

Abstract
Human autoimmune diseases, such as multiple sclerosis, type 1 diabetes, rheumatoid arthritis and systemic lupus erythematosus (SLE), are linked genetically to distinct major histocompatibility complex (MHC) class II molecules and other immune modulators. However, genetic predisposition is only one risk factor for the development of these diseases, and low concordance rates in monozygotic twins as well as geographical distribution of disease risk suggest a critical role for environmental factors in the triggering of these autoimmune diseases. Among potential environmental factors, infections have been implicated in the onset and/or promotion of autoimmunity. This review will discuss human autoimmune diseases with a potential viral cause, and outline potential mechanisms by which pathogens can trigger autoimmune disease as discerned from various animal models of infection-induced autoimmune disease.
Keywords: autoimmune disease, epitope spreading, infection, molecular mimicry
Introduction
The immune system has evolved to distinguish self- from harmful non-self to preserve the integrity of the host. Deficits in this process can result in susceptibility to infections or over-reactivity to potentially harmless antigens, leading to immunopathology (e.g. allergy) and autoimmunity. Thus, it is not surprising that genes that influence the sensitivity, intensity and/or class of immune response that are associated with risk for development of autoimmune diseases might only manifest themselves as regulating autoimmunity following infection. Potential mechanisms by which pathogens may trigger or protect from development of autoimmune diseases will be discussed. The focus of this overview on pathogen-induced activation of autoimmune disease will be on recent studies showing the mouse strain-specific induction of a CD4 T cell-mediated autoimmune central nervous system (CNS) inflammatory model of multiple sclerosis (MS) following CNS infection with Theiler's murine encephalomyelitis virus (TMEV), a positive-strand RNA virus.
Potential mechanisms of infection-induced triggering of autoimmune disease
Multiple mechanisms have been described to explain how viruses may trigger autoimmune diseases [1], including virus-induced general activation of the immune system and provision of viral antigens that specifically stimulate immune responses, which cross-react with self-antigens and therefore cause autoreactive immunopathologies. Figure 1 shows a schematic of these potential mechanisms which are discussed in more detail below.
Fig. 1.
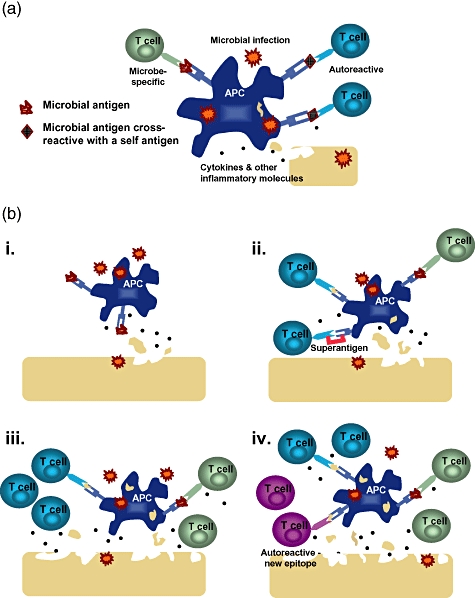
Possible mechanisms of infection-induced autoimmunity. (a) Autoreactive T cells can be activated via molecular mimicry by cross-reactive recognition of a viral antigen that has similarity to self-antigen. (bi) Microbial infection stimulates Toll-like receptors (TLRs) and other pattern-recognition receptors on antigen-presenting cells (APCs), leading to the production of proinflammatory mediators, which can lead in turn to tissue damage. (bii) Self-antigen that is released from damaged tissue can be taken up by activated APCs, processed and presented to autoreactive T cells (concomitant with presentation of virus antigen to virus-specific T cells) in a process known as bystander activation. Alternatively, an infection can lead to microbial superantigen-induced activation of a subset of T cells, some of which could be specific for self-antigen. (biii) Further tissue destruction by activated T cells and inflammatory mediators causes the release of more self-antigen from tissues. (biv) The response can then spread to involve T cells (or antibodies) specific for other self-antigens in a process known as epitope spreading.
