

Abstract
The molecular mechanisms involved in host–microbe interactions during the initial stages of infection are poorly understood. The bacteria-eating nematode Caenorhabditis elegans provides an opportunity to dissect host–microbe interactions in the context of the whole organism, using powerful genomic, genetic and cell-biological tools. Because of the evolutionary conservation of ancient innate host defences and bacterial virulence mechanisms, studies in C. elegans hold great promise to shed light on defences in higher organisms, including mammals. Additionally, C. elegans pathogenesis models provide a platform for the identification of novel classes of anti-infective compounds with therapeutic value.
Keywords: anti-microbial, C. elegans, defence, infection, innate immunity
Introduction
The first metazoans evolved in a world dominated by microbes. There is little doubt that an early requisite for metazoan survival was the acquisition of defensive immune systems to combat microbial infections. As metazoans evolved, their immune systems became increasingly sophisticated. However, many features of immune signalling pathways have been conserved during evolution, and as a result the immune systems of vertebrates are viewed as composites of immune systems that evolved in the invertebrates that existed before them. From this evolutionary perspective, significant insights into the human immune system can be learned from the study of invertebrate immunity. Concomitantly, microbes evolved increasingly sophisticated mechanisms to defend themselves against the metazoan immune response and to exploit chinks in the metazoan armour [1]. Thus, the study of invertebrate pathogenesis models provides new insights into the molecular basis of pathogenesis [1]. As Nobel laureate Thomas Cech famously put it, ‘Because all of biology is connected, one can often make a breakthrough with an organism that exaggerates a particular phenomenon, and later explore the generality’[2]. Here we describe the use of the nematode Caenorhabditis elegans to explore fundamental questions in host–pathogen interactions, with a focus on the mechanisms by which intestinal epithelial cells detect and combat microbial pathogens.
C. elegans is a simple model host to study host–microbe interactions
Nematodes evolved more 600 million years ago [1] and many soil-dwelling species, such as C. elegans, are ‘microbivores’, feeding mainly on a variety of bacterial species. From a microbial perspective, predation avoidance is a highly selected trait that has been postulated to be the evolutionary origin of a variety of virulence-related factors. An ensuing evolutionary arms race led to the evolution of defence mechanisms (immune systems) in microbivores to counteract the detrimental effects of feeding on potential pathogens. This arms race may also be the underlying mechanism leading to the establishment of stable symbiotic relationships such as those between gut microbiota and their human hosts. Soil bacteria that provided nutrients and new metabolic capabilities to primitive animals such as C. elegans may have been the evolutionary precursors to the metazoan microbiota.
C. elegans has been an important resource for biological exploration since its adoption in the 1970s. In the laboratory, C. elegans is simply propagated and maintained on agar plates with lawns of non-pathogenic Escherichia coli as food source [3]. Each adult animal (∼1 mm in length) produces ∼300 genetically identical progeny in its 3-day life cycle, facilitating the establishment and maintenance of large populations of animals. C. elegans is diploid and hermaphroditic, which is an advantage in genetic analysis, because individual hermaphroditic worms automatically self. Gene expression in C. elegans can be knocked down easily via RNA interference (RNAi) by simply feeding worms live E. coli expressing double-stranded RNAs (dsRNAs) corresponding to C. elegans genes (almost 90% of the genome is available as a dsRNA expression library). Transgenic C. elegans can be generated by microinjection of DNA into the adult gonad. C. elegans are transparent, greatly facilitating characterization of gene expression patterns and real-time observation of infectious processes, e.g. by green fluorescent protein (GFP) reporter expression. Moreover, all adult C. elegans have 959 cells, the developmental lineages of which have been traced completely to the fertilized egg.
Many bacterial and fungal pathogens of clinical importance cause intestinal infections in C. elegans that result in death of the animals [4]. C. elegans can be infected in the laboratory by transferring the animals from their normal food source (non-pathogenic E. coli) to agar plates containing lawns of the microbial pathogen that is being studied [3]. Ingestion of the pathogen leads to an intestinal infection characterized by the collapse of the intestinal epithelial cells, the proliferation (or accumulation) of the pathogenic microbe in the C. elegans alimentary tract and premature death of the infected animals. In addition to quantifying nematode survival, pathogenesis can be monitored by using standard microbiological methods to count the number of live microbes in the intestine, by observing the accumulation of the infecting microbes microscopically, or by monitoring changes in host gene expression using quantitative reverse transcription–polymerase chain reaction (qRT–PCR) or transgenic worms carrying GFP reporter constructs. In this review we focus upon recent advances in our understanding of the tissues and organs involved in host defence in C. elegans, as well as the virulence mechanisms employed by some pathogens to defeat those defences.
Guts, skin and brains – tissues involved in the host response
A major advantage of C. elegans as a model system is its relatively simple anatomy. The C. elegans body plan is tubular, with the mouth at the anterior end of the head and the anus at the posterior near the tail. The head contains the pharynx, a muscular organ that contracts rhythmically to pump food into the grinder, a chitinous rigid organ that crushes ingested material before it is pumped through the pharyngeal–intestinal valve into the lumen of the intestine [5].
The intestine proper, which takes up approximately one-third of the midbody transversal section, is a simple organ formed by just 20 non-renewable polarized epithelial cells, organized in nine rings of directly apposed pairs of cells (except for the first ring, which is formed by four cells). These intestinal epithelial cells exhibit many ultrastructural similarities with mammalian intestinal epithelial cells, most conspicuously an apical brush border of microvilli protruding into the intestinal lumen. The microvilli are formed of actin bundles anchored in an intermediate filament terminal web. The intestine is metabolically highly active, with similar functions to the fat body in flies and the liver in mammals [5].
Other major organs include the gonads, which fill up most of the transversal section of the animal and generate oocytes that are fertilized as they pass through the spermathecae near the ventral uterus. Fertilized eggs remain inside the animal until early embryogenesis, at which point they are laid through the ventral vulval opening.
The hypodermis (epidermis) and body wall muscle sheathe the intestine, the gonads and the body cavity (pseudocoelom). The body wall muscle contracts to generate the characteristic sinusoidal movements that allow locomotion and behaviour, co-ordinated by an intricate nervous system that links environmental sensory perception with movement, endocrine signalling and behaviour. The hypodermis, among other functions, deposits the highly impermeable cuticle, the collagenous exoskeleton of the worm.
