposure. Again, no heating was observed in samples that did not contain nanorods. These results indicate precise control over the sample heating through changes in fabrication (i.e., nanorod concentration) and light intensity. Previous work on light-induced changes in polymer networks has focused on molecular switches and interpenetrating molecules that are photoresponsive,[11–14, 35] rather than inclusion of particles that heat with light exposure. An advantage to our approach is that the material can be repeatedly heated without changes in the molecular structure of the material and that it is applicable to many polymers.
Figure 3.
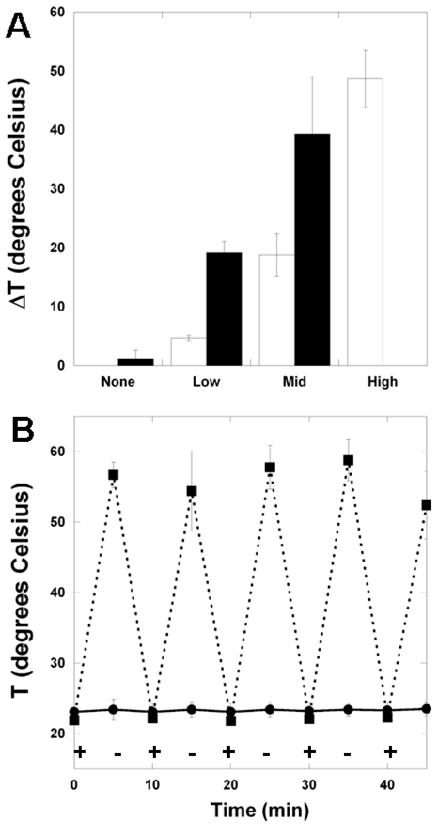
(A) Temperature change for 15:85 A6:tBA networks with various concentrations of nanorods and exposure to 0.3W (white) or 1.0W (black) infrared light (high light intensity and high nanorod concentration was not measured); the temperature changes were statistically different (p<0.05) between all nanorod concentrations and within each concentration between the different light intensities. (B) temperature change in 5 minute intervals of exposure to 0.3W infrared light (“+” denotes light turned on, “−” denotes light turned off) in the 15:85 A6:tBA network with (■) and without (●) nanorods (mid concentration). Anova with Tukey’s post-hoc test was used to determine significant differences among groups, with p < 0.05.
As described above, there are a range of applications where such technology would be useful. We aimed to illustrate the shape-memory effect, where a network is held in a temporary and glassy shape and then changes to a permanent shape when heated past the Tg. Composites were crosslinked into rectangular strips, heated above their Tg, coiled around a small glass cylinder, and cooled to below their Tg. When the room temperature coiled samples were exposed to infrared light, they heated and changed back to their permanent rectangular shape (Figure 4). The process took several minutes to complete, but only because the sample had to be positioned for the laser beam to pass through. When the beam hit areas of the sample, the shape change was nearly instantaneous until the change moved the sample outside of the beam’s path. This shape memory example illustrates the ability of embedded nanorods to heat a polymer sample through its Tg, as well as the spatial control that is possible. However, the shape-memory process will only be repeatable if the system is designed to revert back to the temporary shape when cooled.
Figure 4.

Images of shape transitions of polymers fabricated from 15:85 A6:tBA networks with nanorods (mid concentration) after exposure to 0.3W infrared light. Complete transition took several minutes since the sample would change shape and move from the path of the light, yet shape changes were nearly instantaneous when the light was directed on the sample.
In conclusion, we have developed a versatile method that utilizes infrared light to heat composites of gold nanorods and biodegradable polymers. The heating can change the network from a glassy to rubbery system and alter the polymer from a temporary to permanent shape. The thermal transitions of the networks can be tuned through the crosslinker density and the introduction of nanorods. However, the nanorods do not influence the crosslinking reaction behavior, the cytotoxicity, or the tissue response of the networks. This technology is useful for a wide range of applications, including where temperature transitions are desired in polymers and light provides an optimal means for heating (e.g., transdermal light exposure to heat implants).
