

Abstract
The genomic sequence of Mycoplasma pneumoniae establish this cell-wall-less prokaryote as among the smallest known microorganisms capable of self-replication. However, this genomic simplicity and corresponding biosynthetic austerity are sharply contrasted by the complex terminal structure found in this species. This tip structure (attachment organelle) directs colonization of the human respiratory mucosa, leading to bronchitis and atypical pneumonia. Furthermore, formation of a second tip structure appears to precede cell division, implying temporal regulation. However, the organization, regulation, and assembly of the attachment organelle in M. pneumoniae are poorly understood, and no counterparts have been identified among the walled bacteria. M. pneumoniae possesses a cytoskeleton-like structure required to localize adhesin proteins to the attachment organelle. The cytadherence-associated proteins HMW1, HMW2, and HMW3 are components of the mycoplasma cytoskeleton, with HMW1 localizing strictly along the filamentous extensions from the cell body and HMW3 being a key structural element of the terminal organelle. Disruptions in hmw2 result in the loss of HMW1 and HMW3. However, the hmw1 and hmw3 genes were transcribed and translated at wild-type levels in hmw2 mutants. HMW1 and HMW3 were relatively stable in the wild-type background over 8 h but disappeared in the mutants over this time period. Evaluation of recombinant HMW1 levels in mycoplasma mutants suggested a requirement for the C-terminal domain of HMW1 for turnover. Finally, an apparent defect in the processing of the precursor for the adhesin protein P1 was noted in the HMW− mutants.
Members of the genus Mycoplasma are distinguished by their lack of a cell wall or cell wall precursors, the incorporation of cholesterol in their membranes, the use of UGA as a codon for tryptophan rather than termination, and their extremely small genomes (≥580 kbp). In addition, several mycoplasma species possess a complex terminal structure that directs the colonization of vertebrate host cells and is thought to function in cell division and gliding motility. The presence of this complex organelle stands in marked contrast to the perception of mycoplasmas as otherwise simple wall-less prokaryotes (1). In Mycoplasma pneumoniae, a major cause of primary atypical community-acquired pneumonia, the terminal (attachment) organelle is seen by transmission electron microscopy of thin sections as a membrane-bound extension of the cell body, with an electron-dense core associated with a cytoskeleton-like network of mycoplasma proteins (2). The organization, regulation, and assembly of the terminal organelle of M. pneumoniae is poorly understood, largely due to the stringent nutritional requirements and poor growth yields of mycoplasmas, as well as a general lack of genetic tools for use in these prokaryotes. Finally, no counterparts to the terminal organelle have been identified among the walled bacteria.
Adherence of M. pneumoniae to host respiratory epithelium (cytadherence) is pivotal to successful colonization and requires a functional attachment organelle. The adhesin proteins P1 (3, 4) and P30 (5) are densely clustered at the attachment organelle of wild-type M. pneumoniae cells. Maintenance of this polarity requires the interaction of P1 with the mycoplasma cytoskeleton, including proteins HMW1–HMW3. Spontaneously arising mutants that fail to cytadhere have been classified according to changes in their protein profiles (6). The levels of HMW1 and HMW3 are greatly reduced and HMW2 is completely absent in class I mutants (Table 1). Loss of the HMW proteins renders M. pneumoniae unable to localize the adhesin P1 to the attachment organelle (11). Significantly, HMW3 is found exclusively in the terminal button of the attachment organelle, where it contributes to the architecture of the tip structure and may anchor adhesin proteins in the mycoplasma membrane (12). HMW1 likewise has an unusual subcellular distribution, localizing along the leading and trailing filamentous extensions of the mycoplasma cell (13). Although the subcellular location of HMW2 has not been determined, like HMW1 and HMW3, it partitions in the Triton X-100-insoluble cytoskeleton fraction of M. pneumoniae (13). Furthermore, the coiled-coil structure predicted to dominate HMW2 (7) is characteristic of the filamentous domains of cytoskeletal proteins and, therefore, consistent with a scaffolding role.
Table 1.
M. pneumoniae strains used in this study (1)
| Strain | Relevant cytadherence proteins
|
Mutation | Ref(s). | |||
|---|---|---|---|---|---|---|
| HMW1 | HMW2 | HMW3 | P30 | |||
| M129 (parent strain) | +++* | +++ | +++ | +++ | None | 9 |
| Class I mutants (2, 8 and 10) | +/−† | −‡ | +/− | +++ | Point mutation tentatively identified in hmw2 | 6 |
| M6 | − | +++ | +++ | Δ§ | Frameshift mutation in hmw1; Δ at 3′ end of p30 | 10 |
| crl | +/− | − | +/− | +++ | Tn4001 insertion in hmw2 | 7, 8 |
Protein present at wild-type level.