C. elegans lacks a circulatory system, professional immune cells and macrophage-like phagocytes. Being an invertebrate, it lacks antibody-generating adaptive immunity and relies on epithelial-based innate immunity for defence. Nevertheless, C. elegans mounts a sophisticated immune response, as measured by transcriptional regulation of host defence genes upon infection. In contrast to what is known about flies and mammals, the C. elegans immune response is mostly independent of Toll-like receptor (TLR) signalling [6,7]. Moreover, the apparent lack of a nuclear factor kappa-B (NF-kB) homologue in the C. elegans genome has led to the conclusion that host defence is mediated by transcription factors that differ from the NF-kB/Relish family. The picture emerging from a series of recent studies is that of complex communication between organs to co-ordinate the host response to infection at a systemic level. What are the organs involved in the perception of and defence against infection? What signalling pathways are involved in each organ? What are the systemic signals involved in host defence?
Intestinal epithelium
Pathogen-mediated C. elegans killing correlates typically with accumulation of microorganisms in the intestinal lumen [4]. When C. elegans feeds on non-pathogenic E. coli there are few intact bacteria in the intestine, although this number increases with age – and, presumably, immunesenescence. In contrast, when feeding on pathogenic microbes, large quantities of intact pathogen cells accumulate in the intestinal lumen, which can become grossly distended [4]. A vast majority of pathogen response genes identified by transcriptional profiling of infected animals are expressed in the intestinal epithelium, suggesting that it is a major immune organ [8–10](J. E. Irazoqui, E. R. Troemel and F. M. Ausubel, unpublished). This mirrors recent data showing that mammalian intestinal epithelial cells sense the presence of bacteria and mount a defensive host response [11,12]. What signalling pathways act in the C. elegans intestine for the perception of and response to bacterial pathogens?
The first piece of the puzzle was identified in a forward genetic screen for mutants that exhibited shortened longevity on Pseudomonas aeruginosa (but not on non-pathogenic E. coli). This approach identified the NSY-1/SEK-1/PMK-1 p38 mitogen-activated protein kinase (MAPK) cascade as a key component of the C. elegans immune response [13,14]. NSY-1 (MAPKKK), SEK-1 (MAPKK) and PMK-1 (p38 MAPK) are the C. elegans orthologues of human ASK-1, MKK3/MKK6 and p38, respectively, that are involved in the mammalian cellular immune response [15]. As their counterparts in mammals, NSY-1, SEK-1 and PMK-1 function linearly in a phosphotransfer cascade (Fig. 1a) [13,14]. In insects and mammals the corresponding MAPK pathway acts downstream of TLRs, but the C. elegans TLR homologue TOL-1 does not appear to play a major role in the C. elegans immune response to most pathogens [6], although it is involved in conferring some resistance to Salmonella enterica[16]. Instead, the C. elegans p38 MAPK cascade functions downstream of TIR-1 [17], the only other C. elegans protein that contains a TIR (Toll, interleukin receptor) domain that is a hallmark of TLR-mediated signalling. TIR-1 is homologous to the human SARM protein that functions as a negative regulator of TIR domain-containing adaptor-inducing interferon β (TRIF)-dependent TLR signalling downstream of TLR-3 and TLR-4 [18]. In subsequent studies, the PMK-1 cascade was found to regulate intestinal gene induction in response to infection [19].
Fig. 1.
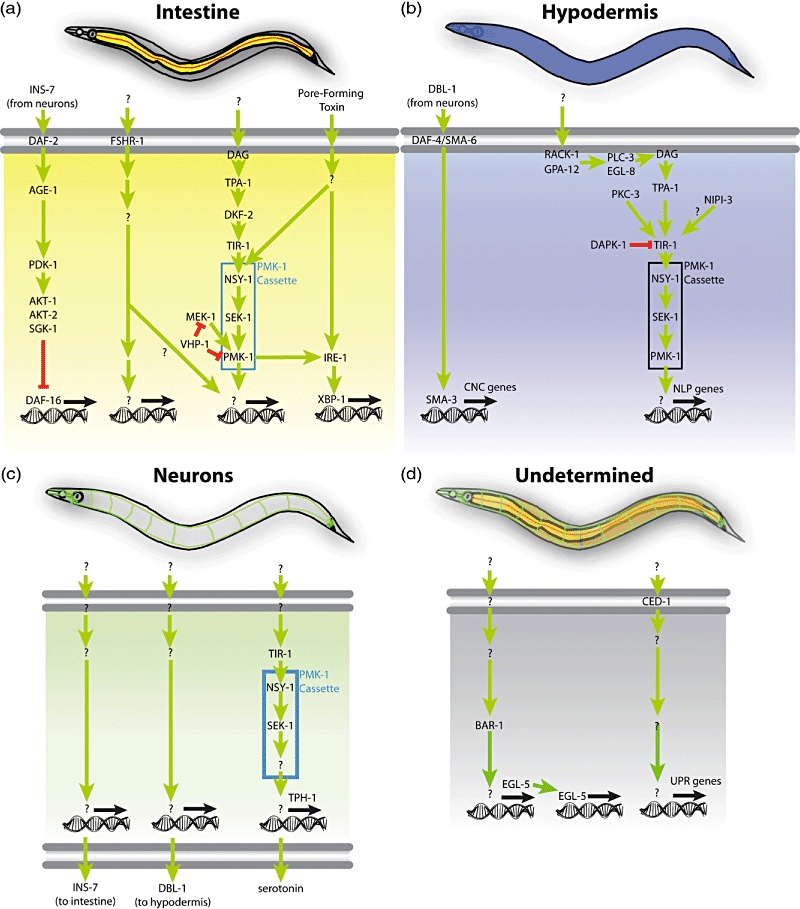
Diverse tissues participate in host defence in Caenorhabditis elegans. (a) The intestine (yellow) is a major immune organ. In it, insulin signalling regulates DAF-16/FOXO, a transcription factor involved in stress responses. A novel putative receptor, follicle stimulating hormone receptor (FSHR-1), acts in the intestine to presumably regulate signal transduction cascades that control the expression of pathogen-induced genes. A signalling pathway originating in diacylglycerol (DAG), produced presumably by phospholipase C, involves the protein kinase C homologue TPA-1 and the protein kinase D homologue DKF-2 to activate the PMK-1/p38 mitogen-activated protein kinase (MAPK) cascade that controls host defence genes. A parallel pathway, including MEK-1 and the protein phosphatase VHP-1, modulates PMK-1 activity. (b) In the hypodermis, an epidermal tissue in C. elegans, a non-canonical transforming growth factor (TGF)-β signalling pathway downstream of the heterodimeric TGF-β receptor formed by DAF-4 and SMA-6 regulates the SMAD homologue SMA-3 to control expression of caenacin genes (CNC) during infection with Drechmeria coniospora. In parallel, a pathway involving RACK-1 and GPA-12 (subunits of heterotrimeric G proteins), PLC-3 and EGL-8 (phospholipase C homologues), TPA-1 and PKC-3 (protein kinase C homologues), the Tribbles-like kinase NIPI-3 and the PMK-1 cassette regulates the expression of neuropeptide-like peptides (NLP) also during D. coniospora infection. In this pathway, the death-associated protein kinase (DAPK-1) regulates the PMK-1 cascade negatively at the level of or upstream of the protein scaffold TIR-1 (Toll, interleukin receptor). (c) In neurones, secretion of insulin INS-7 down-regulates DAF-16 in the intestine. Secretion of TGF-β DBl-1 regulates the SMA-3 in the hypodermis. Also, a poorly understood pathway including TIR-1, NSY-1 and SEK-1 regulates the production of serotonin via TPH-1; this PMK-1-independent pathway thus controls the aversive learning behaviour following infection. (d) It remains unknown where the BAR-1/β-catenin–EGL-5/HOX pathway controlling host response gene induction is required. Similarly, the locus of CED-1 activity for the induction of non-canonical unfolded protein response (UPR) genes remains to be determined.