Experimental
Polymer Synthesis and Characterization
PBAEs were synthesized by the conjugate addition of primary amines to diacrylates by mixing the liquid precursors and reacting overnight (90°C) with stirring. Specifically, macromer A6 was synthesized through the reaction of diethylene glycol diacrylate (A, Scientific Polymer Products, Inc.) and isobutylamine (6, Sigma) in a 1.2:1 molar ratio. The A6 molecular weight was confirmed to be ~1.3 kDa using 1H-NMR (Bruker Advance 360 MHz, Bruker, Billerica, MA).
Nanorod Fabrication, Characterization, and Encapsulation
Nanorods were grown through a mixture of cetyltrimethylammonium bromide (CTAB), HAuCl4•3H2O, silver nitrate, L-ascorbic acid, and a seed solution containing CTAB, sodium borohydride, and HAuCl4H2O. After mixing (2 hours), the nanorods were pegylated to alter their solubility using reaction with PEG-thiol (MW=5kDa)[31] and then transferred to dichloromethane. The nanorods were imaged on a transmission electron microscope (TEM, JEOL 2010F) at an accelerating voltage of 200 kV by placing a drop of the nanorod/dichloromethane solution on a holey carbon-coated grid (SPI Supplies). Upon imaging (Figure S1A), the nanorod dimensions were assessed using NIH Image Analysis. The absorption spectra of the nanorods were also collected (Beckman Coulter DU730 Life Science uv/vis spectrophotometer).
The photoinitiator 2,2-dimethoxy-2-phenyl acetophenone (DMPA, Sigma) was added to mixtures of the multi-functional macromer A6 and the mono-functional tBA at a final concentration of 0.5% (w/w). The nanorods were diluted in methylene chloride and added to the monomer/initiator solutions at a range of final concentrations. The solvent was removed in a vacuum desiccator overnight and all polymerizations were performed in bulk. The polymerization behavior was monitored using attenuated total internal reflectance – Fourier transform infrared (ATR-FTIR, Nicolet 6700, Thermo Electron) spectroscopy with a zinc selenide crystal collecting a spectrum every 17 seconds with a resolution of 3.86 cm−1 for 10 minutes. A drop of the macromer/initiator solution (with or without nanorods) was placed directly on the horizontal crystal, covered with glass, and exposed from above to ultraviolet light (~1.5 mW cm−2, 365 nm, Omnicure Series 1000, Exfo). The change in area of the double bond peak (~1630 cm−1, normalized to the carbonyl peak at ~1730 cm−1) was used to monitor double-bond conversion with light exposure.
Composite Characterization
Polymer films were made by injecting the monomer/initiator solution (without and with nanorods) between two glass slides with a 1 mm spacer and polymerizing with exposure to ultraviolet light (Blak Ray, ~10 mW cm−2, 10 minutes). Cross-sections (~50 nm, Richtheart Jung Ultramicrotome) of polymer slabs embedded in an epoxy resin (LR White Resin, SPI Supplies) were floated onto carbon-coated copper grids for TEM imaging (JEOL 2010, 80 kV). The viscoelastic behavior of the samples was determined using a Dynamic Mechanical Analyzer (Q800 TA Instruments). Rectangular strips (25 mm × 5 mm × 1 mm) of polymer were cut from polymer slabs and tested in a controlled strain mode at 1 Hz, an amplitude of 10 μm, and a heating rate of 3°C min−1 from −20°C to 55°C. The Tg is reported as the peak of the tan δ (the storage modulus over the loss modulus) curve.