Adjuvant effects of pathogens in priming autoreactive immune responses
The innate ability of the host to defend against invading microbes is mediated by germline-encoded receptors known as pattern-recognition receptors (PRRs). These molecules include Toll-like receptors (TLRs), nucleotide-binding and oligomerization domain non-obese diabetic (NOD)-like receptors (NLRs) (RIG-I)-like helicases and a subset of C-type lectin receptors, which together recognize a large number of molecular patterns present in bacteria, viruses and fungi [2]. The signalling pathways triggered by engagement of these molecules lead to cellular activation, which increases the expression of co-stimulatory molecules by antigen-presenting cells (APCs), as well as activating their antigen-presenting capacity and production of type I interferons, proinflammatory cytokines and chemokines, which initiate and direct the immune response against the invading pathogen. By triggering PRRs, stimulating early innate immune responses and increasing the function of APCs, pathogens act as adjuvants for the immune response, while also providing an antigen source to drive T cell and B cell activation and effector function. In this highly inflammatory environment, it is easy to envision how an aberrant destructive immune response can be triggered and/or escalated if autoreactive cells are present. There are a number of postulated mechanisms by which pathogenic infections can trigger autoimmune disease, but most evidence in animal models supports the idea that cross-reactive immune responses cause autoimmunity due to similarities between viral and self-antigens.
Molecular mimicry
Antigen recognition by the T cell receptor (TCR) is degenerate, allowing T cell activation by different peptides bound to one or even multiple major histocompatibility complex (MHC) molecules [3]. Thus, responses to foreign antigens could result in the activation of T cells that are also cross-reactive with self-antigens. Similarly, monoclonal antibodies have been shown to recognize both microbial and self-antigens [4]. This concept, known as molecular mimicry (Fig. 1a), was first put forward by Fujinami and Oldstone [5]. Flexibility of TCR recognition is thought to be central to the ability of the immune system to recognize and respond to the majority of pathogen-derived peptides, but a side effect of this degeneracy is the potential induction of autoimmunity by microbial antigens. In vitro studies have shown clearly that viral peptides with some degree of homology with self-peptides can stimulate autoreactive T cells [6].
There are numerous animal models in which molecular mimicry has been shown to trigger autoimmune diseases (Table 1). These include: Theiler's murine encephalomyelitis virus (TMEV) engineered to express mimics of encephalitogenic myelin epitopes, a model of human multiple sclerosis (MS) [7]; herpes simplex virus (HSV)-associated stromal keratitis, in which HSV infection leads to T cell-mediated blindness in both humans and mice [8]; various models of type 1 diabetes [9]; autoimmune demyelinating disease associated with Semliki Forest virus (SFV) [10]; and autoimmune myocarditis associated with coxsackievirus [11] or murine cytomegalovirus infection [12]. Microbial pathogens have also been implicated in contributing to autoimmune disease by molecular mimicry, e.g. streptococcus infection in rheumatic fever [13].
Table 1.
Selected murine models of infection-induced autoimmune diseases.
| Disease modelled | Infectious agent | Mechanism(s) of disease initiation or exacerbation*† |
|---|---|---|
| Multiple sclerosis | TMEV | Bystander activation and epitope spreading |
| TMEV expressing PLP139 | Molecular identity | |
| TMEV expressing PLP139 mimics | Molecular mimicry | |
| LCMV (in mice expressing LCMV protein in CNS) | Molecular identity | |
| Semliki virus | Molecular mimicry | |
| EAE + bacterial superantigen staphylococcal enterotoxin B (SEB)‡ | Superantigen | |
| Type 1 diabetes | Coxsackie B4 | Bystander activation |
| LCMV (in mice expressing LCMV protein in pancreas) | Molecular identity | |
| Pichinde virus (in mice expressing LCMV protein in pancreas)‡ | Molecular mimicry | |
| Myocarditis | Mouse cytomegalovirus | Bystander activation or molecular mimicry |
| Coxsackie B3 virus | Molecular mimicry | |
| Chlamydia | Molecular mimicry | |
| Stromal keratitis | HSV | Molecular mimicry and/or bystander activation |
| Rheumatoid arthritis | Collagen-induced arthritis (CIA) + murine arthritogenic mycoplasma superantigen (MAM)‡ | Superantigen |
Unless otherwise noted, the infectious agent has been shown to trigger the indicated disease.
References to these murine models of infection-induced autoimmune diseases can be found in Munz et al. [46].
The indicated infectious agent does not cause disease, but results in exacerbation of disease established by other means. TMEV: Theiler's murine encephalomyelitis virus; PLP: proteolipid protein; LCMV: lymphocytic choriomeningitis virus; CNS: central nervous system; EAE: experimental autoimmune encephalomyelitis; HSV: herpes simplex virus.