How do the intestinal cells connect events occurring at the cell surface (the apical side facing the intestinal lumen) with the PMK-1 cascade, to regulate gene transcription in the nucleus? Recent work identified upstream components involved in PMK-1 regulation in the intestine [20–23]. One candidate upstream component is the leucine-rich repeat (LRR)-containing G-protein-coupled receptor (GPCR) follicle-stimulating hormone receptor (FSHR-1), which was identified in a limited reverse genetic screen of 14 candidate transmembrane LRR receptors in C. elegans. RNAi directed against fshr-1 results in a high degree of susceptibility to killing by P. aeruginosa, Staphylococcus aureus and Enterococcus faecalis, but not in a reduced lifespan during infection by non-pathogenic E. coli[24]. Expression of FSHR-1 in intestinal cells is necessary and sufficient for its role in innate immunity.
Genetic analysis indicates that FSHR-1 functions in the intestine in a separate pathway from PMK-1 and DAF-2, the worm insulin receptor that is involved in stress responses (see below) [24]. Further, qRT–PCR analysis shows that FSHR-1 and the PMK-1/p38 MAPK cassette regulate the induction of overlapping, but non-identical, sets of P. aeruginosa-induced genes. Although transcriptional profiling data suggest that FSHR-1 regulates host response genes independently of PMK-1, it is unclear whether the PMK-1/p38 MAPK cassette may be involved partially in signal transduction downstream of FSHR-1 [24]. It is also possible that the FSHR-1 and PMK-1/p38 MAPK pathways function in parallel but converge on common sets of target genes in response to pathogen infection. How is FSHR-1 involved in mediating the C. elegans host response? We currently lack evidence that FSHR-1 can sense infection directly, for instance by binding pathogen-associated molecular patterns (PAMPs). FSHR-1 is the sole C. elegans LGR-type GPCR. In mammals the heterodimeric glycopeptide hormone FSHα/β is the canonical ligand for this class of GPCR. Worms do not have an identifiable FSHα subunit and the endogenous ligands, if any, have not been identified.
As an LGR-type receptor, one might expect FSHR-1 to transduce signals through heterotrimeric G-proteins in the intestinal cell. Recent findings implicate at least one heterotrimeric G-protein in signal transduction events upstream of PMK-1 in a different tissue, the hypodermis (see below). Whether this or other G-proteins mediate FSHR-1 signal transduction in the intestine remains unknown.
Recent findings show that the protein kinase Cδ (PKCδ) TPA-1 activates the protein kinase D DKF-2 upstream of PMK-1/p38 in the intestine [20]. The upstream signals that control TPA-1 activity remain unknown, although by analogy with other systems, a likely candidate is diacylglycerol (DAG, produced by phospholipase C). In addition to their roles in immune signalling, during development, NSY-1 and SEK-1, the upstream components of the PMK-1/p38 MAPK cascade, act downstream of UNC-43 (CaM-kinase II)-mediated Ca2+ signalling for the establishment of neuronal left–right asymmetry [25].
Recent data suggest a central role for the endoplasmic reticulum (ER) in the regulation of the C. elegans response to infection. During exposure to Cry5B, a pore-forming toxin from Bacillus thuringiensis that destroys the C. elegans intestinal epithelium [26], PMK-1 acts in the intestine to activate the canonical unfolded protein response (UPR), an ER stress response pathway [27]. Mutants defective in the UPR exhibit increased susceptibility to killing by Cry5B. Moreover, mutants defective in a non-canonical UPR exhibit increased susceptibility to killing by S. enterica, suggesting that the UPR is important for host defence against intestinal pathogenesis [28]. These results potentially imply the existence of a regulatory feedback loop: during infection, ER homeostasis may be affected by an unknown mechanism, possibly involving phospholipase C activation leading to the IP3-mediated release of Ca2+ from intracellular stores. The increased DAG (and, potentially, Ca2+) levels lead ultimately to PMK-1 activation, causing an up-regulation of the UPR. Increased UPR activity may be necessary to restore the altered balance in the ER, causing the levels of cytosolic Ca2+ to decrease and restoring PMK-1 activity to basal levels. However, several steps in this scenario remain hypothetical; unknowns include whether phospholipase C is activated during infection, how PMK-1 activates the UPRs, and whether Ca2+ levels change during infection and regulate PMK-1 activity in the intestine.