For indirect cytotoxicity assessment, polymer discs (5mm diameter, 1mm thick) were prepared as above, sterilized with exposure to ethanol and a germicidal lamp for 30 minutes, and placed in transwell inserts above cultures of 3T3-fibroblasts (ATCC, seeded 24 hours previously at 5000 cells cm−2). A standard Alamar Blue assay (Invitrogen) following manufacturer’s protocols was used after 1, 2, and 3 days to assess changes in cell viability through mitochondrial activity. For direct cellular interaction studies, films were prepared by dropping the monomer/initiator solution (with and without nanorods) onto a coverslip, photocrosslinking in an inert environment, sterilizing with exposure to a germicidal lamp, and seeding with 3T3-fibroblasts (5000 cells cm−2). At specified time points, the cells were fixed and processed using standard protocols for staining with actin (TRITC-conjugated phalloidin, 0.66 μg ml−1 in PBS, Sigma) or nuclei (DAPI, 1:2500, Sigma). When quantified (counting cells in 10 images per group for each time point),
For in vivo assessment, polymer disks (1 mm thick, 5 mm diameter) were prepared as described above (without and with nanorods), weighed, and sterilized with ethanol and germicidal lamp exposure for 30 minutes. Animals were cared for according to a protocol approved by the University of Pennsylvania Institute for Animal Care and Use Committee. The discs (6 discs per animal) were implanted subcutaneously into the dorsal pocket of male Sprague-Dawley rats. The animals were sacrificed at various time points (4, 8, and 12 weeks) and the polymer samples (n=3) and surrounding tissue were collected and fixed with 10% formalin for 24 hours, paraffin embedded, and processed for standard hematoxylin and eosin stains. Additional samples (n=3) were removed to monitor in vivo degradation behavior. All tissue was excised from the sample prior to lyophilization to obtain the sample dry weight.
Composite Light Exposure
Composites were exposed to light with a Coherent Chameleon Ti:sapphire laser, mode-locked at 80 MHz, running at 770nm with up to 3.3W output power and 140fs pulse width. The beam was directed with gold-coated mirrors to the sample and expanded to about 7mm diameter at the sample position. Power levels used were 0.3 – 1.0W and selected by a combination of waveplate and polarizer. The sample temperature was measured at the back surface (1 mm depth) with a probe (Microtherma 2T thermometer and MT-D thermocouple probe, Thermoworks) immediately upon removal of the light source.
Supplementary Material
Footnotes
This work was supported by a Fellowship in Science and Engineering from the David and Lucille Packard Foundation and NIH grant P41RR001348. We gratefully acknowledge the assistance of Mike Hore for ultramicrotome processing of the composites, Professor Karen Winey for use of her lab’s ultramicrotome, and Dr. Lolita Rotkina and Dr. Doug Yates at the Penn Regional Nanotech Facility and Christopher Rodd for assistance in TEM imaging.
Supporting Information is available online from Wiley InterScience or from the author.
References
- 1.Behl M, Lendlein A. Soft Matter. 2007;3:58–67. doi: 10.1039/b610611k. [DOI] [PubMed] [Google Scholar]
- 2.Lendlein A, Kelch S. Angew Chem Int Edit. 2002;41:2034–2057. [PubMed] [Google Scholar]
- 3.Gall K, Kreiner P, Turner D, Hulse M. J Microelectromech S. 2004;13:472–483. [Google Scholar]
- 4.Yakacki CM, Shandas R, Safranski D, Ortega AM, Sassaman K, Gall K. Adv Funct Mater. 2008;18:2428–2435. doi: 10.1002/adfm.200701049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida M, Langer R, Lendlein A, Lahann J. Polym Rev. 2006;46:347–375. [Google Scholar]
- 6.Lendlein A, Kelch S. Clin Hemorheol Micro. 2005;32:105–116. [PubMed] [Google Scholar]
