

Abstract
Immunological aetiologies of disease are not generally well understood, but have been attributed to intrinsic immunological imbalances, infectious triggers or persistent infections. Evolutionary considerations lead to the formulation of three feasible categories of immunopathology for common diseases. One category of hypotheses presumes that the immune system is exposed to environmental conditions to which the individual is not well adapted. One hypothesis within this category, often referred to as the hygiene hypothesis, proposes that new more hygienic environmental conditions have generated compositions of symbionts that differ from those to which humans have been adapted. A second category of hypotheses proposes that infectious agents act as triggers of immunopathology by shifting the immune system into a self-destructive state. A third category proposes that infectious agents keep the immune in a self-destructive state by causing persistent infections. To evaluate disease causation rigorously and to determine the appropriate interventions, these three categories of causation need to considered for every disease that involves immunopathology. Assessment of the progress in understanding oncogenesis and other chronic diseases emphasizes the value of such integrated assessments.
Keywords: cancer, chronic disease, evolution, immunopathology, infection
Introduction
The causes of most chronic diseases of humans are not well understood. One of the central uncertainties involves chains of causation when immunological processes contribute to pathology. Ambiguity arises in part because of the difficulty in distinguishing between three categories of hypotheses. The first category, which can be labelled ‘rogue immune responses’, hypothesizes that immunological damage is not stimulated by infectious processes although microorganisms could be involved. Specifically, in recent decades, it has been proposed that co-habiting organisms could ameliorate immune-induced pathology, which then occurs when the organisms are absent. The most widely discussed version of this hypothesis has been labelled the ‘hygiene hypothesis’, which proposes that the exposure to symbionts adjusts or tunes the immune system so that it is less likely to cause illness [1–5]. The second category implicates infectious triggers; it hypothesizes that infection causes an imbalance in immunological activities which then have pathological effects that no longer depend upon the presence of the infection [6]. The third category implicates persistent infection; it hypothesizes that continued presence of an infectious agent alters immunological responses in a way that causes ongoing immune-induced pathology.
Distinguishing between these three categories of hypotheses is critically important, because identification of the correct explanations has consequences for treatment and prevention. If immunological dysfunction is not caused by infection, interventions must focus upon altering of immune function and changes in environmental exposures. If an infectious trigger is responsible, such interventions could be supplemented with or replaced by interventions that prevent or cure the infectious trigger. If persistent infections are responsible, prevention of infection is similarly important, but treatment of infections is of increased importance, because positive effects of such treatment can be expected throughout much or all of the of illness. However, if the immune-induced pathology results from persistent infection and the immune response is helping to control the infection, altering the immunological responses could have detrimental effects on disease progression by inhibiting the control of the infectious agent.
Discussions of such topics are often confounded by different definitions, which can compromise communication. For the purposes of this paper I define a symbiont as an organism that lives in intimate association with another organism. Symbionts are therefore defined broadly to include parasites, mutualists and commensals. I define a parasite as an organism that lives in or on another organism, and lowers the host organism's evolutionary fitness. This broad definition subsumes parasites at the cellular or subcellular level of organization (i.e. pathogens) as well as multi-cellular parasites. I define a mutualist as a symbiont that benefits from and provides a net fitness benefit to its host. I define a commensal as an organism that neither increases nor decreases its host's fitness. From this evolutionary perspective, commensalism is best considered a dividing line between parasitism and mutualism rather than a distinct category, because if measurements were sufficiently precise one would always expect to find some net negative or positive effect on the host organism [7]. The usage of the commensalism category is therefore related directly to the imprecision of measurement of net fitness benefit incurred by the symbiont. I define an infection as a parasitism in which the parasite is internal or associated intimately with the organism's external surface. (By this definition, intestinal helminths are agents of infection but ticks and lice are not.) I define immunopathology as injury induced by an immunological response [8].
As pathogens have been associated increasingly with immunological diseases of uncertain cause, immunologists have increasingly incorporated explanations that invoke infectious triggers. This tendency has apparently occurred partly because pathogens are often not isolated during the course of disease. In such cases the infectious trigger hypothesis is considered more parsimonious than the persistent infection hypothesis. The infectious trigger may also be attractive to immunologists because it simplifies the pathological process, making conceptual arguments and experimental tests more tractable. The history of the germ theory of disease, however, has been characterized by a tendency to discover the presence of infectious processes where they were assumed to be absent [9]. This tendency continues to the present, suggesting that it is tenuous – and not scientifically rigorous – to reject hypotheses of persistent infectious influences based on a lack of identification of a persistent infectious agent. Because such agents might have effects on immunological function that are difficult to discern, it is also tenuous to reject a role for persistent infections based on an absence of evidence identifying particular mechanisms by which infectious agents bring about immunological dysfunction.
This paper applies an evolutionary perspective to consider the three categories of disease causation mentioned above. My goals are to suggest how evolutionary considerations help to evaluate the feasibility of alternative hypotheses and to draw attention to hypotheses that have not received the attention that is merited by their feasibility and potential significance.