In addition to the complex PMK-1 pathway, C. elegans insulin signalling is involved in host defence. Loss of function of the insulin receptor DAF-2 triggers the constitutive activation of the downstream target transcription factor DAF-16 [29]. Activated DAF-16 drives the transcription of many target stress-response genes, including intestinal genes involved in anti-microbial responses [30–32]. As a result, daf-2 mutants exhibit DAF-16-dependent enhanced resistance to all pathogens tested to date. Somewhat surprisingly, however, DAF-16 is not normally activated during infection in wild-type animals, suggesting that the damage caused by pathogenesis in the intestine does not trigger DAF-16 activation [9,19,33,34]. The molecular basis of this observation is poorly understood, yet could result from insulin induction during infection with some pathogens [35] (e.g. P. aeruginosa, see below). The one noted exception is the recent description of DAF-16 activation during ‘conditioning’ of animals with attenuated enteropathogenic E. coli (EPEC), which renders animals more resistant to subsequent infection with virulent EPEC [36].
Hypodermis
The hypodermis, the C. elegans epidermis equivalent, was identified recently as an active immune organ. Certain insults, such as wounding with a needle or a laser, or infection by the fungal pathogen Drechmeria coniospora, disrupt the integrity of the hypodermis and trigger a host hypodermal response represented by anti-microbial genes that encode neuropeptide-like peptides (NLPs) and a family of closely related genes called caenacins (CNCs) [21,37]. The PMK-1/p38 MAPK cassette is required for NLP and CNC expression. Although the upstream signals that activate PMK-1 during wounding are unknown, the death-associated protein kinase DAPK-1 functions as an upstream negative regulator of PMK-1 for NLP induction in the hypodermis [22].
During infection and injury, upstream regulation of PMK-1 for NLP induction in the hypodermis involves not only TPA-1/PKCδ (as in the intestine), but also PKC-3/PKCι, EGL-8/PLC and PLC-3/PLC (phospholipase Cs), and GPA-12/Gα12 and RACK-1/GNB2L1/Gβ2 (heterotrimeric G protein subunits). During D. coniospora infection, NLP gene activation by the PMK-1 cassette involves NIPI-3 (related to human Tribbles-like kinase), a different upstream component from that involved in wounding [21,23]. Not all steps in this complex pathway are delineated clearly, although it appears that NIPI-3 acts upstream of, or parallel to, GPA-12/RACK-1 G protein, phospholipase C and PKC to activate PMK-1 [23]. The same study showed that DKF-2, which functions downstream of TPA-1 to regulate PMK-1 in the intestine (see above), is not required for PMK-1 activity in the hypodermis, and neither is its paralogue DKF-1 [23]. Thus, it is possible that TPA-1 regulates PMK-1 in the hypodermis either directly or through some unidentified kinase other than DKF-1 and -2.
CNC gene induction in the hypodermis during D. coniospora requires a non-canonical signalling pathway composed of the heterodimeric TGF-β receptor DAF-4/SMA-6 and the downstream signalling component SMA-3/SMAD. These genes function cell-autonomously in the hypodermis, responding to a DBl-1/TGF-β signal originating in the nervous system [7]. In contrast, NLP induction during infection does not require neurosecretion [23].
Nervous system
As mentioned in the previous section, DBl-1/TGF-β produced in neurones regulates the host response to D. coniospora in the hypodermis. It is unclear what the proximal trigger is that causes an up-regulation of DBl-1 in response to infection. The same can be said for all neuronally originated signals related to host defence. There are additional recent examples of the importance of the nervous system in systemic regulation of the host response to infection.
First, neural secretion is important for the host response. C. elegans mutants that lack dense-core vesicle secretion (and thus are unable to secrete polypeptide signalling molecules) exhibit enhanced resistance to P. aeruginosa intestinal infection [38]. The underlying mechanism appears to be the activation of the insulin-repressed FOXO transcription factor DAF-16: lack of neuronal secretion of insulin causes de-repression of DAF-16, leading to the transcription of anti-microbial genes [38]. In an interesting example of the complex interplay between host and microbe, P. aeruginosa infection causes increased production of insulin INS-7, leading to the down-regulation of DAF-16 during infection [35]. Exposure to an attenuated mutant of P. aeruginosa does not cause increased INS-7 production or DAF-16 repression, leading to the speculation that P. aeruginosa suppresses the host immune response actively [35]. Alternatively, it is also possible that perception of a chemical signal produced by wild-type P. aeruginosa (and absent from the mutant) causes an active host response involving repression of DAF-16, a general stress response factor, to allow for a more specific host response to P. aeruginosa infection. Further work is necessary to distinguish between these alternative hypotheses.
Similarly, recent work showed that mutants lacking the neuronal GPCR NPR-1, defective in oxygen perception, also exhibit defective host defences in response to P. aeruginosa and S. enterica[39]. This effect was suppressed by mutation of the neuronally expressed guanylate cyclase GCY-35 and its targets TAX-2 and TAX-4, subunits of an ion channel, suggesting that certain specific neurones are involved in repressing the host response to P. aeruginosa in the intestine. Data from a different group, however, challenged this interpretation, arguing that mutation of npr-1 had an indirect effect on host response genes due to behavioural avoidance of the pathogen. npr-1 mutants do not avoid P. aeruginosa, thereby spending more time on the pathogenic food and succumbing earlier to infection than their wild-type counterparts [40]. The resolution of this debate is likely to shed more light on neuroendocrine regulation of host responses to intestinal infection.
The upstream components of the PMK-1/p38 MAPK pathway NSY-1 and SEK-1 are required in the nervous system for the regulation of serotonin upon exposure to P. aeruginosa[41]. Intriguingly, PMK-1 itself is dispensable for this function. Serotonin biogenesis is important for pathogen-induced learning and behaviours (see below).
Pharynx
Aballay and co-workers recently found that S. enterica, which establishes intracellular infections in human epithelial cells, also invades the epithelial cells of the C. elegans pharynx [16]. Pharyngeal invasion is most significant in mutants defective in immune defences, such as ced-1 mutants, which are defective in host defence through a non-canonical UPR pathway expressed in the pharynx, and tol-1 mutants, which have been shown to be slightly defective in the induction of anti-microbial peptide abf-2[16,28]. Thus, CED-1 and TOL-1 function upstream of pharyngeal host defence pathways, but whether they act cell-autonomously in the pharynx remains unknown. Other unresolved issues include the identity of the signalling pathways involved in pathogen detection in the pharynx, and the mechanisms responsible for pharyngeal host defence.