There are multiple models of molecular identity, in which an exact microbial protein or epitope is expressed transgenically in a particular tissue. Under these conditions, animals do not develop spontaneous autoimmune disease, but autoimmunity in the transgenic protein-expressing organ often ensues after infection with the microorganism containing the protein [14,15]. Although artificial, these approaches indicate that T cells specific for a ‘self’-antigen can become activated by infection with a microorganism containing an identical antigen, which provides appropriate innate immune signals, resulting in overt autoimmune disease. Even when the transgenic antigen is also expressed in the thymus, so that normal mechanisms of negative selection significantly decrease the number of high-affinity T cells specific for the viral/self-antigen, infection results eventually in autoimmunity [16]. This indicates that even T cells with low affinity for a self-antigen and that have escaped negative selection can be activated through molecular mimicry with a microbial antigen and cause disease. Similarly, in the TMEV model of MS, a severe rapid-onset demyelinating disease in the CNS is induced by intracerebral or peripheral infection of SJL/J mice with TMEV that has been engineered to express the immunodominant self-epitope from myelin, i.e. proteolipid protein (PLP139–151) [17].
Several bacterial and viral peptides that mimic PLP139–151 have been identified [18] and used in animal models that address more directly the possibility of induction of autoimmune disease via infection-induced molecular mimicry. TMEV engineered to express peptides derived from Haemophilus influenzae that mimic PLP139–151 (TMEV-HI, which shares six of 13 amino acids with PLP139–151) or murine hepatitis virus (TMEV-MHV, which shares only three of 13 amino acids with PLP139–151), induces a rapid-onset, severe demyelinating disease that is similar to that induced by infection with TMEV expressing PLP139–151 itself [17,19]. Interestingly, the three critical amino acids conserved in the HI and MHV mimics corresponded to the primary and secondary MHC class II and the primary TCR contacts, again illustrating the remarkable degeneracy of the TCR repertoire. The H. influenzae mimic of PLP139–151 could be processed and presented out of its own native flanking sequences when larger portions of the bacterial protein were expressed in TMEV, which further supports the potential pathological role for molecular mimicry in a natural infection [19]. These studies also illustrate the importance of pathogen-stimulated innate immune responses as infection with TMEV engineered to express the HI PLP139–151 mimic induced autoreactive myelin-specific T cells capable of mediating CNS demyelination, while immunization with this peptide in complete Freund's adjuvant (CFA) was not able to induce clinical CNS disease [19]. Relevant to human MS, bacterial peptide mimics of the human leucocyte antigen (HLA)-DR2-restricted myelin basic protein epitope (MBP85–99), derived from different pathogens such as Mycobacterium tuberculosis, Bacillus subtilis and Staphylococcus aureus, induce demyelinating disease in mice that express transgenically a human MBP85–99-specific TCR as well as the HLA-DR2 restricting molecule [20]. Molecular mimicry was also shown in a model of diabetes in which lymphocytic choriomeningitis virus (LCMV) nucleoprotein was expressed under the control of the rat insulin promoter. Infection with Pichinde virus, which contains an epitope that mimics a subdominant epitope in the LCMV nucleoprotein, accelerated autoimmune disease that had already been established by previous infection with LCMV [9]. HSV-induced stromal keratitis is mediated by corneal-antigen-specific T cell responses that are induced following corneal HSV infection. This illustrates a natural model of autoimmune disease in which molecular mimicry can occur in the absence of genetic manipulation of either the virus or host [8].
Self-reactive lymphocytes from patients with autoimmune diseases such as SLE, rheumatoid arthritis and MS have been shown to have higher frequencies and activation states and/or less requirement for co-stimulation [21–23]. In MS, receptor analysis of T and B cells in CNS tissue and in the cerebrospinal fluid (CSF) showed clonal expansions in both populations, indicating that there is clonal reactivity to a restricted number of disease-relevant antigens [24,25]. In addition, longitudinal studies provided evidence for long-term persistence of individual myelin-specific T cell clones tracked over several years in the blood of MS patients [26], indicating a strong, persisting memory response and/or ongoing autoantigen exposure at least for a subset of myelin-reactive T cells. These memory responses may reflect, at least in part, persisting clonal expansions of polyspecific T cells recognizing both self and virus antigens that have been found associated with human autoimmune diseases (Table 2). For example, high viral loads that occur during symptomatic primary Epstein–Barr virus (EBV) infection, resulting in infectious mononucleosis, are associated with an increased risk of developing multiple sclerosis [27,28], and could prime these polyspecific T cell responses [29]. Accordingly, patients with multiple sclerosis have predominant clonal expansions of T cells specific for the EBV-encoded nuclear antigen 1 (EBNA1), which is the most consistently recognized EBV-derived CD4+ T cell antigen in healthy virus carriers, and EBNA1-specific T cells recognize myelin antigens more frequently than other autoantigens that are not associated with multiple sclerosis [30].