Protein present at greatly reduced level.
Protein absent.
Partial deletion.
Duplication of the tip structure of M. pneumoniae is thought to precede cell division (14), suggesting that assembly of organelle components, including the HMW proteins, is temporally regulated. Regulation is poorly understood in the mycoplasmas, and a particularly striking feature to emerge from the analysis of the sequenced genomes of M. pneumoniae (15) and Mycoplasma genitalium (16) is a general lack of predicted transcriptional regulatory mechanisms. The genes for HMW1 and HMW3 are closely linked to that of P30 (hmw gene cluster, Fig. 1), and the gene for HMW2 is part of the cytadherence regulatory locus (crl), mapping approximately 160 kbp from the hmw gene cluster (7, 8). Transposon insertion in the gene for HMW2 (crl mutant) results in loss of HMW1 and HMW3 and a noncytadhering phenotype indistinguishable from the class I mutants (ref. 7 and Table 1). Reacquisition of HMW1–HMW3 and a cytadherence-positive phenotype accompanies excision of Tn4001 from crl mutants (7, 8). Furthermore, preliminary data identify a mutation in the gene for HMW2 in a class I mutant (M. Fisseha, H. Göehlmann, R. Herrmann, K. Birkhead, and D.C.K., unpublished results). Thus, whether by point mutation or transposon insertion, the loss of HMW2 leads to the disappearance of HMW1 and HMW3. The current study establishes that the loss of HMW1 and HMW3 occurs post-translationally by a likely proteolytic mechanism that targets the C terminus of these proteins. Amino acid sequence similarities were noted with the C terminus of the chemoreceptor McpA of Caulobacter crescentus (19), for which spatial and temporal control by proteolysis has been demonstrated (20), raising the possibility of a role for proteolysis in regulating components of the attachment organelle in M. pneumoniae. Inhibition of P1 processing from precursor to mature adhesin also accompanied the turnover of HMW1 and HMW3 in the class I mutant. As this mutant fails to cluster P1 at the attachment organelle, the delayed processing of P1 may reflect a defect in the trafficking of this membrane protein to that site.
Figure 1.
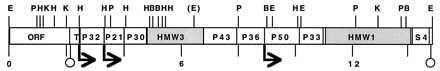
Organization of the hmw gene cluster (17). The genes encoding P30, HMW3, HMW1, and ribosomal protein S4 are indicated. The remaining ORFs have no clear identity to genes of known function and are identified by size (kDa) of their deduced products. Transcriptional start sites (P.L.P. and D.C.K., unpublished results) are indicated by arrows, and predicted terminators are designated by circles. B, BamHI; E, EcoRI; H, HindIII; K, KpnI; P, PstI. The (E) designates an EcoRI site engineered in the original cloning of hmw3 by ligation of a linker to sheared mycoplasma DNA (18).
MATERIALS AND METHODS
Bacterial Strains and Culture Conditions.
Mycoplasma strains used in this study (Table 1) included wild-type M. pneumoniae parent strain M129 [broth passage 17 (9)]; the noncytadhering class I mutant isolates 2, 8, and 10 (6); crl mutants A-3 and D-3 (8); and the noncytadhering mutant M6 (10). Mycoplasmas were cultured at 37°C in Hayflick medium (21) until midlogarithmic phase and harvested as described (13). Mycoplasmas were spread-plated on PPLO agar (22), incubated at 37°C for 6–9 days, and visualized by hemolytic plaques for isolation of individual colonies or enumeration of colony-forming units (8). Escherichia coli Sure strain (Stratagene) was grown for plasmid production in Luria broth and prepared as competent cells for transformation by standard techniques (23). Plasmid DNA was purified by using pZ523 columns (5 Prime–3 Prime, Inc.) according to the manufacturer’s protocol.
RNA Slot Blot Analysis.