Rectal epithelial cells
Infection with Microbacterium nematophilum causes a remarkable swelling response in the anal region, a phenotype that has been termed ‘deformed anal region’ (Dar). Recent work revealed that Dar is an enlargement of rectal epithelial cells K/K′, F, U and B [42]. Genetic analysis has shown that host-encoded sugar transporters and acyltransferases are required for microbial attachment to the anus and induction of the Dar phenotype [43]. In addition, the swelling response requires an extracellular-regulated kinase (ERK) signalling pathway, as does inflammation in mammalian cells [43,44]. These results provided a cellular explanation for the Dar phenotype, and revealed for the first time a role for the rectal epithelium in the host response to infection. Interestingly, forward genetic screens for mutants defective in the swelling response to M. nematophilum identified the HOX gene egl-5. EGL-5 is required in the rectal epithelial cells for the transcription of the ERK homologous gene mpk-1[45]. S. aureus infection also causes a swelling response in the anal region, although in this case the involvement of the rectal epithelial cells is still conjecture. Despite having a defective transcriptional host response to S. aureus infection, egl-5 mutants are not defective in the swelling response to S. aureus[9]. In contrast, the β-catenin gene bar-1, which acts upstream of egl-5 during Wnt signal transduction, is required both for the swelling response and the transcriptional host response to S. aureus infection (J. E. Irazoqui and F. M. Ausubel, unpublished). Thus, even if the same cells were involved in the responses to M. nematophilum and S. aureus, the signalling pathways required for cell swelling are distinct. Further work is required to identify the components of each different pathway.
Rectal gland
Several genes induced during infection with S. aureus or P. aeruginosa are expressed in the rectal gland, a group of cells directly apposed to the rectum that are thought to secrete molecules into the rectal lumen [9,10] (J. E. Irazoqui and F. M. Ausubel, unpublished). This is consistent with a potential role for rectal gland cells in secretion of immune defence molecules into the rectal lumen. Further study is required to test this hypothesis.
Coelomocytes
Although it is clear that C. elegans lacks a bona fide circulatory system with sessile professional phagocytes, C. elegans does have phagocytes that reside in its body cavity, the pseudocoelom. Three pairs of static coelomocytes are located in ventral anterior, ventral posterior and dorsal posterior locations, where they constitutively endocytose pseudocoelomic fluid [46]. The coelomocytes have been proposed to function in immune surveillance, although direct experimental evidence is lacking [46].
Barriers – the cuticle
The collagenous cuticle that encases the C. elegans body provides a highly impermeable physical barrier with the environment. However, some bacteria have learned to exploit this surface to their advantage. Forward genetic analysis has identified components of the cuticle required for M. nematophilum binding and for Yersinia biofilm formation [47,48].
In summary, recent data highlight the involvement and co-ordination of several different organs and tissues during infection with distinct pathogens. Although exactly which organs are involved in all the infection models currently used remains unclear, it is likely that C. elegans benefits from a large arsenal of signalling pathways that function tissue-specifically to produce a physiologically co-ordinated, organism-wide and pathogen-tailored host response to infection.
Higher-order defences: learning to flee
Behavioural avoidance of pathogens is critical for survival in the soil. C. elegans are able to associate chemosensory cues with pathogenesis, and learn to avoid pathogenic bacteria. Avoidance of S. marcescens was shown to require TOL-1, although the mechanism of TOL-1 function for avoidance is unknown [6]. Subsequently, work with P. aeruginosa showed that exposure to the pathogen causes aversive olfactory learning mediated by serotonin signalling [49]. It is likely that other pathogenic bacteria also induce conditioned taste avoidance in C. elegans, although different pathogens (and even different strains of a specific pathogen) may differ in the chemical cues used by C. elegans to sense imminent danger. It is also possible that natural pathogens of C. elegans have evolved strategies to avoid detection as such, or even attract nematodes to a smelly death trap. The characterization of signalling pathways and mechanisms involved in pathogen avoidance in C. elegans has just begun, as in the case of NPR-1 mentioned previously. Further studies will probably shed more light on this matter.
Insights into bacterial virulence
Many pathogen mutations that reduce pathogenesis in mammalian hosts also result in diminished killing of C. elegans. These virulence factors include two-component regulators (gacA/gacS of P. aeruginosa, phoP/phoQ of S. typhimurium), quorum-sensing systems (lasR of P. aeruginosa, agr of S. aureus, fsr of E. faecalis), and alternative sigma factors (rpoN of P. aeruginosa, rpoS of S. typhimurium, and σB of S. aureus). These results showcase C. elegans as a host in which to identify novel pathogen virulence factors required for mammalian pathogenicity. Indeed, our laboratory, for example, has used the C. elegans model to identify novel virulence factors in P. aeruginosa, E. faecalis, S. typhimurium, S. aureus and C. neoformans (see [50] and references therein).
Our laboratory has focused upon a highly virulent clinical P. aeruginosa isolate, strain PA14, which is capable of infecting and causing disease in a variety of model invertebrates including plants, nematodes, slime moulds and insects, in addition to mice [51]. Moreover, many PA14 virulence factors that are important for pathogenesis in these simple hosts are also important virulence factors in mammalian hosts [50], suggesting that the underlying mechanisms of pathogenesis have been conserved, irrespective of the host. P. aeruginosa PA14 kills worms by both infection-associated killing and intoxication [52,53]. The latter mode of pathogenicity does not require live bacteria once the toxins have been released. The role of low molecular weight toxins in pathogenesis is poorly understood, in part because many pathogens such as P. aeruginosa synthesize literally thousands of different metabolites. Interestingly, P. aeruginosa virulence appears to be multi-factorial and combinatorial, the result of a pool of pathogenicity related genes that interact in various combinations in different genetic backgrounds [54].
To facilitate genome-scale study of PA14, our laboratory constructed a non-redundant library of 5850 PA14 transposon mutants in which ∼75% of PA14 genes are represented by a single transposon insertion chosen from a comprehensive library of insertion mutants [55]. A public internet-accessible database (PATIMDB; http://ausubellab.mgh.harvard.edu/cgi-bin/pa14/home.cgi) was developed to facilitate construction, distribution and use of the library. In recent unpublished work, our laboratory has screened the PA14NR Set transposon library (5850 mutants representing about 4600 unique genes) for bacterial virulence factors that affect P. aeruginosa-mediated killing of C. elegans and approximately 100 genes have been identified that are now undergoing further study (R. Feinbaum, N. Liberatti and F. Ausubel, unpublished).