Table 2.
Selected autoimmune diseases and their proposed viral associations and potential mechanisms.
| Disease | Proposed viral association | Proposed mechanism*† |
|---|---|---|
| Multiple sclerosis | Measles | None suggested |
| EBV | Molecular mimicry/bystander activation | |
| HHV-6 | Molecular mimicry | |
| Type 1 diabetes | Coxsackie virus | Bystander activation, molecular mimicry |
| Rubella | Molecular mimicry | |
| Rheumatoid arthritis | EBV | None suggested |
| Parvovirus | None suggested | |
| SLE | EBV | Molecular mimicry |
| HTLV-1-associated myelopathy | HTLV-1 | Molecular mimicry |
| Stromal keratitis | HSV-1 | Molecular mimicry |
For those diseases in which no proposed mechanism has been suggested, evidence for involvement of the indicated virus in disease pathogenesis includes association of increased anti-viral responses or increased detectable virus at site of diseased tissue, and/or epidemiological evidence.
References to these virus-associated human autoimmune diseases can be found in Munz et al. [46]. EBV: Epstein–Barr virus; HHV: human herpesvirus; HTLV: human T cell lymphotropic virus; HSV: herpes simplex virus; SLE: systemic lupus erythematosus.
Bystander activation of autoreactive cells and epitope spreading
APCs that are activated within the inflammatory milieu of a pathogenic infection can stimulate the activation and proliferation of autoreactive T or B cells in a process known as bystander activation. In this case, APCs present self-antigen, obtained subsequent to tissue destruction and/or the uptake of local dying cells, to autoreactive cells [31] (Fig. 1bi). In addition to autoimmune responses that are primed initially by APCs and stimulated by bystander activation, additional autoantigen-specific T or B cells can be primed through epitope spreading [32,33]– wherein an immune response initiated by various stimuli, including microbial infection, trauma, transplanted tissue or autoimmunity, ‘spreads’ to include responses directed against a different portion of the same protein (intramolecular spreading) or a different protein (intermolecular spreading) (Fig. 1biii, iv). Activating a broader set of T cells through epitope spreading is clearly helpful in anti-pathogen or anti-tumour immune responses. However, autoimmune disease is potentially induced and/or exacerbated when spread to and within self-proteins occurs subsequent to the destruction of self-tissue. Epitope spreading in animal models proceeds in an orderly, directed and hierarchical manner, such that more immunodominant epitopes elicit responses first, followed by less dominant responses. This type of spreading has been shown in experimental autoimmune encephalomyelitis (EAE), a non-infectious model of MS [33,34], as well as in TMEV-induced demyelinating disease induced by infection with the non-engineered, wild-type BeAn virus strain [35–37] (Fig. 2) and in the spontaneously arising NOD mouse model of type 1 diabetes [38] (S. D. M., unpublished observations). While these examples document epitope spreading within autoantigens and to additional autoantigens, the inflammatory environment of viral infections could support these immune response cascades by increasing the presentation of self-antigens via provision of signals through PRRs, which has been shown to be required for the activation of myelin peptide-specific T cells via epitope spreading in SJL mice infected with wild-type TMEV [39,40].
Fig. 2.