All solutions and plasticware were rendered RNase-free by diethyl pyrocarbonate treatment (23). RNA was extracted from midlogarithmic-phase cultures of M. pneumoniae. Immediately after harvesting by centrifugation, cell pellets were incubated for 10 min at 68°C in TRI Reagent (Molecular Research Center, Cincinnati; 1 ml per 3.6 ml of cell suspension). Samples were cooled to room temperature, and the organic portion of the TRI reagent was removed by the addition of chloroform to a final concentration of 18%. After centrifugation at 12,000 × g for 15 min at 4°C, we collected the aqueous phase, being particularly careful to avoid the organic phase. Nucleic acids were precipitated from the aqueous phase with isopropanol, resuspended, and treated with RNase-free DNase (3,000 units, Promega) in 0.25 ml of 10 mM sodium acetate/0.05 mM NaCl/1 mM MgCl2, pH 5.0, containing 39 units of RNasin (Promega) for 30 min at 37°C. RNA samples were extracted with phenol, phenol/chloroform (twice), and chloroform and precipitated with ethanol. RNA was quantitated spectrophotometrically and assessed visually after agarose gel electrophoresis and ethidium bromide staining. The latter proved particularly critical to the reproducibility of experimental results with respect to mRNA quality. For slot blot analysis, equal amounts of RNA were diluted in an RNase-free solution containing formamide (17%), formaldehyde (2%), and 10× SSC (1× SSC is 0.15 M NaCl/0.015 M sodium citrate, pH 7.0); heated to 68°C for 15 min; and collected in duplicate on nitrocellulose by vacuum (Minifold II; Schleicher & Schuell). Blots were probed with double-stranded DNA probes (0.2 μg of DNA) cloned from the wild-type M. pneumoniae genes for P1, HMW1, HMW3, or rRNA. Probes were tagged by random-primed labeling (Prob-eze; 5 Prime–3 Prime, Inc.) with [α-32P]dATP. Blots were hybridized overnight at 68°C in 6× SSC/0.1% SDS/2× Denhardt’s reagent, washed for 20 min in 1× SSC/0.1% SDS at 25°C and for three 20-min periods in 0.1× SSC/0.1% SDS at 72°C, air-dried, and exposed to film overnight.
Pulse–Chase Analysis of HMW1 and HMW3 Synthesis.
Mycoplasmas cultured to midlogarithmic phase in Hayflick medium were washed three times with PBS (0.01 M sodium phosphate at pH 7.2) and suspended in HBSS [Hanks’ balanced salts solution (Sigma)] supplemented with 0.004% phenol red and 25 mM Hepes (pH 7.4), containing 10% agamma horse serum previously dialyzed against Tris-buffered saline. Cells were pulse-labeled for 5 or 30 min at 37°C with [35S]methionine (>1,000 Ci/mmol; 1 Ci = 37 GBq; Amersham) at 67 μCi/ml, then centrifuged (16,000 × g) for 10 min, and washed once with HBSS containing 1 mM methoinine. Cells were suspended in Hayflick medium and incubated at 37°C for 0, 1, 4, or 8 h, then collected by centrifugation, and solubilized in TDSET (1% Triton X-100/0.2% sodium deoxycholate/0.1% SDS/10 mM tetrasodium EDTA/10 mM Tris⋅HCl, pH 7.8) containing 1 mM phenylmethylsulfonyl fluoride. Samples were incubated at 37°C for 10 min and centrifuged at 128,000 × g for 30 min at 4°C. The resulting supernatants were either examined directly by SDS/PAGE or evaluated by radioimmunoprecipitation as described in detail elsewhere (24). The preparation and characterization of the antisera to HMW1, HMW3, and P1 used in this study have been reported (11–13).
Analysis of M. pneumoniae Transformants.
Recombinant transposons containing the wild-type gene for HMW1 cloned into transposon Tn4001 have been characterized in detail (25). Wild-type and mutant M. pneumoniae were transformed with recombinant transposons by electroporation. Transformants were selected on PPLO agar plates containing 18 μg of gentamicin per ml, picked with sterile Pasteur pipettes, and filter-cloned three times (26). Transformant protein profiles were evaluated by discontinuous SDS/PAGE using 3% polyacrylamide stacking gels and 4.5% polyacrylamide separating gels. Proteins were visualized by silver staining or evaluated by Western immunoblotting using HMW1-specific antibodies, as detailed (13).
RESULTS
Slot Blot Hybridization Analysis of hmw1 and hmw3 Expression.