Natural C. elegans pathogens
C. elegans is also attacked by natural pathogens. Our laboratory identified Nematocida parisii, an intracellular microsporidian parasite in wild isolates of C. elegans that appears to evade known immune responses [56]. As mentioned previously, infection with the filamentous fungal pathogen D. coniospora, possibly through the vulva, leads to wounding of the hypodermis and whole body colonization [57]. The vulva is also the point of entry for a new subspecies of Leucobacter chromiireducens, a Gram-positive bacterium that forms uterine cysts, inducing a transcriptional host response and nematode death [58]. Continued study of these and other natural pathogens yet to be identified will probably illuminate the multiple strategies that have evolved to exploit weaknesses in host defence systems and, in the process, basic biological questions about the hosts themselves.
High-throughput screening for compounds that block pathogen-mediated killing of C. elegans
In addition to fundamental studies of innate immunity and pathogen virulence, C. elegans has been used in translational research designed to identify novel targets for new generation anti-microbial compounds. Although there is widespread awareness of an imperative to identify new classes of anti-microbials, the rate of new anti-microbial discovery is unlikely to meet the expected need for the foreseeable future [59]. C. elegans can be adapted for use in fully automated high-throughput screens (HTS) to identify novel low molecular weight compounds with anti-microbial or immune enhancing activity [60,61]. High-throughput screening is possible because C. elegans killing assays can be miniaturized and carried out in standard 384-well microtitre plates. These assays provide a unique opportunity to screen for small-molecule inhibitors of infection within the context of a whole animal host. Use of anti-infectives that do not kill bacteria, rather than traditional antibiotics, theoretically lifts the strong selective pressure for the evolution of resistance.
Our laboratory initially developed a manual liquid-based assay for identifying compounds that cure Enterococcus faecalis infection and used the assay to screen ∼6000 compounds in a proof-of-principle experiment [61]. We identified 18 compounds that cured the infection, having in vivo efficacious doses substantially lower than their in vitro minimal inhibitory concentrations (MIC). In contrast, the in vivo effective doses of traditional antibiotics such as tetracycline were much higher than their in vitro MICs. These data showed that, in contrast to traditional antibiotic screens, the C. elegans–E. faecalis curing assay identifies compounds that affect the virulence of the pathogen, that suppress pathogen survival in vivo or that enhance the immune response of the host. Because these latter compounds have activity in vivo only in the whole-animal assay, it provides proof-of-principle for a proposed drug discovery approach that exploits (and blocks) pathogen adaptation to host physiology.
Figure 2 illustrates a newly developed automated scoring assay that discriminates between live and dead worms [62]. The assay uses the fluorescent dye SYTOX that is excluded from living cells and tissues, but stains dead organisms, including C. elegans. To test the assay, a pilot screen of 33 931 small molecules and 3283 natural product extracts has been carried out using the C. elegans–E. faecalis infection model. Of these 37 214 compounds and extracts, 136 and 108 tested positive in primary and secondary screens, respectively. Of the 108 compounds, 28 were not previously known anti-microbials. Nine of these 21 compounds were able to promote nematode survival at concentrations lower than their MIC values in vitro, a hallmark of anti-infective compounds that could be targeting bacterial virulence or host immunity [62]. These nine compounds are now undergoing in-depth chemical and biological characterization.
Fig. 2.
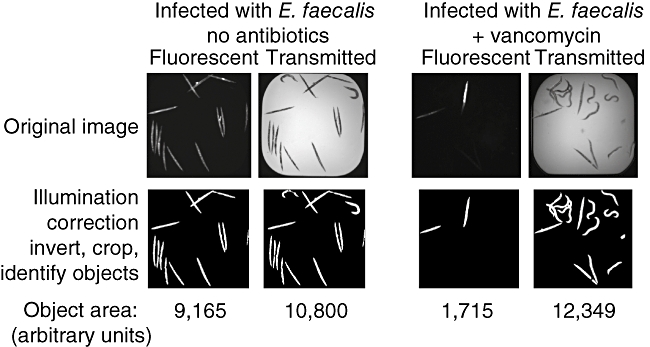
Automated high-throughput Caenorhabditis elegans curing assay in 384-well plates. Image analysis quantifies worm survival. Top row shows raw fluorescent SYTOX orange and bright field images captured from an untreated well and an antibiotic-treated well. The images were analysed using the image analysis software CellProfiler through a pipeline of 29 processing steps, which is shown in the bottom row. The total object area of the fluorescent and the bright field images are measured. The areas of the SYTOX orange objects and bright field objects are used to approximate the number of dead worms and the total number of worms, respectively. Worm survival is calculated as 1-(SYTOX orange area/total bright field worm area).
The future
The next couple of years will probably see fast progress in a number of areas related to host–pathogen interactions in C. elegans and beyond. In C. elegans, important areas that require further study include extensive characterization of the signalling networks that influence the outcome of infection and host response, and the cell types in which they function. At the whole organism level, different tissues and organ functions are co-ordinated during infection through systemic endocrine signals that remain to be delineated precisely. Further insight will be gained by precise examination of the actual mechanisms involved in pathogenesis for each pathogen type and infection process. Because the study of C. elegans immunity highlights the role of epithelial innate immunity, it is important to explore further the generality of such findings. How many features of C. elegans epithelial immunity will apply to mammals as well, and to what extent? What pathways were conserved, which were modified, and which lost altogether in the splits and radiations that constitute the evolutionary journey from the first multi-cellular metazoans to Homo sapiens?
In addition to gaining new insights into the evolution of innate immunity, there are practical benefits to be gained from these efforts. In the same way that the study of bacterial pathogens has provided important insights to mammalian biology, understanding the different host strategies selected throughout evolution to combat infection enhances our understanding of bacterial biology. The novel insights gained from these studies can be applied to the design of better therapeutic approaches, needed desperately in this age of rampant antibiotic resistance and human overpopulation.