Rogue immune responses
Evolutionary considerations raise an important caveat pertaining to hypotheses that presume no role for infectious stimulation of immunopathology. Natural selection should disfavour individuals whose immune systems generate disease without providing compensating benefits, causing such diseases to be rare. The presence of individuals in modern populations who do not suffer from immune-induced disorders shows that human bodies can persist without such disorders. However, the commonness of such disorders (e.g. cardiovascular disease, multiple sclerosis, allergies and asthma) suggests that the causes are so new, evolutionarily, that natural selection has not yet purged the immunopathological response.
The literature on immunological disease rarely addresses this evolutionary problem. Instead, the starting point of analyses generally presumes that inherited predispositions or environmental factors destabilize the immune system. The focus is then shifted to understanding the immunological mechanisms associated with the immunopathology.
One exception to this generalization is molecular mimicry, which is discussed in the following section. The hygiene hypothesis is another exception, because it proposes that the cause of immunopathology is something that is missing from modern environments, namely exposure to particular symbiotic agents. Most versions of the hygiene hypothesis propose that tuning of the immune system requires exposures to such agents during the early years of life, and that the pervasiveness of immune-induced illnesses in modern life results from improvements in hygiene that have occurred during the past century or so, particularly in wealthy countries. It is suggested that an immune system without such tuning is liable to be overly sensitive to stimuli and thus prone to immunopathology. The hygiene hypothesis thus proposes that it is the lack of exposure to symbionts that is responsible for an unbalanced development of the immune system and hence of immune-induced damage [10]. Although the hygiene hypothesis does not exclude hypotheses of infectious causation of immune dysregulation, it focuses upon an effect of symbionts that is essentially the opposite of that proposed by infectious causation: amelioration rather than causation of immune-induced damage.
The evidence put forth to support the hygiene hypothesis has been largely correlations between locations with apparently high microbial exposure and lower levels of immune-mediated disease [2,5,11]. Two confounding problems are ambiguity about the levels of exposure to symbionts and the difficulty in sorting out causation from correlation. With modern travel the entire human population is interconnected much more intimately than at any other time in human history and prehistory, while only a tiny proportion of parasites have been omitted from human populations. In contrast to a fundamental assumption of some versions of the hygiene hypothesis, the exposure to diverse antigens may therefore be much greater now than in the past. The cause of increase immune-induced diseases in recent decades might therefore result from a greater exposure to causal pathogens, rather than a lower exposure. Similarly, the lower rate of asthma and other immune-induced diseases in rural areas might result from a greater exposure to causal pathogens in urban areas. Until the possible causal roles of some microorganisms can be separated from the possible ameliorating role of others, support for the hygiene hypothesis will be ambiguous.
Metaphors can be useful to these arguments, but they must be used carefully so that conclusions drawn are consistent with viable evolutionary processes. With regard to immune effects, the starting point of evolutionary analysis is the recognition that the genetic make-up of symbionts differs from that of their hosts. When the evolutionary fitness of symbionts is entirely dependent upon the reproduction of the hosts (as is the case with mitochondria and chloroplasts), then it is reasonable to assume that the symbiont function will benefit the host and that a seemingly mutually beneficial association is, in fact, a mutualism; but when the fitness of the symbiont has at least some independence from the evolutionary fitness of the host, as is the case for gut microbes that are transmitted from one host to the other (rather than in the germ line), the genetic interests of the symbiont cannot be assumed to coincide with the genetic interests of their hosts. When a symbiont alters the immune function of a host, hypotheses must be based on conflicts of interest as well as confluences of interest. Considering symbionts as ‘old friends’[4] may therefore be misleading, especially when these old associates cause life-threatening infections, as is the case with Toxoplasma, Helicobacter pylori, Salmonella and Mycobacterium[4]. The more balanced metaphor would be to consider long-established symbionts along a continuum from old friends (mututalists) to old enemies (parasites), with individuals that are neither friends nor enemies (commensals) marking the dividing line between these two categories.
These considerations prompt researchers to investigate whether a symbiont suppresses immunity for its own benefit (as is the case for some helminths [12–14]). Such a symbiont would probably still be, on balance, an enemy (a parasite) even though it might have some ameliorating effects on some immunopathological illness. Research on hepatitis A virus [15,16] provides a good example of the need for this broader framework. Effects of the hepatitis A virus through the T cell immunoglobulin and mucin-containing molecule (TIM-1) receptor indicates that hepatitis A has evolved to survive and reproduce more successfully through immune suppression. The ameliorating effects of hepatitis A infections on allergy and asthma are therefore best interpreted as immunosuppressive rather than immune tuning. This distinction is important, because immune suppression may increase vulnerability to the hepatitis A virus and other infectious threats. Hepatitis A viruses inflict substantial morbidity in human populations. The immune suppression by hepatitis A viruses would therefore probably be, on balance, negative. The more conservative action of invoking the mechanism of hepatitis A immune alteration without hepatitis A infection would be safer, but would still be questionable, because it would involve immune suppression rather than immune fine-tuning.