Transcript levels for hmw1 and hmw3 were indistinguishable between wild-type M. pneumoniae and several class I (Fig. 2) or crl mutants (data not shown) by slot blot hybridization. The hybridization signal intensities with each hmw probe were commensurate with mRNA concentration and consistent between duplicate samples. To control for total mRNA amounts, samples were also probed with a DNA fragment from the gene encoding the cytadhesin P1. This protein is produced at similar levels in wild-type and class I or crl mutant M. pneumoniae (6, 8), and hybridization signals with this probe were likewise equivalent in the mutant and wild-type samples. In some studies, mycoplasma RNA was also hybridized with a rRNA-specific DNA probe as an alternative internal control. On the basis of RNA slot blot hybridizations, as well as detailed analysis of mRNA from wild-type and mutant mycoplasmas by primer extension (P.L.P. and D.C.K., unpublished results), we concluded that HMW1 and HMW3 are controlled post-transcriptionally in the class I and crl mutants.
Figure 2.

RNA slot blot analysis of hmw1 and hmw3 transcription in wild-type and class I mutant M. pneumoniae. RNA samples (1-μg, 0.2-μg, or 0.04-μg quantities in duplicate) were probed with internal restriction fragments from the genes for HMW1, HMW3, or P1. Mycoplasma strains tested were the class I mutant isolates 2, 8, and 10 (6) and the parent wild-type strain. The probe for hmw1 was a 900-bp PstI–KpnI fragment (Fig. 1), and the probe for hmw3 was a 1.1-kbp HindIII–EcoRI fragment [Fig. 1, indicated by (E)]. The probe for p1 was a 412-bp HindIII–KpnI fragment (27).
Evaluation of HMW1 and HMW3 Synthesis and Turnover.
The HMW proteins are readily identified by their characteristic electrophoretic mobility in profiles of wild-type M. pneumoniae. All three are absent from profiles of class I (6) and crl mutants (8) by Coomassie blue staining, but HMW1 and HMW3 are detected at low levels in these mutants by more sensitive methods, an observation previously attributed to revertants in the mutant population (28). To examine the synthesis and fate of HMW1 and HMW3 directly, mycoplasmas were pulse-labeled with [35S]methoinine and then examined immediately or at specific times over an 8-h chase period, by SDS/PAGE and autoradiography. Significantly, newly synthesized HMW1 and HMW3 were present at wild-type levels in the mutant profiles immediately after pulse labeling (Fig. 3A, 0 h). The intensities of HMW1 and HMW3 in wild-type profiles remained relatively stable over an 8-h chase. In contrast, HMW1 and HMW3 gradually diminished over the same chase period in the mutants (Fig. 3A). No HMW2 was detected at any time point in the mutant profiles, consistent with transposon insertion or frameshift mutation in hmw2 in crl or class I mutants, respectively.
Figure 3.
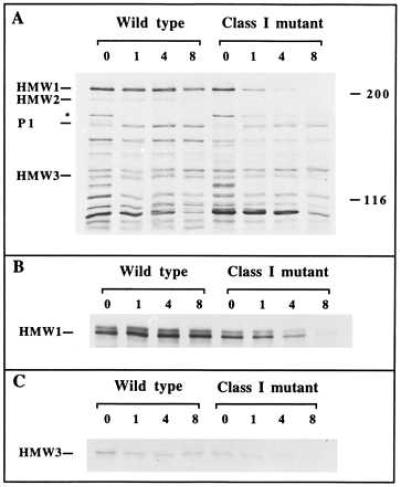
Pulse–chase analysis of HMW1, HMW2, and HMW3 synthesis and turnover in wild-type and class I mutant M. pneumoniae. (A) Total protein profiles at chase times of 0 h, 1 h, 4 h, and 8 h. Wild-type and mutant samples are indicated, as are the bands corresponding to HMW1, HMW2, HMW3, and P1. The asterisk corresponds to the putative P1 precursor. Protein size standards are given in kDa. (B and C) Radioimmunoprecipitation analysis of HMW1 and HMW3, respectively, after pulse–chase labeling.
The identity of HMW1, HMW2, and HMW3 in the mycoplasma profiles in Fig. 3A was confirmed by radioimmunoprecipitation with specific antisera. As in the total-protein profiles, HMW1 and HMW3 (Fig. 3 B and C, respectively) were detected in the mutants immediately after the pulse but disappeared over the 8-h chase. As expected, HMW2 was absent at all time points in the mutant profiles (data not shown). In some experiments discreet lower molecular weight bands were noted in the 0-h and 1-h times with radioimmunoprecipitation with the HMW1-specific serum (data not shown). These were not seen in the later times and may reflect proteolytically truncated derivatives of HMW1. Furthermore, no loss of HMW1 and HMW3 occurred in the mutant profiles over the 8-h chase period when incubated on ice rather than at 37°C, consistent with proteolytic turnover (data not shown).