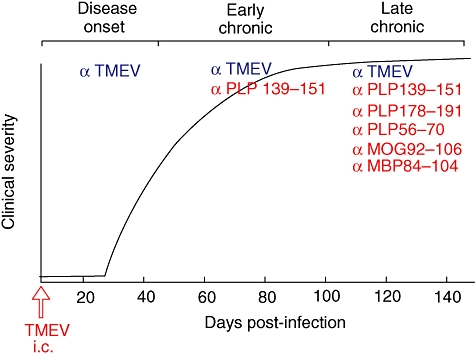
Model of induction of central nervous system (CNS) autoimmunity in SJL mice infected with Theiler's murine encephalomyelitis virus (TMEV) via epitope spreading to released endogenous myelin antigens. Intracerebral (i.c.) infection of SJL mice with the BeAn strain of TMEV leads to a chronic-progressive CNS demyelinating disease, with clinical signs first appearing approximately 30 days post-infection. TMEV establishes a persistent CNS infection in CNS-resident microglia, as well as in CNS-infiltrating macrophages and dendritic cells (DCs). Disease is initiated by TMEV-specific CD4+ T cells which release proinflammatory cytokines in response to viral epitopes presented in the CNS by the various APCs leading to bystander damage to myelin and release of endogenous myelin epitopes. Infiltrating myeloid DCs process and present released myelin epitopes leading to de novo activation of myelin epitope-specific T cells via epitope spreading. The response spreads initially to the immunodominant proteolipid protein (PLP)139–151 epitope and subsequently to other less dominant encephalitogenic myelin peptides in late progressive disease.
An even broader form of bystander activation is achieved by cross-linking MHC class II molecules on APCs with TCRs expressing a certain Vβ domain by superantigens (Fig. 1bii). T cell populations that are stimulated in this manner could potentially contain a subset of T cells specific for a self-antigen [41]. There are multiple examples in which superantigens are involved in diseases such as EAE, arthritis and inflammatory bowel disease, making superantigens another mechanism by which bystander activation could initiate, or at the least exacerbate, autoimmunity in mouse models [42]. In this study, superantigens were shown to amplify, but not initiate, autoimmune T cell responses (Table 1). Furthermore, the association of certain genotypes of the superantigen-encoding endogenous retrovirus HERV-K18, which is transactivated by EBV, with MS has been reported [43]. These various mechanisms are not mutually exclusive, e.g. molecular mimicry might initially prime autoreactive T cells, but these responses could be amplified by epitope spreading [7,17] or by superantigen-mediated expansion and by activation of autoantigen-specific T cells that express a given Vβ chain.
Emerging mechanisms
Infections can affect the immune response in many ways, and mechanisms such as molecular mimicry and bystander activation/epitope spreading are certainly not the only ways in which pathogens might trigger or accelerate autoimmune disease. A recent study showed that in a spontaneous animal model of SLE, lipid raft aggregation on T cells could be induced by several microorganisms or toxins leading to enhanced T cell signalling and exacerbated disease [44]. Furthermore, viral infections could also directly maintain autoreactive effector cells or autoantigen-presenting cells. As one example of the latter, persistent infection of microglia with TMEV has been shown to cause up-regulation of MHC, co-stimulatory molecules as well as cytokines required for CD4 T cell differentiation, and enhance the ability of these cells to function as effective APCs [45]. Lastly, it is possible that virus infections can cause changes in normal immunoregulatory mechanisms [47]. Recent unpublished studies in our laboratory have shown that susceptibility to TMEV-induced demyelinating disease in SJL/J mice is mediated by virus-induced activation of regulatory T cells (Tregs) in the susceptible SJL/J strain, which interfere with CTL-mediated virus clearance leading to persistent CNS infection and later initiation of autoimmunity via epitope spreading. In contrast, infection of disease resistant C57BL/6 mice fails to activate Tregs and thus virus is rapidly cleared.
Conclusions
Results from the multiple animal models summarized above show clearly that various mechanisms including molecular mimicry, bystander activation and epitope spreading can initiate activation of autoreactive T cells that then expand, differentiate and become pathogenic. It is important to realize that these mechanisms are interrelated, non-mutually exclusive and dynamic, so the idea of microbial infection eliciting autoimmunity must be viewed not as a defined event that occurs through a particular mechanism, but as a process that can occur through many pathways simultaneously and/or sequentially (Fig. 1). For example, epitope spreading can be initiated through molecular mimicry, as illustrated by the activation of PLP178–191-specific T cells in SJL mice in which autoimmunity was induced by infection with TMEV expressing either PLP139–151 or a PLP139–151 mimic peptide [17].
Despite the multiple causal relationships between infection and autoimmunity which have been identified in animal models and the correlations drawn in human autoimmune diseases, pathogen-derived triggers of human autoimmunity have been difficult to identify. This could be due to the fact that evidence of autoimmunity is likely to become clinically apparent only after a considerable period of subclinical autoimmune responses, at which time the pathogen might have already been cleared and/or the anti-viral immune responses might have subsided, the so-called ‘hit-and-run’ hypothesis. It is also possible that the initiating infection may take place in a site distal from the target organ of the autoimmune disease. As a result, it can be difficult to distinguish between the postulated mechanisms, even in seemingly simple animal models.