These considerations provide a framework for clarifying fundamentally different hypotheses that pertain to symbiont amelioration of immunopathology. Amelioration could result from tuning of the immune system as specified by the narrowly framed hygiene hypothesis; or it could result from adaptations of symbionts to exploit the host. Immune suppression is one obvious category of a symbiont adaptation for exploitation, but pathogens might also evolve to enhance immune responses to exploit a host. Salmonella enterica, for example, apparently enhances inflammatory responses to increase its ability to infect the intestinal tract in the context of microbial competitors [17]. Hosts may evolve countermeasures to block such immune-enhancing exploitations of parasites. These countermeasures could involve suppressing parts of the immune system [18], an effect that could resemble fine-tuning of the immune system in response to symbionts. These two alternatives, however, implicate different courses of action. If the process is fine-tuning, it would generally be encouraged. If the process is a countermeasure against immunological manipulation by the parasite, it could be associated with immunological vulnerabilities.
Another category of symbiont adaptation may involve interference between symbionts. If a gut symbiont affects the host adversely, the host can mobilize defences such as immunoglobulin (Ig)A secretion, non-specific defensive proteins or diarrhoea to destroy or protect against the offending symbiont [19,20]. By eliminating problem symbionts that stimulate strong immune responses, this process would tend to generate associations with benign parasitic or mutualistic symbionts that compete effectively with other symbionts using means that do not harm the host or trigger host immunological defences, giving the appearance of symbiont-induced tuning of the immune system.
These considerations emphasize the need for controlled experiments that distinguish the various alternative hypotheses. Much attention has been given to experimental infections with nematodes, which cause reductions in asthma symptoms. This effect has been advanced as support for the hygiene hypothesis. However, immune suppression by nematodes is well documented [12–14], and amelioration of asthma through immune suppression is not the same as amelioration due to tuning of the immune system. Although subtle, this distinction is important because immune suppression could exacerbate any underlying infectious cause of the asthma or other infections in the body. Immune tuning would not carry the same risk.
Infectious triggers and persistent infection
General evolutionary argument
Infectious agents co-evolve with hosts and therefore may be continuously generating novel characteristics that could be causing self-destructive immune responses. The resulting evolutionary arms race may lead to immune responses from which immunopathology cannot be purged by natural selection.
One specific hypothesis of infection-induced immunopathology has been labelled ‘molecular mimicry’[21]. In this case the evolutionary arms race forces pathogens to evolve antigens that increasingly resemble host molecules. To deal with such changes the host immune system may mount defences against the modified antigens that cross-react with the host molecules, thereby initiating immunopathology. Molecular mimicry may help to explain a variety of autoimmune disease, such as Sydenham's chorea and PANDAS (paediatric autoimmune disorders associated with strep) resulting from Streptococcus pyogenes infection, and multiple sclerosis resulting from Chlamydiophila (= Chlamydia) pneumoniae and viruses [22–24]. Molecular mimicry can be contrasted with hypotheses that require persistent infection, because autoimmunity induced by molecular mimicry could persist after the instigating pathogen is cleared [23,6]; but persistent infection would be a more powerful cause of autoimmunity due to molecular mimicry, because persistent infection poses a persistent immunological stimulation. Accordingly, pathogens that have been linked most strongly to immunopathology through antigenic cross reactivity, such as S. pyogenes and C. pneumoniae, tend to cause persistent infections.
Hypotheses that do not incorporate some direct role for infection must posit some other evolutionarily new provoking factor. Immunological responses to non-infectious agents such as allergens show that non-infectious agents can trigger immune-mediated conditions, but such associations do not rule out the involvement of an infectious agent. C. pneumoniae, for example, probably contributes to the immunopathology of asthma [25–28].
Oncogenesis
One of the great challenges in the study of immune-mediated disease is to identify whether pathogens correlated with immunopathology are playing a causal role and, if so, whether the causal role is primary (i.e. essential) or secondary (i.e. exacerbating). In either case it is important to identify whether the influence is as a trigger or a persistent infection with a persistent influence. The prolonged development of chronic disease, the character of candidate infections and ethical constraints hamper this process severely by limiting the use of Koch's postulates [9]. One hope has been that understanding details of pathogenesis down to the molecular level will reveal the causal mechanisms. The complexity of immunological and infectious processes, however, has hindered the ability to obtain a sufficiently complete understanding at the molecular level for most chronic diseases.