The cytadhesin P1 is thought to be synthesized as a precursor, from which processing removes a large leader peptide (6.5 kDa) to yield the mature protein (27). Pulse–chase studies combined with radioimmunoprecipitation confirmed the processing of pre-P1 (data not shown). Significantly, the processing of pre-P1 to its mature form occurred more slowly in the class I mutant than in wild-type M. pneumoniae. This is seen in Fig. 3A in the longer half-life of pre-P1 and the delayed accumulation of mature P1 in the class I mutant.
Synthesis of Recombinant HMW1 in Wild-Type and Mutant M. pneumoniae.
We recently described the delivery and expression of the cloned gene for HMW1 into wild-type and class I mutant M. pneumoniae through the use of a recombinant transposon (25). This approach resulted in both a resident and recombinant copy of the hmw1 allele in mycoplasma transformants. A recombinant hmw1 gene slightly truncated at the 3′ end was included in those studies to distinguish between the products of the resident and recombinant hmw1 alleles. Surprisingly, full-length recombinant HMW1 was not produced at wild-type levels in the class I mutant, but the truncated HMW1 (HMW1′) was. It was not clear at the time whether the failure to produce full-length recombinant HMW1 at wild-type levels in the class I mutant was a function of the mycoplasma background or the recombinant transposon construction. Recently, an M. pneumoniae mutant was described (10) with a frameshift mutation in the hmw1 gene and a deletion in the 3′ end of the p30 gene (mutant M6). This mutant provided an alternative background (one which was HMW2+ and HMW3+), in which to evaluate production of recombinant HMW1. We reasoned that if failure to synthesize full-length recombinant HMW1 at wild-type levels in the class I mutant were due to the class I mutant background and not the recombinant transposon construction, then HMW1 should accumulate at wild-type levels in the M6 transformants. Because synthesis of HMW1′ is influenced by genes flanking the recombinant transposon insertion site, considerable variability is seen among individual transformants of each type by this technique (25). Therefore, for this study, 25 transformants were isolated for each construction in each mycoplasma background. Representative isolates are shown in Fig. 4A, where full-length recombinant HMW1 was clearly produced at wild-type levels in M6 but not class I transformants. In contrast, the levels of HMW1′ were similar in wild-type and both mutant backgrounds. These results are summarized in Fig. 4B and strongly suggest that the loss of HMW1 in the class I mutant requires amino acid sequences present in the C-terminal domain of HMW1.
Figure 4.

(A) Western immunoblotting analysis of representative wild-type and class I or M6 mutant M. pneumoniae transformed with recombinant Tn4001 containing either the full-length or truncated hmw1 gene (see B). Mycoplasma protein profiles were probed with anti-HMW1 serum (1:2,000 dilution). Lanes: a, wild-type M. pneumoniae; b, class I mutant 2; c, M6 mutant; d–f and g–i, M6 and class I transformants, respectively, containing full-length hmw1; j–l, m–o, and p–r, wild type, class I, or M6 transformants, respectively, containing truncated hmw1. HMW1 and HMW1′ are indicated on the right, and the protein size standard is shown on the left. In some transformants (lanes o and q), HMW1′ migrated larger than expected, probably the result of translational fusion with mycoplasma genomic sequence flanking the transposon. (B) Summary of the production of full-length and truncated recombinant HMW1 in wild-type and class I or M6 mutant M. pneumoniae. The restriction map indicates fragments subcloned into Tn4001 for transformation into M. pneumoniae. E, EcoRI; B, BamHI; ++, protein present at wild-type levels; −, protein present at mutant levels; NT, not tested. The deduced amino acid sequence of HMW1 (17) from the BamHI site to the 3′ end of the gene, with the regions emphasized in 4C underlined. Single-letter amino acid designations are used for space considerations. (C) Deduced amino acid sequences of the Ser-Ser motifs at the C terminus of HMW1 and HMW3, the cleavage site (inverted arrowhead) of the M. pneumoniae ORF6 product to yield proteins B and C (29), and the C terminus of C. crescentus McpA (19), aligned by the Ser-Ser motifs (vertical box). The motif Pro-Xaa-Lys/Arg-Xaa(1–5)-Ser-Ser is boxed horizontally.