Conversely, it is imperative that the molecular mechanisms used by pathogens to exploit their hosts be understood fully. The use of model hosts will be instrumental in understanding the molecular functions of virulence factors and their regulation during infection in vivo. C. elegans provides a means to test quickly hypotheses related to general features of host epithelial cells in a whole organism context, and identify the ‘Achilles heels’ that bacteria have evolved to exploit so expertly. Genetic and chemical screens can be performed to identify new ways to neutralize those poisoned arrows and the means to deploy them, thereby depriving pathogenic bacteria of the tools to cause infection and disease.
Disclosure
The authors declare no competing financial interests.
References
- 1.Waterfield NR, Wren BW, Ffrench-Constant RH. Invertebrates as a source of emerging human pathogens. Nat Rev Microbiol. 2004;2:833–41. doi: 10.1038/nrmicro1008. [DOI] [PubMed] [Google Scholar]
- 2.Cech T. Funding above and beyond scientific research. An interview with Thomas R. Cech, President of the Howard Hughes Medical Institute in Chevy Chase, MD. Interviewed by Holger Breithaupt. EMBO Rep. 2001;2:751–4. doi: 10.1093/embo-reports/kve192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powell JR, Ausubel FM. Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol Biol. 2008;415:403–27. doi: 10.1007/978-1-59745-570-1_24. [DOI] [PubMed] [Google Scholar]
- 4.Sifri CD, Begun J, Ausubel FM. The worm has turned – microbial virulence modeled in Caenorhabditis elegans. Trends Microbiol. 2005;13:119–27. doi: 10.1016/j.tim.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 5.McGhee JD. The C. elegans intestine. WormBook. 2007:1–36. doi: 10.1895/wormbook.1.133.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pujol N, Link EM, Liu LX, et al. A reverse genetic analysis of components of the Toll signaling pathway in Caenorhabditis elegans. Curr Biol. 2001;11:809–21. doi: 10.1016/s0960-9822(01)00241-x. [DOI] [PubMed] [Google Scholar]
- 7.Zugasti O, Ewbank JJ. Neuroimmune regulation of antimicrobial peptide expression by a noncanonical TGF-beta signaling pathway in Caenorhabditis elegans epidermis. Nat Immunol. 2009;10:249–56. doi: 10.1038/ni.1700. [DOI] [PubMed] [Google Scholar]
- 8.O'rourke D, Baban D, Demidova M, Mott R, Hodgkin J. Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum. Genome Res. 2006;16:1005–16. doi: 10.1101/gr.50823006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irazoqui JE, Ng A, Xavier RJ, Ausubel FM. Role for beta-catenin and HOX transcription factors in Caenorhabditis elegans and mammalian host epithelial–pathogen interactions. Proc Natl Acad Sci USA. 2008;105:17469–74. doi: 10.1073/pnas.0809527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alper S, McBride SJ, Lackford B, Freedman JH, Schwartz DA. Specificity and complexity of the Caenorhabditis elegans innate immune response. Mol Cell Biol. 2007;27:5544–53. doi: 10.1128/MCB.02070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host–microbial interface. Proc Natl Acad Sci USA. 2008;105:20858–63. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–30. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim DH, Feinbaum R, Alloing G, et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–6. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 14.Kim DH, Liberati NT, Mizuno T, et al. of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc Natl Acad Sci USA. 2004;101:10990–4. doi: 10.1073/pnas.0403546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol. 2008;48:199–225. doi: 10.1146/annurev.pharmtox.48.113006.094606. [DOI] [PubMed] [Google Scholar]
- 16.Tenor JL, Aballay A. A conserved Toll-like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep. 2008;9:103–9. doi: 10.1038/sj.embor.7401104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liberati NT, Fitzgerald KA, Kim DH, Feinbaum R, Golenbock DT, Ausubel FM. Requirement for a conserved Toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc Natl Acad Sci USA. 2004;101:6593–8. doi: 10.1073/pnas.0308625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carty M, Goodbody R, Schröder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006;7:1074–81. doi: 10.1038/ni1382. [DOI] [PubMed] [Google Scholar]
- 19.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genetics. 2006;2:e183. doi: 10.1371/journal.pgen.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren M, Feng H, Fu Y, Land M, Rubin CS. Protein kinase D is an essential regulator of C. elegans innate immunity. Immunity. 2009;30:521–32. doi: 10.1016/j.immuni.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pujol N, Cypowyj S, Ziegler K, et al. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr Biol. 2008;18:481–9. doi: 10.1016/j.cub.2008.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong A, Lynn G, Ngo V, et al. Negative regulation of Caenorhabditis elegans epidermal damage responses by death-associated protein kinase. Proc Natl Acad Sci USA. 2009;106:1457–61. doi: 10.1073/pnas.0809339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziegler K, Kurz CL, Cypowyj S, et al. Antifungal innate immunity in C. elegans: PKCdelta links G protein signaling and a conserved p38 MAPK cascade. Cell Host Microbe. 2009;5:341–52. doi: 10.1016/j.chom.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Powell JR, Kim DH, Ausubel FM. The G protein-coupled receptor FSHR-1 is required for the Caenorhabditis elegans innate immune response. Proc Natl Acad Sci USA. 2009;106:2782–7. doi: 10.1073/pnas.0813048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka-Hino M, Sagasti A, Hisamoto N, et al. SEK-1 MAPKK mediates Ca2+ signaling to determine neuronal asymmetric development in Caenorhabditis elegans. EMBO Rep. 2002;3:56–62. doi: 10.1093/embo-reports/kvf001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borgonie G, Claeys M, Leyns F, Arnaut G, DE Waele D, Coomans A. Effect of nematicidal Bacillus thuringiensis strains on free-living nematodes : 2. Ultrastructural analysis of the intoxication process in Caenorhabditis elegans. Fundam Appl Nematol. 1996;19:407–14. [Google Scholar]