One of the best prospects for this approach is the pathogenesis of cancer. The history of controversy over oncogenesis and the emerging resolution of this controversy may therefore provide a useful example of how a broad consideration of hypotheses and evidence may resolve the role of infectious processes in chronic diseases. Since the discovery of the structure of DNA the dominant paradigm of oncogenesis has been that mutations dysregulate cellular control of cell proliferation and spread, with normal cells being transformed progressively into metastatic cancer cells. Infectious agents (broadly defined here to include multi-cellular, unicellular and subcellular parasites) were first accepted as causes of human cancer during the 1970s and 1980s. A role for infection was accommodated by the mutational paradigm by assuming that infection contributed to cancer by elevating the rate of mutations. Inflammation in particular has been, and is still, considered to be important contributor to oncogenesis by increasing mutation rates through the generation of highly reactive intermediates [29,30]. Oncogenic effects of infection could therefore be interpreted as occurring through infection-induced inflammation. Because the role of infection was considered exacerbating and peripheral, the possibility that they acted as a trigger or a persistent source of cellular malfunction was generally not addressed.
Over the past two decades hypotheses based upon persistent infection began to emerge and be tested. These hypotheses proposed that infectious agents specifically compromised barriers to cancer that are known to be compromised or destroyed by mutation. These hypotheses for infectious causation may have seemed redundant and unparsimonious, because evidence had documented the presence of such mutations in cells derived from advanced cancers. However, hypotheses that do not rely on persistent infection are associated with an important conceptual problem; namely, the difficulty in generating the necessary sequence of oncogenic mutations without first having large populations of dysregulated cells. If, for example, cell cycle arrest is compromised by a mutation in the retinoblastoma gene in a cell in which telomerase is being repressed, a mutation causing telomerase expression would be needed within the restricted number of divisions specified by the telomere length of the cell, without having mutations in the far greater number of other genes that could destroy the viability of the cell. Because infections can compromise simultaneously more than one barrier to oncogenesis, this problem of generating a sequence of specific mutations in a small number of divisions may be abrogated by hypotheses that invoke persistent infection.
Molecular mechanisms of oncogenesis have been understood sufficiently well for some cancers to allow these hypotheses to be distinguished. Cells have four critical barriers to cancer: (a) cell cycle arrest keeps the cell from dividing; (b) apoptosis (cell suicide) can destroy proliferating cells before they progress to metastatic cancer; (c) restriction of telomerase can block oncogenesis by placing an upper limit on the total number of divisions that a cell lineage undergoes; and (d) cell adhesion can provide a barrier to metastasis.
Viruses that are known to cause human cancer also are known to compromise all of these barriers. The viruses have evolved different mechanisms for interfering with these barriers, but all barriers are known to be compromised by each of the best-studied viruses: human T lymphotropic virus type 1, hepatitis B virus, Epstein–Barr virus (EBV), human papillomavirus (HPV) and human herpes virus 8 [31].
These viruses have apparently evolved to compromise these barriers because these effects foster persistence and spread of the viral genomes within the human body. By causing the cells that they infect to divide, the viral genetic material can replicate in concert, while incurring little exposure to the immune system. By interfering with apoptosis, viruses can keep the cell from destroying the virus via cell suicide. By increasing telomerase activity, the virus can push the infected cell towards immortality, perpetuating the exploitation of the host cell for resources and protection. By altering cell-to-cell adhesion, infected cells can spread to other parts of the body to facilitate further viral proliferation and transmission. This argument does not imply that pathogens benefit from lethal cancer. Rather, their compromising of the barriers to cancer nudge infected cells towards cancer as a side effect of the selection for persistence and spread within the host.
Only a small proportion of the people who are infected with any one of the known oncogeneic viruses will develop cancer. Other causes must therefore contribute to oncogenesis. The common occurrence in virally induced cancers of mutations that influence proliferation and adhesion confirms that mutations contribute to virus-driven oncogenesis. In fact, virus-driven oncogenesis leaves the standard paradigm of mutation-driven oncogenesis largely intact for the later stages of oncogenesis, because the compromising of barriers to cancer by infection is proposed to be the initiating step of oncogenesis (right side of Fig. 1).
Fig. 1.
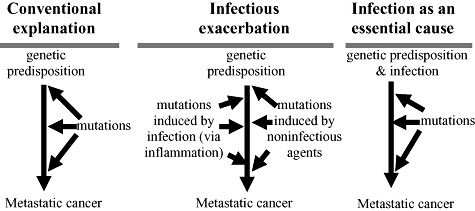
Alternative hypotheses for carcinogenesis.
This expectation follows from consideration of the improbability of generating mutational dysregulation of all three barriers to proliferation without incurring mutations that make the cell lineage dysfunctional. Specifically, when only one barrier to cancer is compromised by mutation, opportunities for oncogenesis are extremely limited. Activation of cell replication without inhibition of cellular senescence, for example, would generate only a limited number of cellular divisions unless the cell type expressed telomerase constitutively. Similarly, inhibition of cellular senescence would be of limited value without inhibition of apoptosis, which would act to destroy the infected cell. Without infectious causation, an unlikely sequence of specific mutations would generally have to compromise these barriers without destroying the cell's viability. In contrast, oncogenic viruses generally compromise all the barriers to metastatic cancer simultaneously. This simultaneous dysregulation allows infected cells to proliferate greatly. Once large numbers of precancerous infected cells are generated, the standard arguments about cancer evolution apply: oncogenic mutations that inhibit further these or other barriers to cancer would favour evolution of subsets of the precancerous cells toward cancer. Even if the vast majority of these mutations collectively cause large numbers of precancerous cells to become non-functional, oncogenesis can proceed because many other actively dividing infected cells remain to generate sequentially the rare oncogenic mutations that confer a competitive advantage through additional dysregulation of proliferation and adhesion.