HMW1 and HMW3 are structurally similar, with an internal domain that is dominated by repeating patterns rich in proline and acidic residues (17, 30). Analysis of the C terminus of HMW1 revealed a paired serine motif repeated four times over a 100-amino acid stretch (Fig. 4 B and C). Furthermore, this motif was repeated four times in the C terminus of HMW3 (Fig. 4C). Significantly, the paired serine motif is also found just downstream of a known M. pneumoniae protein processing site. The product of ORF6 of the P1 operon of M. pneumoniae is a 130-kDa polypeptide that is subsequently cleaved to yield proteins B and C (29). Like HMW3, these proteins are both localized to the terminal structure (31, 32), where they are required to anchor the adhesin P1 to the attachment organelle (11). Further examination revealed that the sequence Pro-Xaa-Lys/Arg-Xaa(0–5)-Ser-Ser was shared between the processing site for B and C and the C-terminal domains of HMW1 and HMW3 (Fig. 4C). In addition, the C terminus of the deduced HMW1 and HMW3 homologs in M. genitalium (16) contains similar motifs (data not shown). Thus, the target site for protease recognition may lie within one or more of these repeated motifs in HMW1 and HMW3.
DISCUSSION
The loss of HMW1–HMW3 in Class I mutants is associated with the inability to cluster the adhesin P1 at the attachment organelle and a cytadherence-negative phenotype. The hmw1 and hmw3 genes are closely linked (17) but separated from the hmw2 gene by approximately 160 kbp (15). Although the loss of HMW1 and HMW3 has been traced to frameshift mutation or transposon insertion in hmw2 (M. Fisseha, H. Göehlmann, R. Herrmann, K. Birkhead, and D.C.K., unpublished results and ref. 7, respectively), it was not clear how the loss of HMW2 and HMW1 and HMW3 were related. In the current study, analysis by RNA slot blot hybridization revealed no difference in transcript levels for hmw1 and hmw3 between wild-type and class I or crl mutant M. pneumoniae, establishing that control of HMW1 and HMW3 levels occurred post-transcriptionally. Furthermore, pulse–chase experiments demonstrated that HMW1 and HMW3 are actually synthesized at wild-type levels in the mutants but are subsequently turned over.
The turnover rates for HMW1 and HMW3 differed (half-lives of 1.7 h and 8.7 h, respectively), and the slower turnover of HMW3 may account for conflicting reports regarding its loss in crl insertion mutants (8, 33). HMW3 appears to occur in a polymerized form in the tip of the attachment organelle (12), and compartmentalization, polymerization, or both, might contribute to the greater stability of HMW3 in these mutants. The instability of HMW1 and HMW3 in the class I mutant probably accounts for our failure to restore each protein to wild-type levels by transforming with the wild-type recombinant hmw1 or hmw3 allele (ref. 25 and C. Romero-Arroyo and D.C.K., unpublished data). In contrast, full-length HMW1 could clearly be restored in the M6 mutant by the same approach (Fig. 4), underscoring the unusual, but as yet, incompletely defined nature of the class I mutant background. Furthermore, the stability of the truncated recombinant HMW1 in the class I mutant implicates a C-terminal domain of the protein in its differential turnover. Loss of HMW1 and HMW3 probably occurs via a proteolytic cascade where the first cleavage may be the most important, channeling the protein to complete proteolysis. Without that initial cleavage, the truncated recombinant HMW1 remains stable at wild-type levels. The presence of shared motifs at the C terminus of HMW1 and HMW3 raises the possibility that the C-terminal domain of HMW3 may likewise be important in proteolytic targeting.
At least three scenarios could account for proteolytic removal of HMW1 and HMW3 (34). Housekeeping proteases remove abnormally folded or otherwise defective proteins. There is no indication that HMW1 and HMW3 are defective in the class I mutant, however. For example, HMW1 is a phosphoprotein (24), and newly synthesized HMW1 appears to be phosphorylated normally in the class I mutant (data not shown). Although a homolog of the Lon protease of Bacillus was identified in the M. pneumoniae genome (15), turnover of abnormal proteins generally occurs rapidly, in contrast to the slow turnover noted herein for HMW1 and HMW3.