Acknowledgments
Supported in part by grants from NIH, the National Multiple Sclerosis Society and the Myelin Repair Foundation.
Disclosure
M.T.G. and S.D.M. have no conflicts of interest relative to the material discussed in this review.
References
- 1.Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol. 2009;155:1–15. doi: 10.1111/j.1365-2249.2008.03834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishii KJ, Koyama S, Nakagawa A, et al. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe. 2008;3:352–63. doi: 10.1016/j.chom.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Marrack P, Scott-Browne JP, Dai S, et al. Evolutionarily conserved amino acids that control TCR–MHC interaction. Annu Rev Immunol. 2008;26:171–203. doi: 10.1146/annurev.immunol.26.021607.090421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujinami RS, Oldstone MB, Wroblewska Z, et al. Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc Natl Acad Sci USA. 1983;80:2346–50. doi: 10.1073/pnas.80.8.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–5. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 6.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson JK, Ercolini AM, Miller SD. A virus-induced molecular mimicry model of multiple sclerosis. Curr Top Microbiol Immunol. 2005;296:39–53. doi: 10.1007/3-540-30791-5_3. [DOI] [PubMed] [Google Scholar]
- 8.Zhao ZS, Granucci F, Yeh L, et al. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279:1344–7. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 9.Christen U, Edelmann KH, McGavern DB, et al. A viral epitope that mimics a self antigen can accelerate but not initiate autoimmune diabetes. J Clin Invest. 2004;114:1290–8. doi: 10.1172/JCI22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mokhtarian F, Zhang Z, Shi Y, et al. Molecular mimicry between a viral peptide and a myelin oligodendrocyte glycoprotein peptide induces autoimmune demyelinating disease in mice. J Neuroimmunol. 1999;95:43–54. doi: 10.1016/s0165-5728(98)00254-9. [DOI] [PubMed] [Google Scholar]
- 11.Gauntt CJ, Tracy SM, Chapman N, et al. Coxsackievirus-induced chronic myocarditis in murine models. Eur Heart J. 1995;16(Suppl. O):56–8. doi: 10.1093/eurheartj/16.suppl_o.56. [DOI] [PubMed] [Google Scholar]
- 12.Lawson CM. Evidence for mimicry by viral antigens in animal models of autoimmune disease including myocarditis. Cell Mol Life Sci. 2000;57:552–60. doi: 10.1007/PL00000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham MW, Antone SM, Gulizia JM, et al. Cytotoxic and viral neutralizing antibodies crossreact with streptococcal M protein, enteroviruses, and human cardiac myosin. Proc Natl Acad Sci USA. 1992;89:1320–4. doi: 10.1073/pnas.89.4.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oldstone MB, Nerenberg M, Southern P, et al. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–31. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 15.Ohashi PS, Oehen S, Buerki K, et al. Ablation of ‘tolerance’ and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–17. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 16.von Herrath MG, Dockter J, Oldstone MB. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity. 1994;1:231–42. doi: 10.1016/1074-7613(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 17.Olson JK, Croxford JL, Calenoff M, et al. A virus-induced molecular mimicry model of multiple sclerosis. J Clin Invest. 2001;108:311–8. doi: 10.1172/JCI13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrizosa AM, Nicholson LB, Farzan M, et al. Expansion by self antigen is necessary for the induction of experimental autoimmune encephalomyelitis by T cells primed with a cross-reactive environmental antigen. J Immunol. 1998;161:3307–14. [PubMed] [Google Scholar]
- 19.Croxford JL, Olson JK, Anger HA, et al. Initiation and exacerbation of autoimmune demyelination of the central nervous system via virus-induced molecular mimicry: implications for the pathogenesis of multiple sclerosis. J Virol. 2005;79:8581–90. doi: 10.1128/JVI.79.13.8581-8590.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greene MT, Ercolini AM, Degutes M, et al. Differential induction of experimental autoimmune encephalomyelitis by myelin basic protein molecular mimics in mice humanized for HLA-DR2 and an MBP(85-99)-specific T cell receptor. J Autoimmun. 2008;31:399–407. doi: 10.1016/j.jaut.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yurasov S, Wardemann H, Hammersen J, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201:703–11. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samuels J, Ng YS, Coupillaud C, et al. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med. 2005;201:1659–67. doi: 10.1084/jem.20042321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovett-Racke AE, Trotter JL, Lauber J, et al. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients – a marker of activated/memory T cells. J Clin Invest. 1998:725–30. doi: 10.1172/JCI1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baranzini SE, Jeong MC, Butunoi C, et al. B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J Immunol. 1999;163:5133–44. [PubMed] [Google Scholar]