This argument challenges the mutational dysregulation paradigm because it assigns a primary causal role to infection in many and perhaps most cancers. This primary causal role is not through the generation of mutation, but rather through the compromising of the barriers to cancer. The conclusion, however, does not rule out a role for infection-induced inflammation. Reactive intermediates associated with inflammation could be an important cause of the mutations that eventually transform cell lineages from a precancerous state to cancerous. The important point is that the available evidence confirms a direct compromising of the barriers to cancer but does not yet confirm a major contribution by inflammation-induced mutations.
This argument predicts that cancers will occur without infection only for those cells that have the cellular barriers to cancer suppressed or shut down (e.g. stem cell or other cell types for which long series of cell divisions are necessary) or when a single mutation can compromise multiple barriers simultaneously. The retinoblastoma protein, for example, regulates cell cycle arrest, but can also facilitate apoptosis and delay cellular senescence [32,33]. Mutations in the retinoblastoma gene may therefore compromise simultaneously three critical barriers to cancer. This unusual tendency may explain why infection may not be necessary for the oncogenesis of retinoblastoma. For infection-induced cancers, infectious dysregulation and mutational dysregulation are both best considered essential causes because oncogenesis requires both processes. In terms of practical public health benefits, infectious dysregulation may be more important when infection can be prevented (e.g. through vaccination), but the causes of mutation cannot.
A steady rise in the acceptance of infectious causation of human cancers has occurred over the past three decades (see Table 1). Before 1970 no human cancer was generally accepted as caused by infection. Currently, about 20% of all human cancer is accepted as caused by infection [34]. The large number of cancers for which infectious causation is suspected, but not yet accepted (Table 2), suggests that we are in the midst of the overall process of recognition of the actual role of infectious causation. Many of the viruses discussed above that are now accepted as causes of some cancers (Table 1) are also candidate causes of other cancers (Table 2). The compromising of barriers by pathogens will therefore probably turn out to be important for cancers that are not yet accepted as caused by infection.
Table 1.
Acceptance of parasitic causes of human cancers.
| Cancer | Parasite | ∼ Year accepted |
|---|---|---|
| Cholangioma liver cancers | Opisthorchid trematodes† | 1970 |
| Bladder cancer | Schistosome trematodes† | 1970 |
| Burkitt's lymphoma | Epstein–Barr virus† jointly with Plasmodium falciparum | 1975 |
| Adult T cell leukaemia | Human T lymphotropic virus I† | 1980 |
| Cervical cancer | Human papilloma virus (HPV)† | 1985 |
| Nasopharyngeal cancer | Epstein–Barr virus (EBV)† | 1990 |
| Liver cancer | Hepatitis B† & C† viruses | 1995 |
| Kaposi's sarcoma | Human herpesvirus 8† | 1995 |
| Stomach cancer | Helicobacter pylori† | 2000 |
| Oropharyngeal cancer | HPV† | 2005 |
Persistent parasites. Years of acceptance are approximate because the transition to acceptance generally has been gradual and controversial.
Table 2.
Cancers that have been associated with particular parasites for which a causal role has not yet been generally accepted.
| Cancer | Candidate parasites |
|---|---|
| Colorectal cancer | Schistosoma japonicum† |
| Liver cancer | Schistosoma japonicum† |
| Merkel cell cancer | Merkel cell polyomavirus† |
| Mesothelioma | Simian virus 40 (SV40)† |
| Breast cancer | MMTV†, EBV†, HPV† |
| Acute lymphoblastic leukaemia | EBV† |
| Hodgkin's lymphoma | EBV† |
| Non-Hodgkin's lymphomas | EBV†, SV40† |
| Skin cancers | HPV† |
| Oesophageal cancer | HPV† |
| Colon cancer | JC virus† |
| Ovarian cancer | Unknown retrovirus, EBV† |
| Prostrate | XMRV, BK virus† |
Known to be persistent parasites.
EBV, Epstein–Barr virus; HPV, human papilloma virus; MMTV, mouse mammary tumour virus; JC virus, John Cunningham virus; XMRV, xenotropic murine leukemia virus-related virus.