- 7.Lendlein A, Langer R. Science. 2002;296:1673–1676. doi: 10.1126/science.1066102. [DOI] [PubMed] [Google Scholar]
- 8.Bikram M, Gobin AM, Whitmire RE, West JL. J Control Release. 2007;123:219–227. doi: 10.1016/j.jconrel.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Sershen SR, Westcott SL, Halas NJ, West JL. J Biomed Mater Res. 2000;51:293–298. doi: 10.1002/1097-4636(20000905)51:3<293::aid-jbm1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 10.Mohr R, Kratz K, Weigel T, Lucka-Gabor M, Moneke M, Lendlein A. Proc Natl Acad Sci U S A. 2006;103:3540–3545. doi: 10.1073/pnas.0600079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irie M. Adv Polym Sci. 1990;94:27–67. [Google Scholar]
- 12.Lendlein A, Jiang HY, Junger O, Langer R. Nature. 2005;434:879–882. doi: 10.1038/nature03496. [DOI] [PubMed] [Google Scholar]
- 13.Natansohn A, Rochon P. Chem Rev. 2002;102:4139–4175. doi: 10.1021/cr970155y. [DOI] [PubMed] [Google Scholar]
- 14.Yu YL, Nakano M, Ikeda T. Pure Appl Chem. 2004;76:1467–1477. [Google Scholar]
- 15.Weissleder R. Nat Biotechnol. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 16.Dickerson EB, Dreaden EC, Huang XH, El-Sayed IH, Chu HH, Pushpanketh S, McDonald JF, El-Sayed MA. Cancer Lett. 2008;269:57–66. doi: 10.1016/j.canlet.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. P Natl Acad Sci USA. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang XH, El-Sayed IH, Qian W, El-Sayed MA. J Am Chem Soc. 2006;128:2115–2120. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 19.Huang XH, Jain PK, El-Sayed IH, El-Sayed MA. Nanomedicine-Uk. 2007;2:681–693. doi: 10.2217/17435889.2.5.681. [DOI] [PubMed] [Google Scholar]
- 20.Loo C, Lowery A, Halas N, West J, Drezek R. Nano Lett. 2005;5:709–711. doi: 10.1021/nl050127s. [DOI] [PubMed] [Google Scholar]
- 21.O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Cancer Lett. 2004;209:171–176. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Gall K, Yakacki CM, Liu YP, Shandas R, Willett N, Anseth KS. J Biomed Mater Res A. 2005;73A:339–348. doi: 10.1002/jbm.a.30296. [DOI] [PubMed] [Google Scholar]
- 23.Ortega AM, Kasprzak SE, Yakacki CM, Diani J, Greenberg AR, Gall K. J Appl Polym Sci. 2008;110:1559–1572. [Google Scholar]
- 24.Alteheld A, Feng Y, Kelch S, Lendlein A. Angew Chem Int Ed Engl. 2005;44:1188–1192. doi: 10.1002/anie.200461360. [DOI] [PubMed] [Google Scholar]
- 25.Kelch S, Steuer S, Schmidt AM, Lendlein A. Biomacromolecules. 2007;8:1018–1027. doi: 10.1021/bm0610370. [DOI] [PubMed] [Google Scholar]
- 26.Anderson DG, Tweedie CA, Hossain N, Navarro SM, Brey DM, Van Vliet KJ, Langer R, Burdick JA. Adv Mater. 2006;18:2614-+. [Google Scholar]
- 27.Brey DM, Erickson I, Burdick JA. J Biomed Mater Res A. 2008;85A:731–741. doi: 10.1002/jbm.a.31494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brey DM, Ifkovits JL, Mozia RI, Katz JS, Burdick JA. Acta Biomater. 2008;4:207–217. doi: 10.1016/j.actbio.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan AR, Ifkovits JL, Baker BM, Brey DM, Mauck RL, Burdick JA. J Biomed Mater Res A. 2008;87A:1034–1043. doi: 10.1002/jbm.a.31853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sau TK, Murphy CJ. Langmuir. 2004;20:6414–6420. doi: 10.1021/la049463z. [DOI] [PubMed] [Google Scholar]
- 31.Liao HW, Hafner JH. Chem Mater. 2005;17:4636–4641. [Google Scholar]
- 32.Safranski DL, Gall K. Polymer. 2008;49:4446–4455. [Google Scholar]
- 33.Lee KJ, Lee DK, Kim YW, Choe WS, Kim JH. J Polym Sci Pol Phys. 2007;45:2232–2238. [Google Scholar]
- 34.Rittigstein P, Torkelson JM. J Polym Sci Pol Phys. 2006;44:2935–2943. [Google Scholar]
- 35.Small W, Wilson TS, Benett WJ, Loge JM, Maitland DJ. Opt Express. 2005;13:8204–8213. doi: 10.1364/opex.13.008204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.