Failure to interact with chaperones or other proteins in higher-ordered structures could also account for loss of HMW1 and HMW3 in the mutants. This seems particularly relevant considering their assembly into the Triton X-100-insoluble mycoplasma cytoskeleton (13). Loss of an intermediate in the ordered assembly could prevent incorporation of HMW1 and HMW3, rendering each now susceptible to proteolysis. HMW2 has emerged as a likely intermediate in this scenario, as recent studies have established a pivotal role for HMW2 in the crl/class I mutant phenotype (7, 8). Nevertheless, newly synthesized HMW1 and HMW3 both partitioned in the Triton X-100-insoluble fraction of the class I mutant (data not shown), suggesting their incorporation into the mycoplasma cytoskeleton.
Proteolysis is also increasingly recognized for its role in regulating developmental circuits. The distinct subcellular distribution of HMW1 and HMW3, as well as their likely proteolytic regulation in the class I and crl mutants, are reminiscient of the McpA chemoreceptor protein of C. crescentus. McpA is found at the flagellated pole of the Caulobacter swarmer cell and is degraded during differentiation to the stalked cell type prior to cell division (20). In M. pneumoniae the formation of a second tip structure is thought to precede cell division (14), yet the nucleation or regulatory events in this process are not known. In Caulobacter, the degradation of McpA requires a defined C-terminal motif, as a 14-amino acid deletion at the C terminus results in the stable maintenance of McpA during differentiation (19). The C terminus of McpA exhibits striking similarity to domains in the C terminus of HMW1 and HMW3 (Fig. 4C). Twin serine residues lie just upstream of the required 14-amino acid C-terminal domain of McpA and are preceded by the Pro-Xaa-Lys/Arg motif (19) described here for HMW1 and HMW3. Although these sequence similarities are compelling, it is not known whether proteolysis actually represents one level of control in wild-type M. pneumoniae or whether shared amino acid motifs may merely reflect common protease recognition sites without extending to regulatory circuits. Verification of a role in protease recognition and evaluation of the contribution that proteolysis may play in controlling assembly of the attachment organelle will require identification and detailed analysis of the protease(s) involved. The genes for a putative zinc protease (H03_orf 193o) and a Lon homolog (F10_orf 795) have been identified from the M. pneumoniae genome (15).
The HMW proteins are required collectively to localize the cytadhesin P1 to the attachment organelle (11), but the sequence of events from its synthesis to its localization to the tip structure are not known. P1 is predicted to be synthesized with a 59-amino acid leader that is cleaved to yield the mature protein (27). Pulse–chase labeling and radioimmunoprecipitation confirmed the synthesis of a P1 precursor (data not shown), the processing of which is partially inhibited in the class I mutant (Fig. 2). A similar phenotype was reported in a cytadherence mutant of M. genitalium (35). It is not clear whether this defect is a direct consequence of loss of HMW1–HMW3 or whether the stability of HMW1 and HMW3 and the processing of P1 might share a required function, such as a chaperone to restrict folding until assembly begins. This unexpected finding might help elucidate the function of one or more of the HMW proteins in P1 localization to the tip, as well as the complex interactions that lead to assembly of the attachment organelle.
Acknowledgments
We thank Karen Birkhead and Kyungok Lee for technical help and Frank Gherardini for helpful suggestions. This work was supported by Public Health Service Research Grants AI23362 and AI33396 from the National Institute of Allergy and Infectious Diseases to D.C.K.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Krause D C. Mol Microbiol. 1996;20:247–253. doi: 10.1111/j.1365-2958.1996.tb02613.x. [DOI] [PubMed] [Google Scholar]
- 2.Biberfield G, Biberfield P. J Bacteriol. 1970;102:855–861. doi: 10.1128/jb.102.3.855-861.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu P,-C, Collier A M, Baseman J B. J Exp Med. 1977;145:1328–1343. doi: 10.1084/jem.145.5.1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Razin S, Jacobs E. J Gen Microbiol. 1992;138:407–422. doi: 10.1099/00221287-138-3-407. [DOI] [PubMed] [Google Scholar]
- 5.Baseman J B, Morrison-Plummer J, Drouillard D, Puelo-Scheppke B, Tryon V V, Holt S C. Isr J Med Sci. 1987;23:474–479. [PubMed] [Google Scholar]
- 6.Krause D C, Leith D K, Wilson R M, Baseman J B. Infect Immun. 1982;35:809–817. doi: 10.1128/iai.35.3.809-817.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krause D C, Proft T, Hedreyda C T, Hilbert H, Plagens H, Herrmann R. J Bacteriol. 1997;179:2668–2677. doi: 10.1128/jb.179.8.2668-2677.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedreyda C T, Krause D C. Infect Immun. 1995;63:3479–3483. doi: 10.1128/iai.63.9.3479-3483.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipman R P, Clyde W A, Jr, Denny F W. J Bacteriol. 1969;100:1037–1043. doi: 10.1128/jb.100.2.1037-1043.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Layh-Schmitt G, Hilbert H, Pirkl E. J Bacteriol. 1995;177:843–846. doi: 10.1128/jb.177.3.843-846.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baseman J B, Cole R M, Krause D C, Leith D K. J Bacteriol. 1982;151:1514–1522. doi: 10.1128/jb.151.3.1514-1522.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevens M K, Krause D C. J Bacteriol. 1992;174:4265–4274. doi: 10.1128/jb.174.13.4265-4274.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens M K, Krause D C. J Bacteriol. 1991;173:1041–1050. doi: 10.1128/jb.173.3.1041-1050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biberfield, G. (1972) Pathogenic Mycoplasmas, Ciba Foundation Symposium 1972, p. 322.