This matter has not yet been resolved for bacterial and helminth associations with cancer (see Tables 1 and 2). Bacterial and eukaryotic parasites, especially extracellular ones, might not benefit from compromising the barriers that are compromised by oncogenic viruses. In this case cellular parasites might act in concert with viruses. This sort of interaction is accepted for Burkitt's lymphoma, but in this case it appears that the cellular pathogen (Plasmodium falciparum) activates the activity of the oncogenic virus (EBV), rather than fostering oncogenesis through enhancement of mutation rate.
The mechanism by which trematodes contribute to cancer is still unclear. Most attention has focused upon the elevation of reactive compounds during inflammation [11,35]. Opisthorchid trematodes, which are acquired from eating raw fish, have been linked to cholangiocarcinoma, a liver cancer originating from the bile duct cells, and schistosome trematodes have been recognized as a cause of bladder cancer. Over the past decade, studies have implicated hepatitis B and C viruses as risk factors for intrahepatic cholangiocarcinoma in western and eastern countries, although the relative importance seems to vary from region to region [36–39]. Human immunodeficiency virus (HIV) is a risk factor for cholangiocarcinoma [40] and for cancers with known and suspected infectious causes.
A similar argument applies to bladder cancer. Co-factors emphasized in the literature on bladder cancer have been restricted largely to mutagenic chemicals such as those generated by inflammation and nitrosamines [41], but oncogenic serotypes of human papilloma virus (HPV) have been associated with a bladder cancer [42,43]. This viral association raises the possibility that Schistosoma may interact with viruses to generate bladder cancer, although the viruses tested seem to account for, at most, only a small proportion of bladder cancer [42,43]. Taken together, these findings lend credence to the possibility that trematodes may have oncogenic effects in synergy with viral infection, much as P. falciparum contributes to Burkitt's lymphoma in synergy with EBV.
Other chronic diseases of uncertain cause
This same need for broad analysis applies across the spectrum of chronic diseases associated with immunological damage. Systemic lupus erythomatosus (SLE), for example, appears to be almost always associated with EBV infection. A causal hypothesis for this association emphasizes that SLE is an antibody complex disease and that EBV causes uncontrolled proliferation of B cells by facilitating cellular replication, inhibiting apoptosis and facilitating telomerase expression. Persistent EBV infection of B cells that are specific for antibodies involved in SLE antibody complexes could thus be essential for SLE pathogenesis.
The pathology of multiple sclerosis involves destruction of the myelin basic protein that envelops and insulates axons. Inflammation, destruction by T cells and antibody attachment all seem to participate, and the hygiene hypothesis has been invoked [44]. As is the case with SLE, however, multiple sclerosis is almost always associated with EBV infection. Multiple sclerosis has also been associated with C. pneumoniae by two different groups of investigators [45–47], although a C. pneumoniae-specific peptide cross-reacts serologically with a portion of myelin basic protein and causes a multiple-sclerosis-like disease in rats [22]. This raises the possibility that persistent infection may persistently stimulate immunological damage, with C. pneumoniae inducing immune-mediated damage, and EBV dysregulating the immune response to enhance this damage to the point of clinical illness.
Like multiple sclerosis, the pathogenesis of sporadic Alzheimer's disease and atherosclerosis are associated with inflammation and with C. pneumoniae. Controversy centres on whether inflammation depends upon persistent infection. The most pervasive known genetic influence for both diseases (as well as multiple sclerosis) is the epsilon 4 allele of the apolipoprotein E gene. Recent evidence indicates that entry of C. pneumoniae into cells is enhanced by attachment to the epsilon 4 protein. This connection adds strength to the hypothesis that persistent C. pneumoniae infection is a primary cause of both diseases through a mechanism that stimulates inflammatory damage.
Persistent infections are implicated similarly across the spectrum of common chronic diseases of uncertain cause that involve immune-mediated damage. Crohn's disease, various categories of arthritis and type 1 diabetes are some examples. Although the quality of evidence implicating persistent infectious causes varies, the imperfect state of knowledge indicates that a balanced analysis of causal hypotheses must consider persistent infections as well as infectious triggers.
Acknowledgments
The Rena Shulsky Foundation has generously supported the work on cancer as part of a project to develop a unified theory of oncogenesis.
Disclosure
The author has no conflicts of interest relevant to the content of this article.