- 25.Babbe H, Roers A, Waisman A, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goebels N, Hofstetter H, Schmidt S, et al. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain. 2000;123:508–18. doi: 10.1093/brain/123.3.508. [DOI] [PubMed] [Google Scholar]
- 27.Thacker EL, Mirzaei F, Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol. 2006;59:499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen TR, Rostgaard K, Nielsen NM, et al. Multiple sclerosis after infectious mononucleosis. Arch Neurol. 2007;64:72–5. doi: 10.1001/archneur.64.1.72. [DOI] [PubMed] [Google Scholar]
- 29.Ascherio A, Munger KL. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Epstein–Barr virus and multiple sclerosis: epidemiological evidence. Clin Exp Immunol. 2010;160:120–4. doi: 10.1111/j.1365-2249.2010.04121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lunemann JD, Jelcic I, Roberts S, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med. 2008;205:1763–73. doi: 10.1084/jem.20072397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zipris D, Lien E, Xie JX, et al. TLR activation synergizes with Kilham rat virus infection to induce diabetes in BBDR rats. J Immunol. 2005;174:131–42. doi: 10.4049/jimmunol.174.1.131. [DOI] [PubMed] [Google Scholar]
- 32.Lehmann PV, Forsthuber T, Miller A, et al. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–7. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 33.McRae BL, Vanderlugt CL, Dal Canto MC, et al. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu M, Johnson JM, Tuohy VK. Generation of autonomously pathogenic neo-autoreactive Th1 cells during the development of the determinant spreading cascade in murine autoimmune encephalomyelitis. J Neurosci Res. 1996;45:463–70. doi: 10.1002/(SICI)1097-4547(19960815)45:4<463::AID-JNR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 35.Miller SD, Vanderlugt CL, Begolka WS, et al. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–6. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 36.Katz-Levy Y, Neville KL, Girvin AM, et al. Endogenous presentation of self myelin epitopes by CNS-resident APCs in Theiler's virus-infected mice. J Clin Invest. 1999;104:599–610. doi: 10.1172/JCI7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katz-Levy Y, Neville KL, Padilla J, et al. Temporal development of autoreactive Th1 responses and endogenous antigen presentation of self myelin epitopes by CNS-resident APCs in Theiler's virus-infected mice. J Immunol. 2000;165:5304–14. doi: 10.4049/jimmunol.165.9.5304. [DOI] [PubMed] [Google Scholar]
- 38.Kaufman DL, Clare-Salzler M, Tian J, et al. Spontaneous loss of T cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 1993;366:69–72. doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olson JK, Girvin AM, Miller SD. Direct activation of innate and antigen presenting functions of microglia following infection with Theiler's virus. J Virol. 2001;75:9780–9. doi: 10.1128/JVI.75.20.9780-9789.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMahon EJ, Bailey SL, Castenada CV, et al. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–9. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 41.Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest. 2001;108:1097–104. doi: 10.1172/JCI14235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brocke S, Gaur A, Piercy C, et al. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigen. Nature. 1993;365:642–4. doi: 10.1038/365642a0. [DOI] [PubMed] [Google Scholar]
- 43.Tai AK, O'Reilly EJ, Alroy KA, et al. Human endogenous retrovirus-K18 Env as a risk factor in multiple sclerosis. Mult Scler. 2008;14:1175–80. doi: 10.1177/1352458508094641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng GM, Tsokos GC. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J Immunol. 2008;181:4019–26. doi: 10.4049/jimmunol.181.6.4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olson JK, Ludovic CJ, Miller SD. Innate and adaptive immune requirements for induction of autoimmune demyelinating disease by molecular mimicry. Mol Immunol. 2004;40:1103–8. doi: 10.1016/j.molimm.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 46.Munz C, Lunemann JD, Getts MT, et al. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–58. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Filippi CM, von Herrath MG. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Viruses, autoimmunity and immunoregulation. Clin Exp Immunol. 2010;160:113–19. doi: 10.1111/j.1365-2249.2010.04128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]