- 27.Bischof LJ, Kao C-Y, Los FCO, et al. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008;4:e1000176. doi: 10.1371/journal.ppat.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haskins KA, Russell JF, Gaddis N, Dressman HK, Aballay A. Unfolded protein response genes regulated by CED-1 are required for Caenorhabditis elegans innate immunity. Dev Cell. 2008;15:87–97. doi: 10.1016/j.devcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–22. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 30.McElwee JJ, Schuster E, Blanc E, Thomas JH, Gems D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J Biol Chem. 2004;279:44533–43. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- 31.Murphy CT, McCarroll SA, Bargmann CI, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–83. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 32.Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–7. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- 33.Miyata S, Begun J, Troemel ER, Ausubel FM. DAF-16-dependent suppression of immunity during reproduction in Caenorhabditis elegans. Genetics. 2008;178:903–18. doi: 10.1534/genetics.107.083923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans EA, Chen WC, Tan M-W. The DAF-2 insulin-like signaling pathway independently regulates aging and immunity in C. elegans. Aging Cell. 2008;7:879–93. doi: 10.1111/j.1474-9726.2008.00435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans EA, Kawli T, Tan M-W. Pseudomonas aeruginosa suppresses host immunity by activating the DAF-2 insulin-like signaling pathway in Caenorhabditis elegans. PLoS Pathog. 2008;4:e1000175. doi: 10.1371/journal.ppat.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anyanful A, Easley KA, Benian GM, Kalman D. Conditioning protects C. elegans from lethal effects of enteropathogenic E. coli by activating genes that regulate lifespan and innate immunity. Cell Host Microbe. 2009;5:450–62. doi: 10.1016/j.chom.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pujol N, Zugasti O, Wong D, et al. Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 2008;4:e1000105. doi: 10.1371/journal.ppat.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawli T, Tan MW. Neuroendocrine signals modulate the innate immunity of Caenorhabditis elegans through insulin signaling. Nat Immunol. 2008;9:1415–24. doi: 10.1038/ni.1672. [DOI] [PubMed] [Google Scholar]
- 39.Styer KL, Singh V, Macosko E, Steele SE, Bargmann CI, Aballay A. Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science. 2008;322:460–4. doi: 10.1126/science.1163673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy KC, Andersen EC, Kruglyak L, Kim DH. A polymorphism in npr-1 is a behavioral determinant of pathogen susceptibility in C. elegans. Science. 2009;323:382–4. doi: 10.1126/science.1166527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shivers RP, Kooistra T, Chu SW, Pagano DJ, Kim DH. Tissue-specific activities of an immune signaling module regulate physiological responses to pathogenic and nutritional bacteria in C. elegans. Cell Host Microbe. 2009;6:321–30. doi: 10.1016/j.chom.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodgkin J, Kuwabara PE, Corneliussen B. A novel bacterial pathogen, Microbacterium nematophilum, induces morphological change in the nematode C. elegans. Curr Biol. 2000;10:1615–18. doi: 10.1016/s0960-9822(00)00867-8. [DOI] [PubMed] [Google Scholar]
- 43.Gravato-Nobre MJ, Hodgkin J. The acyltransferase gene bus-1 exhibits conserved and specific expression in nematode rectal cells and reveals pathogen-induced cell swelling. Dev Dyn. 2008;237:3762–76. doi: 10.1002/dvdy.21792. [DOI] [PubMed] [Google Scholar]
- 44.Nicholas HR, Hodgkin J. The ERK MAP kinase cascade mediates tail swelling and a protective response to rectal infection in C. elegans. Curr Biol. 2004;14:1256–61. doi: 10.1016/j.cub.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 45.Nicholas HR, Hodgkin J. The C. elegans Hox gene egl-5 is required for correct development of the hermaphrodite hindgut and for the response to rectal infection by Microbacterium nematophilum. Dev Biol. 2009;329:16–24. doi: 10.1016/j.ydbio.2009.01.044. [DOI] [PubMed] [Google Scholar]
- 46.Fares H, Greenwald I. Genetic analysis of endocytosis in Caenorhabditis elegans: coelomocyte uptake defective mutants. Genetics. 2001;159:133–45. doi: 10.1093/genetics/159.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darby C, Chakraborti A, Politz SM, Daniels CC, Tan L, Drace K. Caenorhabditis elegans mutants resistant to attachment of Yersinia biofilms. Genetics. 2007;176:221–30. doi: 10.1534/genetics.106.067496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gravato-Nobre MJ, Nicholas HR, Nijland R, et al. Multiple genes affect sensitivity of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum. Genetics. 2005;171:1033–45. doi: 10.1534/genetics.105.045716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Lu H, Bargmann CI. Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature. 2005;438:179–84. doi: 10.1038/nature04216. [DOI] [PubMed] [Google Scholar]
- 50.Sifri CD, Begun J, Ausubel FM, Calderwood SB. Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect Immun. 2003;71:2208–17. doi: 10.1128/IAI.71.4.2208-2217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 52.Darby C, Cosma CL, Thomas JH, Manoil C. Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 1999;96:15202–7. doi: 10.1073/pnas.96.26.15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa–Caenorhabditis elegans pathogenesis model. Cell. 1999;96:47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 54.Lee DG, Urbach JM, Wu G, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006;7:R90. doi: 10.1186/gb-2006-7-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberati NT, Urbach JM, Miyata S, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci USA. 2006;103:2833–8. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Troemel ER, Félix M-A, Whiteman NK, Barrière A, Ausubel FM. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol. 2008;6:2736–52. doi: 10.1371/journal.pbio.0060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jansson HB. Adhesion of conidia of Drechmeria coniospora to Caenorhabditis elegans wild type and mutants. J Nematol. 1994;26:430–5. [PMC free article] [PubMed] [Google Scholar]
- 58.Muir RE, Tan MW. Virulence of Leucobacter chromiireducens subsp. solipictus to Caenorhabditis elegans: characterization of a novel host–pathogen interaction. Appl Environ Microbiol. 2008;74:4185–98. doi: 10.1128/AEM.00381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh C. Where will new antibiotics come from? Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 60.Breger J, Fuchs BB, Aperis G, Moy TI, Ausubel FM, Mylonakis E. Antifungal chemical compounds identified using a C. elegans pathogenicity assay. PLoS Pathog. 2007;3:e18. doi: 10.1371/journal.ppat.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moy TI, Ball AR, Anklesaria Z, Casadei G, Lewis K, Ausubel FM. Identification of novel antimicrobials using a live-animal infection model. Proc Natl Acad Sci USA. 2006;103:10414–19. doi: 10.1073/pnas.0604055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moy TI, Conery AL, Larkins-Ford J, et al. High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chem Biol. 2009;4:527–33. doi: 10.1021/cb900084v. [DOI] [PMC free article] [PubMed] [Google Scholar]