References
- 1.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–60. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bremner SA, Carey IM, DeWilde S, et al. Infections presenting for clinical care in early life and later risk of hay fever in two UK birth cohorts. Allergy. 2008;63:274–83. doi: 10.1111/j.1398-9995.2007.01599.x. [DOI] [PubMed] [Google Scholar]
- 3.Okada H, Kuhn C, Feillet H, Bach J-F. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rook GAW. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Darwinian medicine and the ‘hygiene’ or ‘old friends’ hypothesis. Clin Exp Immunol. 2010;160:70–9. doi: 10.1111/j.1365-2249.2010.04133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Mutius. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Farm lifestyles and the hygiene hypothesis. Clin Exp Immunol. 2010;160:130–5. doi: 10.1111/j.1365-2249.2010.04138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Getts MT, Miller SD. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Triggering of autoimmune diseases by infections. Clin Exp Immunol. 2010;160:15–21. doi: 10.1111/j.1365-2249.2010.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ewald PW. Transmission modes and evolution of the parasitism–mutualism continuum. Ann NY Acad Sci. 1987;503:295–306. doi: 10.1111/j.1749-6632.1987.tb40616.x. [DOI] [PubMed] [Google Scholar]
- 8.Mosby's medical dictionary. 8th edn. St Louis, MO: Mosby; 2009. [Google Scholar]
- 9.Cochran GM, Ewald PW, Cochran KD. Infectious causation of disease: an evolutionary perspective. Perspect Biol Med. 2000;43:406–48. doi: 10.1353/pbm.2000.0016. [DOI] [PubMed] [Google Scholar]
- 10.Holt PG, van den Biggelaar A. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: The role of infections in allergy: atopic asthma as a paradigm. Clin Exp Immunol. 2010;160:22–6. doi: 10.1111/j.1365-2249.2010.04129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ball TM, Castro-Rodriguez JA, Griffith KA, et al. Siblings, day-care attendance, and the risk of asthma and wheezing during childhood. N Engl J Med. 2000;343:538–43. doi: 10.1056/NEJM200008243430803. [DOI] [PubMed] [Google Scholar]
- 12.Wilson MS, Maizels RM. Regulation of allergy and autoimmunity in helminth infection. Clin Rev Allergy Immunol. 2004;26:35–50. doi: 10.1385/CRIAI:26:1:35. [DOI] [PubMed] [Google Scholar]
- 13.Maizels RM, Balic A, Gomez-Escobar N, et al. Helminth parasites – masters of regulation. Immunol Rev. 2004;201:89–116. doi: 10.1111/j.0105-2896.2004.00191.x. [DOI] [PubMed] [Google Scholar]
- 14.Babu S, Blauvelt CP, Kumaraswami V, et al. Regulatory networks induced by live parasites impair both Th1 and Th2 pathways in patent lymphatic filariasis: implications for parasite persistence. J Immunol. 2006;176:3248–56. doi: 10.4049/jimmunol.176.5.3248. [DOI] [PubMed] [Google Scholar]
- 15.McIntire JJ, Umetsu SE, Macaubas C, et al. Immunology: hepatitis A virus link to atopic disease. Nature. 2003;425:576. doi: 10.1038/425576a. [DOI] [PubMed] [Google Scholar]
- 16.Umetsu DT, DeKruyff RH. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Microbes, apoptosis and TIM-1 in the development of asthma. Clin Exp Immunol. 2010;160:125–9. doi: 10.1111/j.1365-2249.2010.04136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stecher B, Robbiani R, Walker AW, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–89. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hooper LV. Do symbiotic bacteria subvert host immunity? Nat Rev Microbiol. 2009;7:367–74. doi: 10.1038/nrmicro2114. [DOI] [PubMed] [Google Scholar]
- 19.Ewald PW. Evolutionary biology and the treatment of signs and symptoms of infectious disease. J Theor Biol. 1980;86:169–76. doi: 10.1016/0022-5193(80)90073-9. [DOI] [PubMed] [Google Scholar]
- 20.Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity. 2009;31:368–76. doi: 10.1016/j.immuni.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Oldstone MB. Molecular mimicry, microbial infection, and autoimmune disease: evolution of the concept. Curr Top Microbiol Immunol. 2005;296:1–17. doi: 10.1007/3-540-30791-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lenz DC, Lu L, Conant SB, et al. A Chlamydia pneumoniae-specific peptide induces experimental autoimmune encephalomyelitis in rats. J Immunol. 2001;167:1803–8. doi: 10.4049/jimmunol.167.3.1803. [DOI] [PubMed] [Google Scholar]
- 23.Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol. 2009;155:1–15. doi: 10.1111/j.1365-2249.2008.03834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ascherio A, Munger KL. 99th Dahlem Conference on Infection, Inflammation and Chronic Inflammatory Disorders: Epstein–Barr virus and multiple sclerosis: epidemiological evidence. Clin Exp Immunol. 2010;160:120–4. doi: 10.1111/j.1365-2249.2010.04121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutherland ER, Martin RJ. Asthma and atypical bacterial infection. Chest. 2007;132:1962–6. doi: 10.1378/chest.06-2415. [DOI] [PubMed] [Google Scholar]