- 15.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li B-C, Herrmann R. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser C, Gocayne J D, White O, Adams M D, Clayton R A, et al. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 17.Dirksen L B, Proft T, Hilbert H, Plagens H, Herrmann R, Krause D C. Gene. 1996;171:19–25. doi: 10.1016/0378-1119(96)00050-9. [DOI] [PubMed] [Google Scholar]
- 18.Ogle K F, Lee K K, Krause D C. Gene. 1991;97:69–75. doi: 10.1016/0378-1119(91)90011-y. [DOI] [PubMed] [Google Scholar]
- 19.Alley M R K, Maddock J R, Shapiro L. Science. 1993;259:1754–1757. doi: 10.1126/science.8456303. [DOI] [PubMed] [Google Scholar]
- 20.Shapiro L. Cell. 1993;73:841–855. doi: 10.1016/0092-8674(93)90266-s. [DOI] [PubMed] [Google Scholar]
- 21.Hayflick, L. (1965) Tex. Rep. Biol. Med. 23 (Suppl. 1), 285–303. [PubMed]
- 22.Lipman R P, Clyde W A., Jr Proc Soc Exp Biol Med. 1969;131:1163–1167. doi: 10.3181/00379727-131-34061. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 24.Dirksen L B, Krebes K A, Krause D C. J Bacteriol. 1994;176:7499–7505. doi: 10.1128/jb.176.24.7499-7505.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hahn T-W, Krebes K A, Krause D C. Mol Microbiol. 1996;19(5):1085–1093. doi: 10.1046/j.1365-2958.1996.455985.x. [DOI] [PubMed] [Google Scholar]
- 26.Tully J G. In: Methods in Mycoplasmology. Razin S, Tully J, editors. Vol. 1. New York: Academic; 1983. pp. 173–177. [Google Scholar]
- 27.Su C J, Tryon V V, Baseman J B. Infect Immun. 1987;55:3023–3029. doi: 10.1128/iai.55.12.3023-3029.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krause D C, Leith D K, Baseman J B. Infect Immun. 1983;39:830–836. doi: 10.1128/iai.39.2.830-836.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sperker B, Hu P-C, Herrmann R. Mol Microbiol. 1991;5:299–306. doi: 10.1111/j.1365-2958.1991.tb02110.x. [DOI] [PubMed] [Google Scholar]
- 30.Ogle K F, Lee K K, Krause D C. Infect Immun. 1992;60:1633–1641. doi: 10.1128/iai.60.4.1633-1641.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Layh-Schmitt G, Herrmann R. Infect Immun. 1994;62:974–979. doi: 10.1128/iai.62.3.974-979.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franzoso G, Hu P-C, Meloni G A, Barile M F. Infect Immun. 1993;61:1523–1530. doi: 10.1128/iai.61.4.1523-1530.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy S P, Rasmussen W G, Baseman J B. FEMS Immunol Med Microbiol. 1996;15:199–211. doi: 10.1111/j.1574-695X.1996.tb00086.x. [DOI] [PubMed] [Google Scholar]
- 34.Gottesman S, Maurizi M R. Microbiol Rev. 1992;56:592–621. doi: 10.1128/mr.56.4.592-621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mernaugh, G. R., Dallo, S. F., Holt, S. C. & Baseman, J. B. (1993) Clin. Infect. Dis. 17 (Suppl. 1), S69–S78. [DOI] [PubMed]