- 26.Agarwal A, Chander Y. Chronic Chlamydia pneumoniae infection and bronchial asthma: is there a link? Indian J Med Microbiol. 2008;26:338–41. doi: 10.4103/0255-0857.43567. [DOI] [PubMed] [Google Scholar]
- 27.Ahmadi Torshizi A, Tohidi M, Attaran D, et al. Role of Chlamydia pneumoniae infection in asthma in Northeast of Iran. Iran J Allergy Asthma Immunol. 2008;7:45–6. [PubMed] [Google Scholar]
- 28.Cosentini R, Tarsia P, Canetta C, et al. Severe asthma exacerbation: role of acute Chlamydophila pneumoniae and Mycoplasma pneumoniae infection. Respir Res. 2008;9:48. doi: 10.1186/1465-9921-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slattery ML, Fitzpatrick FA. Convergence of hormones, inflammation, and energy-related factors: a novel pathway of cancer etiology. Cancer Prev Res (Philadelphia, PA) 2009;2:922–30. doi: 10.1158/1940-6207.CAPR-08-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vennervald BJ, Polman K. Helminths and malignancy. Parasite Immunol. 2009;31:686–96. doi: 10.1111/j.1365-3024.2009.01163.x. [DOI] [PubMed] [Google Scholar]
- 31.Ewald PW. An evolutionary perspective on parasitism as a cause of cancer. Adv Parasitol. 2009;68:21–43. doi: 10.1016/S0065-308X(08)00602-7. [DOI] [PubMed] [Google Scholar]
- 32.Sage J, Miller AL, Pérez-Mancera PA, et al. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 33.Wagner S, Roemer K. Retinoblastoma protein is required for efficient colorectal carcinoma cell apoptosis by histone deacetylase inhibitors in the absence of p21Waf. Biochem Pharmacol. 2005;69:1059–67. doi: 10.1016/j.bcp.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 34.zur Hausen H. Weinheim: Wiley-VCH; 2006. Infections causing human cancer. [Google Scholar]
- 35.Holzinger F, Z'graggen K, Büchler MW. Mechanisms of biliary carcinogenesis: a pathogenetic multi-stage cascade towards cholangiocarcinoma. Ann Oncol. 1999;10(Suppl. 4):122–6. [PubMed] [Google Scholar]
- 36.Donato F, Gelatti U, Tagger A, et al. Intrahepatic cholangiocarcinoma and hepatitis C and B virus infection, alcohol intake, and hepatolithiasis: a case–control study in Italy. Cancer Causes Control. 2001;12:959–64. doi: 10.1023/a:1013747228572. [DOI] [PubMed] [Google Scholar]
- 37.Shaib YH, l-Serag HB, Nooka AK, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a hospital-based case–control study. Am J Gastroenterol. 2007;102:1016–21. doi: 10.1111/j.1572-0241.2007.01104.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhou YM, Yin ZF, Yang JM, et al. Risk factors for intrahepatic cholangiocarcinoma: a case–control study in China. World J Gastroenterol. 2008;14:632–5. doi: 10.3748/wjg.14.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee TY, Lee SS, Jung S, et al. Hepatitis B virus infection and intrahepatic cholangiocarcinoma in Korea: a case–control study. Am J Gastroenterol. 2008;103:1716–20. doi: 10.1111/j.1572-0241.2008.01796.x. [DOI] [PubMed] [Google Scholar]
- 40.Shaib Y, El-Serag H, Davila J, et al. Risk factors of intrahepatic cholangiocarcinoma in the United States: a case–control study. Gastroenterology. 2005;128:620–6. doi: 10.1053/j.gastro.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 41.Mostafa MH, Sheweita SA, O'Connor PJ. Relationship between schistosomiasis and bladder cancer. Clin Microbiol Rev. 1999;12:97–111. doi: 10.1128/cmr.12.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim KH, Kim YS. Analysis of p53 tumor suppressor gene mutations and human papillomavirus infection in human bladder cancers. Yonsei Med J. 1995;36:322–31. doi: 10.3349/ymj.1995.36.4.322. [DOI] [PubMed] [Google Scholar]
- 43.Moonen PM, Bakkers JM, Kiemeney LA, et al. Human papilloma virus DNA and p53 mutation analysis on bladder washes in relation to clinical outcome of bladder cancer. Eur Urol. 2007;52:464–8. doi: 10.1016/j.eururo.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 44.Fleming J, Fabry Z. The hygiene hypothesis and multiple sclerosis. Ann Neurol. 2007;61:85–9. doi: 10.1002/ana.21092. [DOI] [PubMed] [Google Scholar]
- 45.Sriram S, Mitchell W, Stratton C. Multiple sclerosis associated with Chlamydia pneumoniae infection of the CNS. J Long Form Workform. 1998;50:571–2. doi: 10.1212/wnl.50.2.571. [DOI] [PubMed] [Google Scholar]
- 46.Munger KL, Peeling RW, Hernan MA, et al. Infection with Chlamydia pneumoniae and risk of multiple sclerosis. Epidemiology. 2003;14:141–7. doi: 10.1097/01.EDE.0000050699.23957.8E. [DOI] [PubMed] [Google Scholar]
- 47.Munger KL, DeLorenze GN, Levin LI, et al. A prospective study of Chlamydia pneumoniae infection and risk of MS in two US cohorts. J Long Form Workform. 2004;62:1799–803. doi: 10.1212/01.wnl.0000125193.58601.2c. [DOI] [PubMed] [Google Scholar]